A Preliminary Study of Cu Exposure Effects upon Alzheimer’s Amyloid Pathology

 ,
, 
Abstract
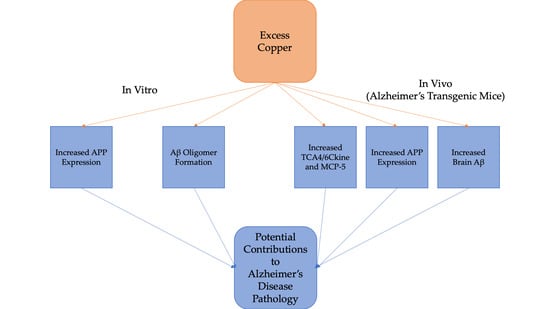
1. Introduction
2. Materials and Methods
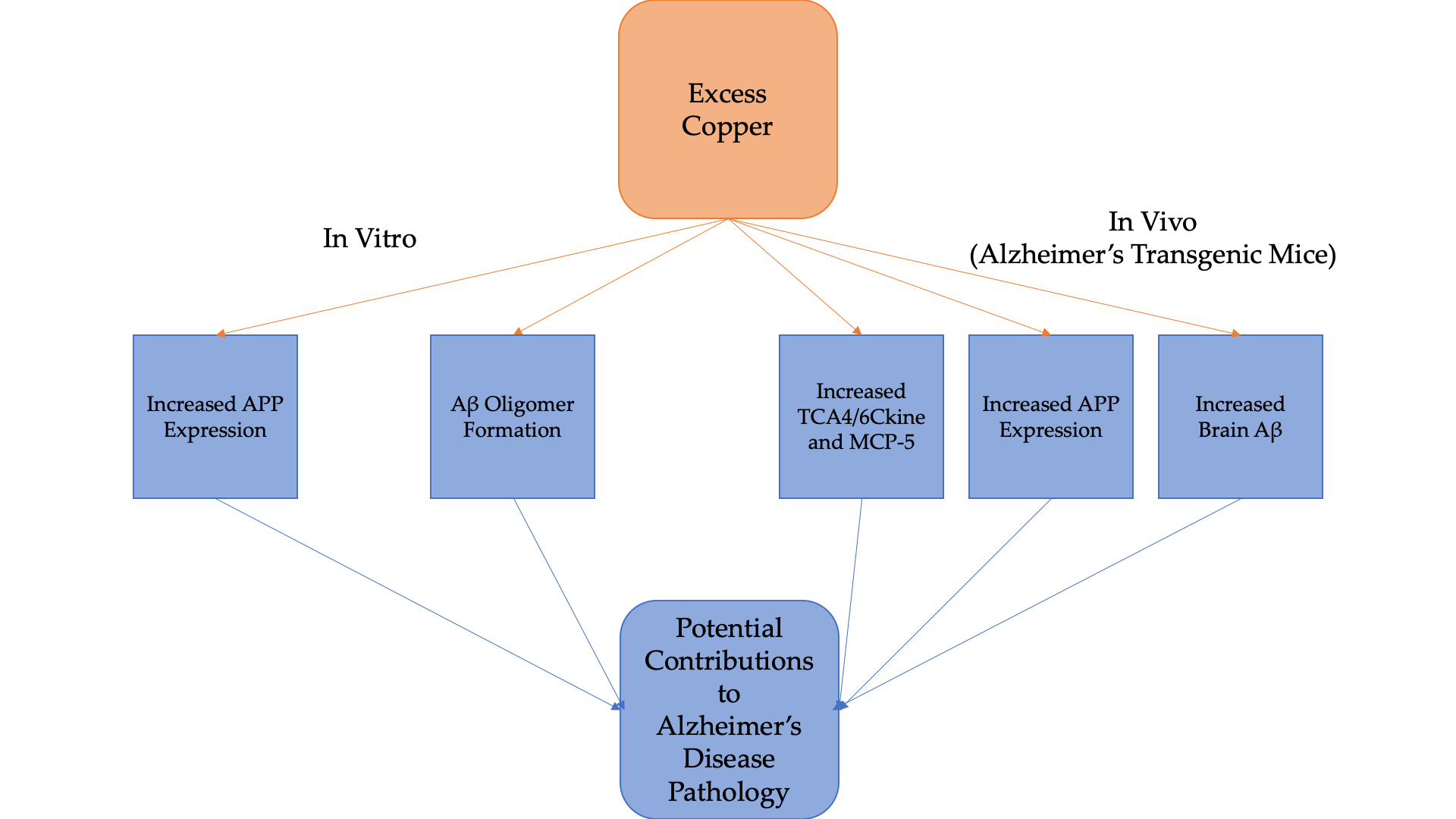
2.1. Effects of Cu and H2O2 upon Aβ oligomerization In Vitro
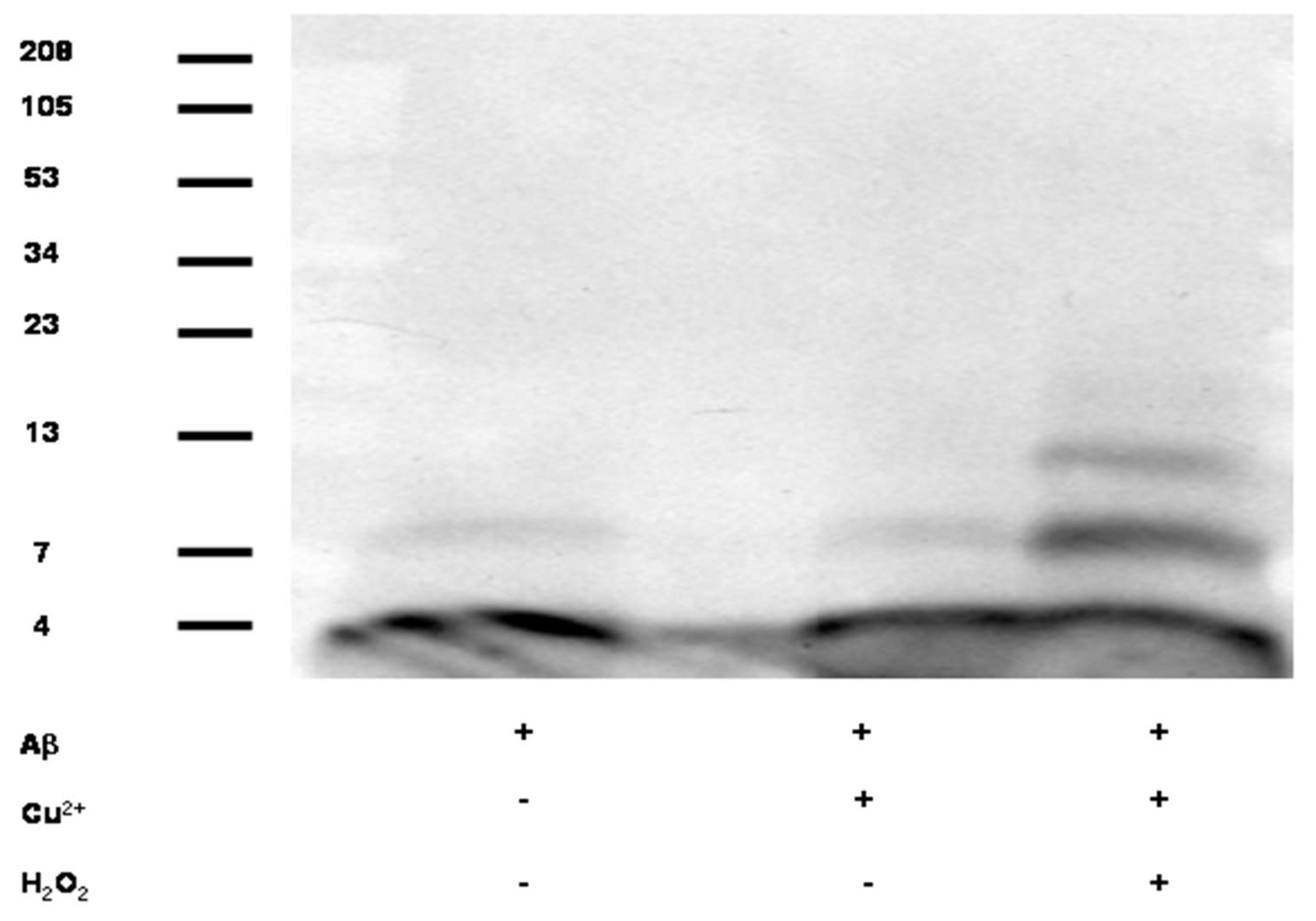
2.2. Effects of Cu Treatment Upon APP Expression
2.2.1. Effects of Cu Treatment Upon APP mRNA 5′UTR Translation
2.2.2. SDS-PAGE Analysis of Cu Effects on APP Protein Expression in Human SH-SY5Y Neuroblastoma Cells
2.3. Effects of Dietary Cu Exposure on Cerebral Aβ Amyloid Pathology in APP/PS1 Transgenic Mice
2.3.1. Treatment of APP/PS1 Transgenic Mice by Cu and Preparation of Mouse Brain Tissue Sections
2.3.2. Cerebral Aβ Measurements by ELISA
2.3.3. SDS-PAGE Analysis of Cu Effects on APP Protein Expression in The APP/PS1 Transgenic Mouse Brain Lysates
2.4. ICP-OES Analysis of Dietary Cu Effects on Cerebral Biometal Levels in APP/PS1 Transgenic Mice
2.5. Proteomic Analysis of Dietary Cu Effects on Cerebral pro-Inflammatory Cytokines in APP/PS1 Transgenic Mice
2.5.1. Data Collection Using Murine Cytokine Microarray
2.5.2. Data Analysis for Murine Cytokine Expression
3. Results
3.1. Cu/H2O2 Exacerbated Aβ Oligomerization In Vitro and Cu Treatment Increased APP Protein Expression Via Its mRNA 5′UTR Translation
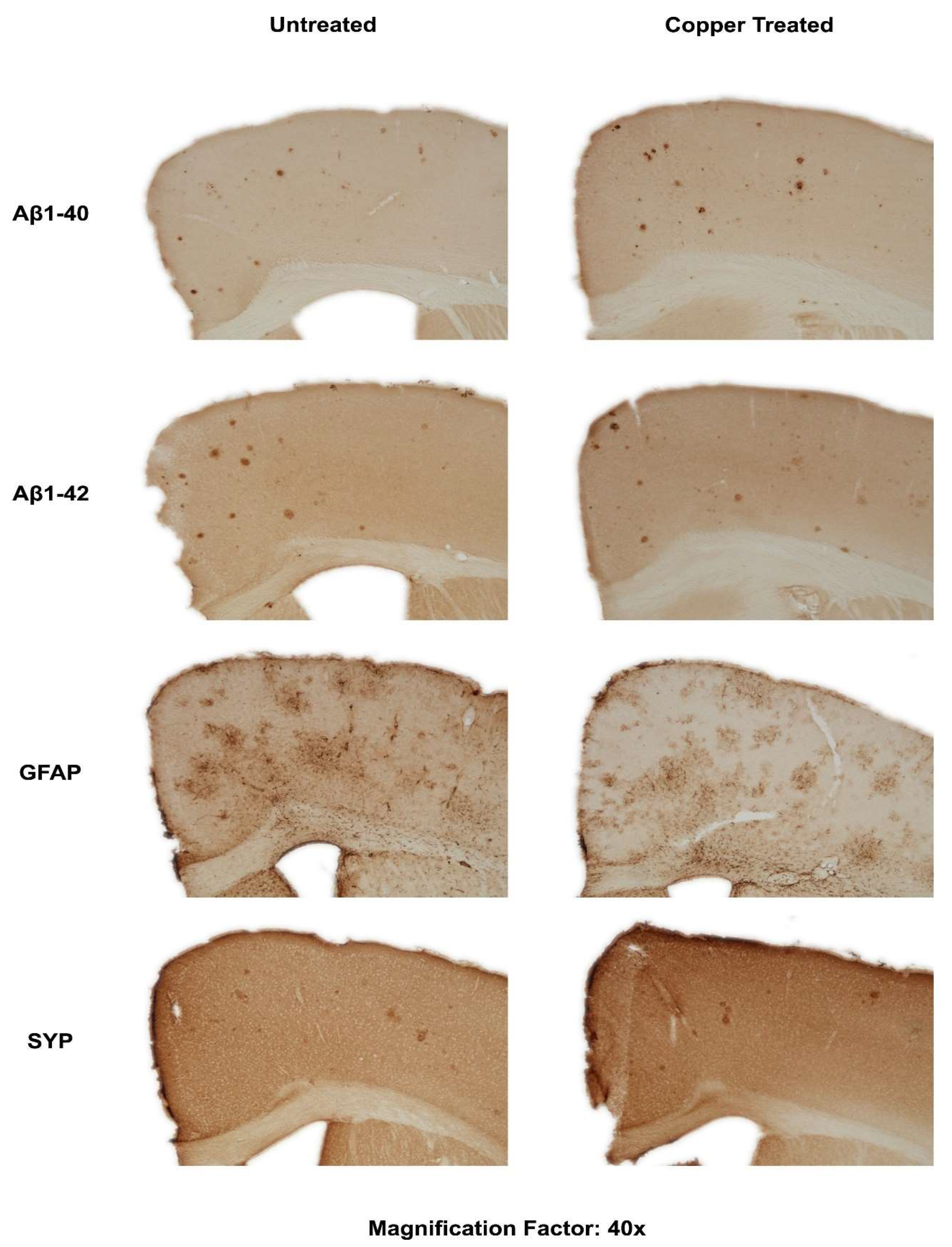
3.2. Dietary Cu Exposure Enhanced Cerebral Aβ Amyloid Pathology in APP/PS1 Transgenic Mice
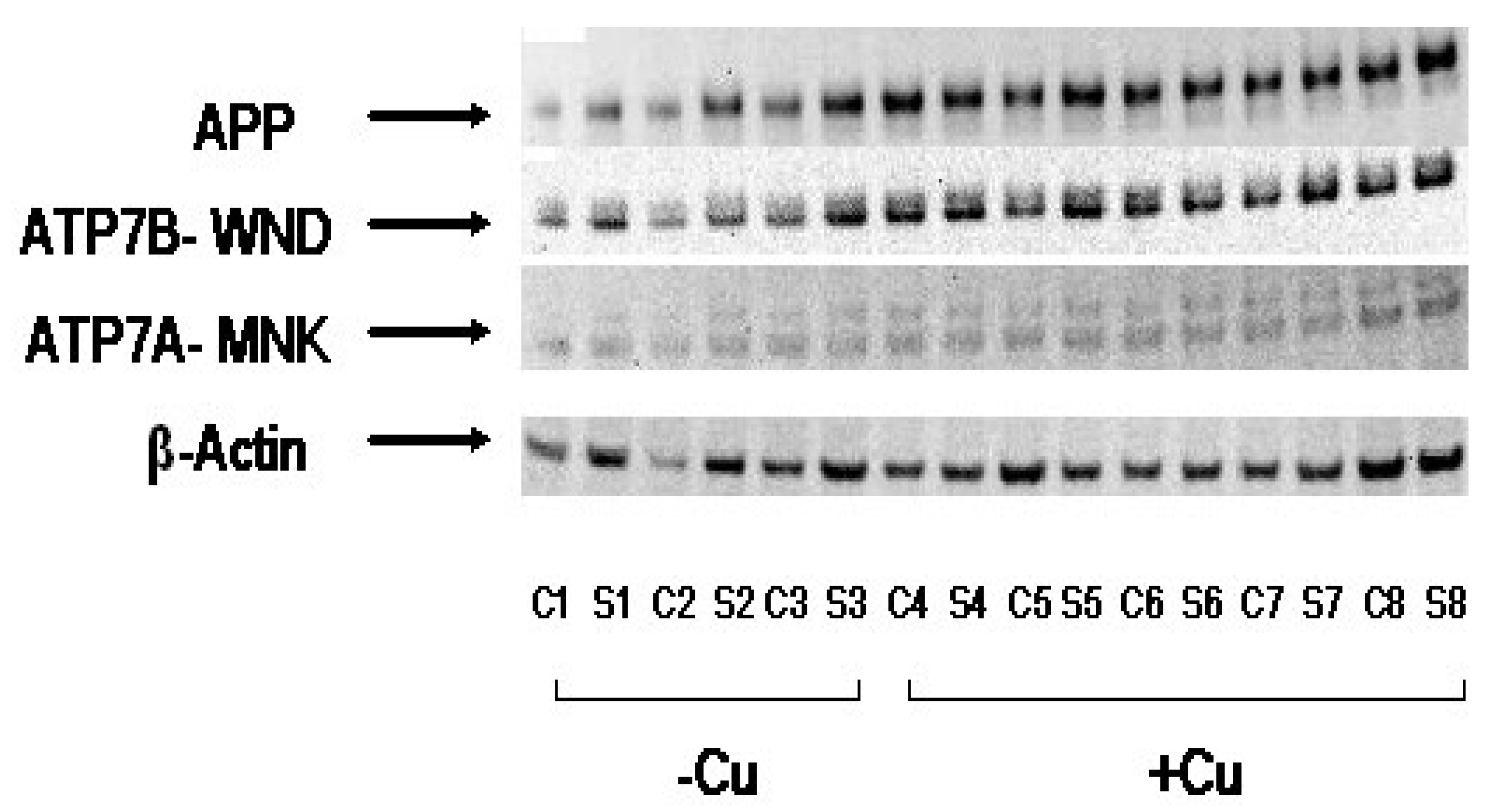
3.3. Cu Treatment Increased APP Protein Expression in Both APP/PS1 Transgenic Mouse Brain and Human SH-SY5Y Neuroblastoma cells
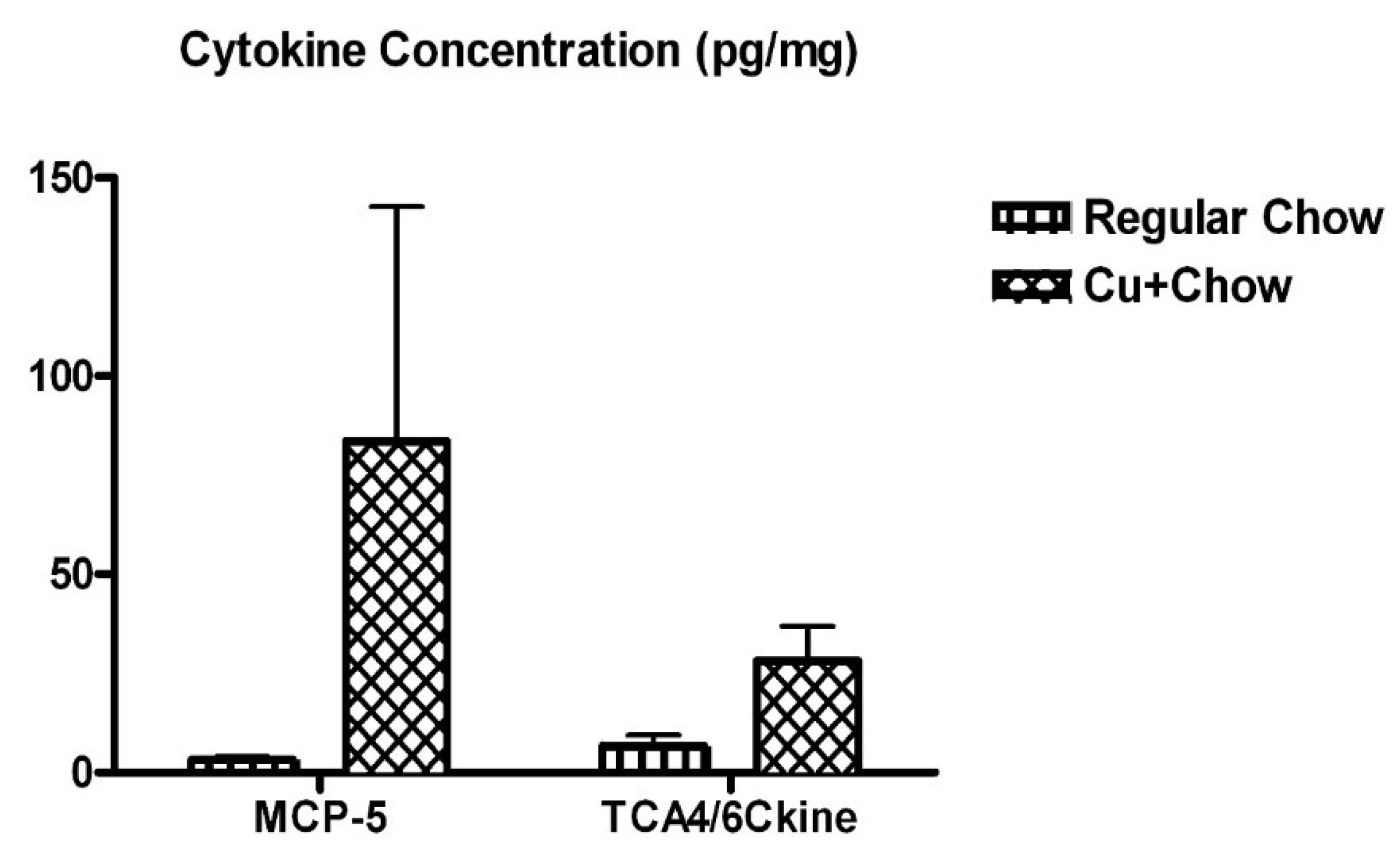
3.4. Dietary Cu Exposure Heightened Cerebral pro-Inflammatory Cytokines in APP/PS1 Transgenic Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | β-amyloid |
| AD | Alzheimer’s Disease |
| APP | Amyloid Precursor Protein |
| DMEM | Dulbecco’s Modified Eagle Medium |
| GFAP | Glial Fibrillary Acidic Protein |
| ICP-OES | Inductively Coupled Plasma Optical Emission Spectroscopy |
| SDS-PAGE | Sodium Dodecyl Sulphate-Polyacrylamide Gel Electrophoresis |
| SYP | Synaptophysin |
References
- Alzheimer’s-Association. 2019 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Koseoglu, E.; Koseoglu, R.; Kendirci, M.; Saraymen, R.; Saraymen, B. Trace metal concentrations in hair and nails from Alzheimer’s disease patients: Relations with clinical severity. J. Trace Elem. Med. Biol. 2017, 39, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Squitti, R.; Ghidoni, R.; Simonelli, I.; Ivanova, I.D.; Colabufo, N.A.; Zuin, M.; Benussi, L.; Binetti, G.; Cassetta, E.; Rongioletti, M.; et al. Copper dyshomeostasis in Wilson disease and Alzheimer’s disease as shown by serum and urine copper indicators. J. Trace Elem. Med. Biol. 2018, 45, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Vaz, F.N.C.; Fermino, B.L.; Haskel, M.V.L.; Wouk, J.; de Freitas, G.B.L.; Fabbri, R.; Montagna, E.; Rocha, J.B.T.; Bonini, J.S. The relationship between copper, iron, and selenium levels and alzheimer disease. Biol. Trace Elem. Res. 2018, 181, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Mercer, S.W.; Wang, J.; Burke, R. In vivo modeling of the pathogenic effect of copper transporter mutations that cause menkes and wilson diseases, motor neuropathy, and susceptibility to alzheimer’s disease. J. Biol. Chem. 2017, 292, 4113–4122. [Google Scholar] [CrossRef]
- Gerber, H.; Wu, F.; Dimitrov, M.; Garcia Osuna, G.M.; Fraering, P.C. Zinc and copper differentially modulate amyloid precursor protein processing by gamma-secretase and amyloid-beta peptide production. J. Biol. Chem. 2017, 292, 3751–3767. [Google Scholar] [CrossRef]
- Stelmashook, E.V.; Isaev, N.K.; Genrikhs, E.E.; Amelkina, G.A.; Khaspekov, L.G.; Skrebitsky, V.G.; Illarioshkin, S.N. Role of zinc and copper ions in the pathogenetic mechanisms of Alzheimer’s and Parkinson’s diseases. Biochemistry (Mosc) 2014, 79, 391–396. [Google Scholar] [CrossRef]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- James, S.A.; Churches, Q.I.; de Jonge, M.D.; Birchall, I.E.; Streltsov, V.; McColl, G.; Adlard, P.A.; Hare, D.J. Iron, copper, and zinc concentration in abeta plaques in the app/ps1 mouse model of alzheimer’s disease correlates with metal levels in the surrounding neuropil. ACS Chem. Neurosci. 2017, 8, 629–637. [Google Scholar] [CrossRef]
- Wang, H.; Wang, M.; Wang, B.; Li, M.; Chen, H.; Yu, X.; Yang, K.; Chai, Z.; Zhao, Y.; Feng, W. Immunogold labeling and X-ray fluorescence microscopy reveal enrichment ratios of Cu and Zn, metabolism of APP and amyloid-beta plaque formation in a mouse model of Alzheimer’s disease. Metallomics 2012, 4, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Cherny, R.A.; Legg, J.T.; McLean, C.A.; Fairlie, D.P.; Huang, X.; Atwood, C.S.; Beyreuther, K.; Tanzi, R.E.; Masters, C.L.; Bush, A.I. Aqueous dissolution of Alzheimer’s disease Abeta amyloid deposits by biometal depletion. J. Biol. Chem. 1999, 274, 23223–23228. [Google Scholar] [CrossRef] [PubMed]
- Cherny, R.A.; Atwood, C.S.; Xilinas, M.E.; Gray, D.N.; Jones, W.D.; McLean, C.A.; Barnham, K.J.; Volitakis, I.; Fraser, F.W.; Kim, Y.; et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 2001, 30, 665–676. [Google Scholar] [CrossRef]
- Robert, A.; Liu, Y.; Nguyen, M.; Meunier, B. Regulation of copper and iron homeostasis by metal chelators: A possible chemotherapy for Alzheimer’s disease. Acc. Chem. Res. 2015, 48, 1332–1339. [Google Scholar] [CrossRef]
- Li, D.D.; Zhang, W.; Wang, Z.Y.; Zhao, P. Serum copper, zinc, and iron levels in patients with alzheimer’s disease: A meta-analysis of case-control studies. Front. Aging Neurosci. 2017, 9, 300. [Google Scholar] [CrossRef]
- Drew, S.C. The case for abandoning therapeutic chelation of copper ions in alzheimer’s disease. Front. Neurosci. 2017, 11, 317. [Google Scholar] [CrossRef]
- Squitti, R.; Salustri, C.; Rongioletti, M.; Siotto, M. Commentary: The case for abandoning therapeutic chelation of copper ions in alzheimer’s disease. Front. Neurol. 2017, 8, 503. [Google Scholar] [CrossRef]
- Wang, P.; Wang, Z.Y. Metal ions influx is a double edged sword for the pathogenesis of Alzheimer’s disease. Ageing Res. Rev. 2017, 35, 265–290. [Google Scholar] [CrossRef]
- Prakash, A.; Dhaliwal, G.K.; Kumar, P.; Majeed, A.B. Brain biometals and Alzheimer’s disease—Boon or bane? Int. J. Neurosci. 2017, 127, 99–108. [Google Scholar] [CrossRef]
- Bush, A.I.; Pettingell, W.H.; Multhaup, G.; Paradis, M.D.; Vonsattel, J.P.; Gusella, J.F.; Beyreuther, K.; Masters, C.L.; Tanzi, R.E. Rapid induction of alzheimer a beta amyloid formation by zinc. Science 1994, 265, 1464–1467. [Google Scholar] [CrossRef]
- Huang, X.; Atwood, C.S.; Moir, R.D.; Hartshorn, M.A.; Vonsattel, J.P.; Tanzi, R.E.; Bush, A.I. Zinc-induced Alzheimer’s Abeta1–40 aggregation is mediated by conformational factors. J. Biol. Chem. 1997, 272, 26464–26470. [Google Scholar] [CrossRef] [PubMed]
- Atwood, C.S.; Moir, R.D.; Huang, X.; Scarpa, R.C.; Bacarra, N.M.; Romano, D.M.; Hartshorn, M.A.; Tanzi, R.E.; Bush, A.I. Dramatic aggregation of Alzheimer abeta by Cu(II) is induced by conditions representing physiological acidosis. J. Biol. Chem. 1998, 273, 12817–12826. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Atwood, C.S.; Hartshorn, M.A.; Multhaup, G.; Goldstein, L.E.; Scarpa, R.C.; Cuajungco, M.P.; Gray, D.N.; Lim, J.; Moir, R.D.; et al. The A beta peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 1999, 38, 7609–7616. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Cuajungco, M.P.; Atwood, C.S.; Hartshorn, M.A.; Tyndall, J.D.; Hanson, G.R.; Stokes, K.C.; Leopold, M.; Multhaup, G.; Goldstein, L.E.; et al. Cu(II) potentiation of alzheimer abeta neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. J. Biol. Chem. 1999, 274, 37111–37116. [Google Scholar] [CrossRef] [PubMed]
- Prosdocimi, T.; De Gioia, L.; Zampella, G.; Bertini, L. On the generation of OH(.) radical species from H2O2 by Cu(I) amyloid beta peptide model complexes: A DFT investigation. J. Biol. Inorg. Chem. 2016, 21, 197–212. [Google Scholar] [CrossRef]
- Cuajungco, M.P.; Goldstein, L.E.; Nunomura, A.; Smith, M.A.; Lim, J.T.; Atwood, C.S.; Huang, X.; Farrag, Y.W.; Perry, G.; Bush, A.I. Evidence that the beta-amyloid plaques of Alzheimer’s disease represent the redox-silencing and entombment of abeta by zinc. J. Biol. Chem. 2000, 275, 19439–19442. [Google Scholar] [CrossRef]
- Opazo, C.; Huang, X.; Cherny, R.A.; Moir, R.D.; Roher, A.E.; White, A.R.; Cappai, R.; Masters, C.L.; Tanzi, R.E.; Inestrosa, N.C.; et al. Metalloenzyme-like activity of Alzheimer’s disease beta-amyloid. Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H(2)O(2). J. Biol. Chem. 2002, 277, 40302–40308. [Google Scholar] [CrossRef]
- Atwood, C.S.; Perry, G.; Zeng, H.; Kato, Y.; Jones, W.D.; Ling, K.Q.; Huang, X.; Moir, R.D.; Wang, D.; Sayre, L.M.; et al. Copper mediates dityrosine cross-linking of Alzheimer’s amyloid-beta. Biochemistry 2004, 43, 560–568. [Google Scholar] [CrossRef]
- Al-Hilaly, Y.K.; Williams, T.L.; Stewart-Parker, M.; Ford, L.; Skaria, E.; Cole, M.; Bucher, W.G.; Morris, K.L.; Sada, A.A.; Thorpe, J.R.; et al. A central role for dityrosine crosslinking of Amyloid-β in Alzheimer’s disease. Acta Neuropathol. Commun. 2013, 1, 83. [Google Scholar] [CrossRef]
- Smith, M.A.; Perry, G.; Richey, P.L.; Sayre, L.M.; Anderson, V.E.; Beal, M.F.; Kowall, N. Oxidative damage in Alzheimer’s. Nature 1996, 382, 120–121. [Google Scholar] [CrossRef]
- Spisni, E.; Valerii, M.C.; Manerba, M.; Strillacci, A.; Polazzi, E.; Mattia, T.; Griffoni, C.; Tomasi, V. Effect of copper on extracellular levels of key pro-inflammatory molecules in hypothalamic GN11 and primary neurons. Neurotoxicology 2009, 30, 605–612. [Google Scholar] [CrossRef]
- Arnal, N.; Castillo, O.; de Alaniz, M.J.; Marra, C.A. Effects of copper and/or cholesterol overload on mitochondrial function in a rat model of incipient neurodegeneration. Int. J. Alzheimers Dis. 2013, 2013, 645379. [Google Scholar] [CrossRef] [PubMed]
- Bittner, H.J.; Guixa-Gonzalez, R.; Hildebrand, P.W. Structural basis for the interaction of the beta-secretase with copper. Biochim. Biophys. Acta 2018, 1860, 1105–1113. [Google Scholar] [CrossRef] [PubMed]
- Siblerud, R.; Mutter, J.; Moore, E.; Naumann, J.; Walach, H. A hypothesis and evidence that mercury may be an etiological factor in alzheimer’s disease. Int. J. Environ. Res. Public Health 2019, 16, 5152. [Google Scholar] [CrossRef] [PubMed]
- Bakulski, K.M.; Rozek, L.S.; Dolinoy, D.C.; Paulson, H.L.; Hu, H. Alzheimer’s disease and environmental exposure to lead: The epidemiologic evidence and potential role of epigenetics. Curr. Alzheimer Res. 2012, 9, 563–573. [Google Scholar] [CrossRef]
- Wallin, C.; Sholts, S.B.; Österlund, N.; Luo, J.; Jarvet, J.; Roos, P.M.; Ilag, L.; Gräslund, A.; Wärmländer, S.K.T.S. Alzheimer’s disease and cigarette smoke components: Effects of nicotine, PAHs, and Cd(II), Cr(III), Pb(II), Pb(IV) ions on amyloid-β peptide aggregation. Sci. Rep. 2017, 7, 14423. [Google Scholar] [CrossRef]
- Ashok, A.; Rai, N.K.; Tripathi, S.; Bandyopadhyay, S. Exposure to As-, Cd-, and Pb-mixture induces Abeta, amyloidogenic APP processing and cognitive impairments via oxidative stress-dependent neuroinflammation in young rats. Toxicol. Sci. 2015, 143, 64–80. [Google Scholar] [CrossRef]
- Rogers, J.T.; Randall, J.D.; Cahill, C.M.; Eder, P.S.; Huang, X.; Gunshin, H.; Leiter, L.; McPhee, J.; Sarang, S.S.; Utsuki, T.; et al. An iron-responsive element type II in the 5’-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J. Biol. Chem. 2002, 277, 45518–45528. [Google Scholar] [CrossRef]
- Multhaup, G.; Schlicksupp, A.; Hesse, L.; Beher, D.; Ruppert, T.; Masters, C.L.; Beyreuther, K. The amyloid precursor protein of Alzheimer’s disease in the reduction of copper(II) to copper(I). Science 1996, 271, 1406–1409. [Google Scholar] [CrossRef]
- Colangelo, V.; Schurr, J.; Ball, M.J.; Pelaez, R.P.; Bazan, N.G.; Lukiw, W.J. Gene expression profiling of 12633 genes in Alzheimer hippocampal CA1: Transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inflammatory signaling. J. Neurosci. Res. 2002, 70, 462–473. [Google Scholar] [CrossRef]
- Uchida, Y.; Takio, K.; Titani, K.; Ihara, Y.; Tomonaga, M. The growth inhibitory factor that is deficient in the Alzheimer’s disease brain is a 68 amino acid metallothionein-like protein. Neuron 1991, 7, 337–347. [Google Scholar] [CrossRef]
- Yu, W.H.; Lukiw, W.J.; Bergeron, C.; Niznik, H.B.; Fraser, P.E. Metallothionein III is reduced in Alzheimer’s disease. Brain Res. 2001, 894, 37–45. [Google Scholar] [CrossRef]
- Crapper McLachlan, D.R.; Dalton, A.J.; Kruck, T.P.A.; Bell, M.Y.; Smith, W.L.; Kalow, W.; Andrews, D.F. Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 1991, 337, 1304–1308. [Google Scholar] [CrossRef]
- Guo, C.; Wang, T.; Zheng, W.; Shan, Z.Y.; Teng, W.P.; Wang, Z.Y. Intranasal deferoxamine reverses iron-induced memory deficits and inhibits amyloidogenic APP processing in a transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 562–575. [Google Scholar] [CrossRef]
- De Lima, M.N.; Presti-Torres, J.; Caldana, F.; Grazziotin, M.M.; Scalco, F.S.; Guimaraes, M.R.; Bromberg, E.; Franke, S.I.; Henriques, J.A.; Schroder, N. Desferoxamine reverses neonatal iron-induced recognition memory impairment in rats. Eur. J. Pharmacol. 2007, 570, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Fine, J.M.; Forsberg, A.C.; Stroebel, B.M.; Faltesek, K.A.; Verden, D.R.; Hamel, K.A.; Raney, E.B.; Crow, J.M.; Haase, L.R.; Knutzen, K.E.; et al. Intranasal deferoxamine affects memory loss, oxidation, and the insulin pathway in the streptozotocin rat model of Alzheimer’s disease. J. Neurol. Sci. 2017, 380, 164–171. [Google Scholar] [CrossRef]
- Regland, B.; Lehmann, W.; Abedini, I.; Blennow, K.; Jonsson, M.; Karlsson, I.; Sjogren, M.; Wallin, A.; Xilinas, M.; Gottfries, C.G. Treatment of Alzheimer’s disease with clioquinol. Dement. Geriatr. Cogn. Disord. 2001, 12, 408–414. [Google Scholar] [CrossRef]
- Ritchie, C.W.; Bush, A.I.; Mackinnon, A.; Macfarlane, S.; Mastwyk, M.; MacGregor, L.; Kiers, L.; Cherny, R.; Li, Q.X.; Tammer, A.; et al. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: A pilot phase 2 clinical trial. Arch. Neurol. 2003, 60, 1685–1691. [Google Scholar] [CrossRef]
- Sampson, E.L.; Jenagaratnam, L.; McShane, R. Metal protein attenuating compounds for the treatment of Alzheimer’s dementia. Cochrane Database Syst. Rev. 2014. [Google Scholar] [CrossRef]
- Bulcke, F.; Dringen, R.; Scheiber, I.F. Neurotoxicity of copper. Adv. Neurobiol. 2017, 18, 313–343. [Google Scholar] [CrossRef]
- Chiorcea-Paquim, A.M.; Enache, T.A.; Oliveira-Brett, A.M. Electrochemistry of alzheimer disease amyloid beta peptides. Curr. Med. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Cole, T.B.; Palmiter, R.D.; Suh, S.W.; Koh, J.Y. Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 7705–7710. [Google Scholar] [CrossRef] [PubMed]
- Sparks, D.L.; Schreurs, B.G. Trace amounts of copper in water induce beta-amyloid plaques and learning deficits in a rabbit model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 11065–11069. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J. Copper-2 hypothesis for causation of the current alzheimer’s disease epidemic together with dietary changes that enhance the epidemic. Chem. Res. Toxicol. 2017, 30, 763–768. [Google Scholar] [CrossRef]
- Morris, M.C.; Evans, D.A.; Tangney, C.C.; Bienias, J.L.; Schneider, J.A.; Wilson, R.S.; Scherr, P.A. Dietary copper and high saturated and trans fat intakes associated with cognitive decline. Arch. Neurol. 2006, 63, 1085–1088. [Google Scholar] [CrossRef]
- Xu, J.; Church, S.J.; Patassini, S.; Begley, P.; Waldvogel, H.J.; Curtis, M.A.; Faull, R.L.M.; Unwin, R.D.; Cooper, G.J.S. Evidence for widespread, severe brain copper deficiency in Alzheimer’s dementia. Metallomics 2017, 9, 1106–1119. [Google Scholar] [CrossRef]
- Singh, I.; Sagare, A.P.; Coma, M.; Perlmutter, D.; Gelein, R.; Bell, R.D.; Deane, R.J.; Zhong, E.; Parisi, M.; Ciszewski, J.; et al. Low levels of copper disrupt brain amyloid-beta homeostasis by altering its production and clearance. Proc. Natl. Acad. Sci. USA 2013, 110, 14771–14776. [Google Scholar] [CrossRef]
- Kitazawa, M.; Hsu, H.W.; Medeiros, R. Copper exposure perturbs brain inflammatory responses and impairs clearance of amyloid-beta. Toxicol. Sci. 2016, 152, 194–204. [Google Scholar] [CrossRef]
- White, A.R.; Multhaup, G.; Maher, F.; Bellingham, S.; Camakaris, J.; Zheng, H.; Bush, A.I.; Beyreuther, K.; Masters, C.L.; Cappai, R. The alzheimer’s disease amyloid precursor protein modulates copper-induced toxicity and oxidative stress in primary neuronal cultures. J. Neurosci. 1999, 19, 9170–9179. [Google Scholar] [CrossRef]
- Bellingham, S.A.; Lahiri, D.K.; Maloney, B.; La Fontaine, S.; Multhaup, G.; Camakaris, J. Copper depletion down-regulates expression of the Alzheimer’s disease amyloid-beta precursor protein gene. J. Biol. Chem. 2004, 279, 20378–20386. [Google Scholar] [CrossRef]
- Bellingham, S.A.; Ciccotosto, G.D.; Needham, B.E.; Fodero, L.R.; White, A.R.; Masters, C.L.; Cappai, R.; Camakaris, J. Gene knockout of amyloid precursor protein and amyloid precursor-like protein-2 increases cellular copper levels in primary mouse cortical neurons and embryonic fibroblasts. J. Neurochem. 2004, 91, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Bayer, T.A.; Schafer, S.; Simons, A.; Kemmling, A.; Kamer, T.; Tepest, R.; Eckert, A.; Schussel, K.; Eikenberg, O.; Sturchler-Pierrat, C.; et al. Dietary Cu stabilizes brain superoxide dismutase 1 activity and reduces amyloid Abeta production in APP23 transgenic mice. Proc. Natl. Acad. Sci. USA 2003, 100, 14187–14192. [Google Scholar] [CrossRef] [PubMed]
- Phinney, A.L.; Drisaldi, B.; Schmidt, S.D.; Lugowski, S.; Coronado, V.; Liang, Y.; Horne, P.; Yang, J.; Sekoulidis, J.; Coomaraswamy, J.; et al. In vivo reduction of amyloid-beta by a mutant copper transporter. Proc. Natl. Acad. Sci. USA 2003, 100, 14193–14198. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.W.; Bondy, S.C.; Kitazawa, M. Environmental and dietary exposure to copper and its cellular mechanisms linking to alzheimer’s disease. Toxicol. Sci. 2018, 163, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Mold, M.; Ouro-Gnao, L.; Wieckowski, B.M.; Exley, C. Copper prevents amyloid-beta(1–42) from forming amyloid fibrils under near-physiological conditions in vitro. Sci. Rep. 2013, 3, 1256. [Google Scholar] [CrossRef]
- Rembach, A.; Hare, D.J.; Lind, M.; Fowler, C.J.; Cherny, R.A.; McLean, C.; Bush, A.I.; Masters, C.L.; Roberts, B.R. Decreased copper in Alzheimer’s disease brain is predominantly in the soluble extractable fraction. Int. J. Alzheimers Dis. 2013, 2013, 623241. [Google Scholar] [CrossRef]
- Solioz, M. Low copper-2 intake in Switzerland does not result in lower incidence of Alzheimer’s disease and contradicts the Copper-2 Hypothesis. Exp. Biol. Med. (Maywood) 2020. [Google Scholar] [CrossRef]
- Bagheri, S.; Squitti, R.; Haertle, T.; Siotto, M.; Saboury, A.A. Role of copper in the onset of alzheimer’s disease compared to other metals. Front. Aging Neurosci. 2017, 9, 446. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Huang, X.; Cho, H.; Greig, N.H.; Youdim, M.B.; Rogers, J.T. Metal specificity of an iron-responsive element in Alzheimer’s APP mRNA 5’untranslated region, tolerance of SH-SY5Y and H4 neural cells to desferrioxamine, clioquinol, VK-28, and a piperazine chelator. J. Neural Transm. Suppl. 2006, 237–247. [Google Scholar] [CrossRef]
- Kowall, N.W.; Hantraye, P.; Brouillet, E.; Beal, M.F.; McKee, A.C.; Ferrante, R.J. MPTP induces alpha-synuclein aggregation in the substantia nigra of baboons. Neuroreport 2000, 11, 211–213. [Google Scholar] [CrossRef]
- Peluso, P.; Wilson, D.S.; Do, D.; Tran, H.; Venkatasubbaiah, M.; Quincy, D.; Heidecker, B.; Poindexter, K.; Tolani, N.; Phelan, M.; et al. Optimizing antibody immobilization strategies for the construction of protein microarrays. Anal. Biochem. 2003, 312, 113–124. [Google Scholar] [CrossRef]
- Feezor, R.J.; Baker, H.V.; Xiao, W.; Lee, W.A.; Huber, T.S.; Mindrinos, M.; Kim, R.A.; Ruiz-Taylor, L.; Moldawer, L.L.; Davis, R.W.; et al. Genomic and proteomic determinants of outcome in patients undergoing thoracoabdominal aortic aneurysm repair. J. Immunol. 2004, 172, 7103–7109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Huang, W.; Moir, R.D.; Vanderburg, C.R.; Lai, B.; Peng, Z.; Tanzi, R.E.; Rogers, J.T.; Huang, X. Metal exposure and Alzheimer’s pathogenesis. J. Struct. Biol. 2006, 155, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Moir, R.D.; Tanzi, R.E.; Bush, A.I.; Rogers, J.T. Redox-active metals, oxidative stress, and Alzheimer’s disease pathology. Ann. N.Y. Acad. Sci. 2004, 1012, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Hyman, B.T.; Van Horsen, G.W.; Damasio, A.R.; Barnes, C.L. Alzheimer’s disease: Cell-specific pathology isolates the hippocampal formation. Science 1984, 225, 1168–1170. [Google Scholar] [CrossRef]
- Zheng, W.; Tsai, M.-Y.; Wolynes, P.G. Comparing the aggregation free energy landscapes of amyloid beta(1–42) and amyloid beta(1–40). J. Am. Chem. Soc. 2017, 139, 16666–16676. [Google Scholar] [CrossRef]
- Jin, L.; Wu, W.H.; Li, Q.Y.; Zhao, Y.F.; Li, Y.M. Copper inducing Abeta42 rather than Abeta40 nanoscale oligomer formation is the key process for Abeta neurotoxicity. Nanoscale 2011, 3, 4746–4751. [Google Scholar] [CrossRef]
- Li, Y.-Q.; Yin, J.-Y.; Liu, Z.-Q.; Li, X.-P. Copper efflux transporters ATP7A and ATP7B: Novel biomarkers for platinum drug resistance and targets for therapy. IUBMB Life 2018, 70, 183–191. [Google Scholar] [CrossRef]
- Sarafi, M.N.; Garcia-Zepeda, E.A.; MacLean, J.A.; Charo, I.F.; Luster, A.D. Murine monocyte chemoattractant protein (MCP)-5: A novel CC chemokine that is a structural and functional homologue of human MCP-1. J. Exp. Med. 1997, 185, 99–109. [Google Scholar] [CrossRef]
- Zhang, K.; Luo, J. Role of MCP-1 and CCR2 in alcohol neurotoxicity. Pharmacol. Res. 2019, 139, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Pelus, L.M.; Appelbaum, E.; Johanson, K.; Anzai, N.; Broxmeyer, H.E. CCR7 ligands, SLC/6Ckine/Exodus2/TCA4 and CKbeta-11/MIP-3beta/ELC, are chemoattractants for CD56(+)CD16(-) NK cells and late stage lymphoid progenitors. Cell Immunol. 1999, 193, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Pham, E.; Crews, L.; Ubhi, K.; Hansen, L.; Adame, A.; Cartier, A.; Salmon, D.; Galasko, D.; Michael, S.; Savas, J.N.; et al. Progressive accumulation of amyloid-beta oligomers in Alzheimer’s disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. FEBS J. 2010, 277, 3051–3067. [Google Scholar] [CrossRef] [PubMed]
- Lesne, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef]
- Revett, T.J.; Baker, G.B.; Jhamandas, J.; Kar, S. Glutamate system, amyloid ß peptides and tau protein: Functional interrelationships and relevance to Alzheimer disease pathology. J. Psychiatry Neurosci. 2013, 38, 6–23. [Google Scholar] [CrossRef]
- Haywood, S. The effect of excess dietary copper on the liver and kidney of the male rat. J. Comp. Pathol. 1980, 90, 217–232. [Google Scholar] [CrossRef]
- Haywood, S. Copper toxicosis and tolerance in the rat. I--Changes in copper content of the liver and kidney. J. Pathol. 1985, 145, 149–158. [Google Scholar] [CrossRef]
- Ghiso, J.; Shayo, M.; Calero, M.; Ng, D.; Tomidokoro, Y.; Gandy, S.; Rostagno, A.; Frangione, B. Systemic catabolism of Alzheimer’s Abeta40 and Abeta42. J. Biol. Chem. 2004, 279, 45897–45908. [Google Scholar] [CrossRef]
- Wang, J.; Gu, B.J.; Masters, C.L.; Wang, Y.-J. A systemic view of Alzheimer disease - insights from amyloid-β metabolism beyond the brain. Nat. Rev. Neurol. 2017, 13, 612–623. [Google Scholar] [CrossRef]
- Maarouf, C.L.; Walker, J.E.; Sue, L.I.; Dugger, B.N.; Beach, T.G.; Serrano, G.E. Impaired hepatic amyloid-beta degradation in Alzheimer’s disease. PLoS ONE 2018, 13, e0203659. [Google Scholar] [CrossRef]
- Wang, Y.-R.; Wang, Q.-H.; Zhang, T.; Liu, Y.-H.; Yao, X.-Q.; Zeng, F.; Li, J.; Zhou, F.-Y.; Wang, L.; Yan, J.-C.; et al. Associations between hepatic functions and plasma amyloid-beta levels-implications for the capacity of liver in peripheral amyloid-beta clearance. Mol. Neurobiol. 2017, 54, 2338–2344. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.J.; Frazer, D.M. Current understanding of iron homeostasis. Am. J. Clin. Nutr. 2017, 106, 1559S–1566S. [Google Scholar] [CrossRef] [PubMed]
- Gulec, S.; Collins, J.F. Molecular mediators governing iron-copper interactions. Annu. Rev. Nutr. 2014, 34, 95–116. [Google Scholar] [CrossRef] [PubMed]
- Doguer, C.; Ha, J.-H.; Collins, J.F. Intersection of iron and copper metabolism in the mammalian intestine and liver. Compr. Physiol. 2018, 8, 1433–1461. [Google Scholar] [CrossRef]
- Feng, C.; Wang, X.; Liu, T.; Zhang, M.; Xu, G.; Ni, Y. Expression of CCL2 and its receptor in activation and migration of microglia and monocytes induced by photoreceptor apoptosis. Mol. Vis. 2017, 23, 765–777. [Google Scholar]
- Wang, W.-Y.; Tan, M.-S.; Yu, J.-T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [CrossRef]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Invest. 2017, 127, 3240–3249. [Google Scholar] [CrossRef]
- Lee, W.-J.; Liao, Y.-C.; Wang, Y.-F.; Lin, I.F.; Wang, S.-J.; Fuh, J.-L. Plasma MCP-1 and cognitive decline in patients with alzheimer’s disease and mild cognitive impairment: A two-year follow-up study. Sci. Rep. 2018, 8, 1280. [Google Scholar] [CrossRef]
- Galimberti, D.; Fenoglio, C.; Lovati, C.; Venturelli, E.; Guidi, I.; Corra, B.; Scalabrini, D.; Clerici, F.; Mariani, C.; Bresolin, N.; et al. Serum MCP-1 levels are increased in mild cognitive impairment and mild Alzheimer’s disease. Neurobiol. Aging 2006, 27, 1763–1768. [Google Scholar] [CrossRef]
- Kiyota, T.; Yamamoto, M.; Schroder, B.; Jacobsen, M.T.; Swan, R.J.; Lambert, M.P.; Klein, W.L.; Gendelman, H.E.; Ransohoff, R.M.; Ikezu, T. AAV1/2-mediated CNS gene delivery of dominant-negative CCL2 mutant suppresses gliosis, beta-amyloidosis, and learning impairment of APP/PS1 mice. Mol. Ther. 2009, 17, 803–809. [Google Scholar] [CrossRef]
- Stein, J.V.; Rot, A.; Luo, Y.; Narasimhaswamy, M.; Nakano, H.; Gunn, M.D.; Matsuzawa, A.; Quackenbush, E.J.; Dorf, M.E.; von Andrian, U.H. The CC chemokine thymus-derived chemotactic agent 4 (TCA-4, secondary lymphoid tissue chemokine, 6Ckine, exodus-2) triggers lymphocyte function-associated antigen 1-mediated arrest of rolling T lymphocytes in peripheral lymph node high endothelial venules. J. Exp. Med. 2000, 191, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Herz, J.; Alme, M.N.; Salvador, A.F.; Dong, M.Q.; Viar, K.E.; Herod, S.G.; Knopp, J.; Setliff, J.C.; Lupi, A.L.; et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat. Neurosci. 2018, 21, 1380–1391. [Google Scholar] [CrossRef] [PubMed]
- Belikan, P.; Bühler, U.; Wolf, C.; Pramanik, G.K.; Gollan, R.; Zipp, F.; Siffrin, V. CCR7 on CD4(+) T Cells Plays a Crucial Role in the Induction of Experimental Autoimmune Encephalomyelitis. J. Immunol. (Baltimore, Md.: 1950) 2018, 200, 2554–2562. [Google Scholar] [CrossRef] [PubMed]
- Lue, L.-F.; Beach, T.G.; Walker, D.G. Alzheimer’s disease research using human microglia. Cells 2019, 8, 838. [Google Scholar] [CrossRef]
- Cai, Z.; Hussain, M.D.; Yan, L.J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef]
- Alasmari, F.; Alshammari, M.A.; Alasmari, A.F.; Alanazi, W.A.; Alhazzani, K. Neuroinflammatory cytokines induce amyloid beta neurotoxicity through modulating amyloid precursor protein levels/metabolism. Biomed. Res. Int. 2018, 2018, 3087475. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time Point | Start | Middle | End |
|---|---|---|---|
| Regular chow (n = 3) | 23.8 ± 0.9 | 24.2 ± 1.4 | 23.2 ± 1.0 |
| Cu-enhanced chow (n = 5) | 24.5 ± 0.8 | 24.7 ± 0.6 | 25.1 ± 1.0 |
| Diet | n | Cu (pgmg-1) | Fe (pgmg-1) | Zn (pgmg-1) |
|---|---|---|---|---|
| Regular Chow | 3 | 25.2 ± 1.3 | 124.5 ± 25.5 | 203.0 ± 28.3 |
| Cu-enhanced Chow | 5 | 37.2 ± 2.4 | 90.3 ± 6.7 | 177.7 ± 4.6 |
| Diet | n | Aβ1-40 Area*10−3 (µm2) | Aβ1-42 Area*10−3 (µm2) | Aβ1-40 (% of Total Cortex Area Fraction) | Aβ1-42 (% of Total Cortex Area Fraction) | Aβ1-40 Plaque Count | Aβ1-42 Plaque Count | Aβ1-42/Aβ1-40 Area Quotient |
|---|---|---|---|---|---|---|---|---|
| Regular Chow | 3 | 118.3 ± 27.4 | 177.6 ± 27.0 | 0.52 ± 0.11 | 0.82 ± 0.09 | 165 ± 38 | 193 ± 31 | 1.50 ± 0.13 |
| Cu-enhanced Chow | 5 | 134.3 ± 12.6 | 190.3 ± 17.2 | 0.62 ± 0.07 | 0.92 ± 0.10 | 181 ± 22 | 232 ± 32 | 1.42 ± 0.07 |
| Diet | n | Aβ1-40 (pmolg-1) | Aβ1-42 (pmolg-1) | Total Aβ (Aβ1-40 + Aβ1-42) (pmolg-1) | Aβ1-42(% of Total Aβ) | Aβ1-42/Aβ1-40 Quotient |
|---|---|---|---|---|---|---|
| Regular Chow | 3 | 39.31 ± 2.60 | 15.44 ± 3.42 | 54.74 ± 6.01 | 27.40 ± 3.63 | 0.38 ± 0.07 |
| Cu-enhanced Chow | 5 | 66.32 ± 6.45 | 18.55 ± 3.80 | 84.87 ± 9.46 | 21.14 ± 2.67 | 0.27 ± 0.04 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pilozzi, A.; Yu, Z.; Carreras, I.; Cormier, K.; Hartley, D.; Rogers, J.; Dedeoglu, A.; Huang, X. A Preliminary Study of Cu Exposure Effects upon Alzheimer’s Amyloid Pathology. Biomolecules 2020, 10, 408. https://doi.org/10.3390/biom10030408
Pilozzi A, Yu Z, Carreras I, Cormier K, Hartley D, Rogers J, Dedeoglu A, Huang X. A Preliminary Study of Cu Exposure Effects upon Alzheimer’s Amyloid Pathology. Biomolecules. 2020; 10(3):408. https://doi.org/10.3390/biom10030408
Chicago/Turabian StylePilozzi, Alexander, Zhanyang Yu, Isabel Carreras, Kerry Cormier, Dean Hartley, Jack Rogers, Alpaslan Dedeoglu, and Xudong Huang. 2020. "A Preliminary Study of Cu Exposure Effects upon Alzheimer’s Amyloid Pathology" Biomolecules 10, no. 3: 408. https://doi.org/10.3390/biom10030408
APA StylePilozzi, A., Yu, Z., Carreras, I., Cormier, K., Hartley, D., Rogers, J., Dedeoglu, A., & Huang, X. (2020). A Preliminary Study of Cu Exposure Effects upon Alzheimer’s Amyloid Pathology. Biomolecules, 10(3), 408. https://doi.org/10.3390/biom10030408