Oxidative-Antioxidant Imbalance and Impaired Glucose Metabolism in Schizophrenia


 ,
,
Abstract
1. Oxidative Stress, Impaired Glucose Metabolism, Schizophrenia
2. Glycation and Oxidative Damage in Schizophrenia
3. Oxidative, Nitrosative, and Sulfuric Stress in Schizophrenia
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Vita, A.; De Peri, L.; Deste, G.; Sacchetti, E. Progressive loss of cortical gray matter in schizophrenia: A meta-analysis and meta-regression of longitudinal MRI studies. Transl. Psychiatry 2012, 2, e190. [Google Scholar] [CrossRef]
- Qin, J.; Sui, J.; Ni, H.; Wang, S.; Zhang, F.; Zhou, Z.; Tian, L. The shared and distinct white matter networks between drug-naive patients with obsessive-compulsive disorder and schizophrenia. Front. Neurosci. 2019, 13, 96. [Google Scholar] [CrossRef] [PubMed]
- Velásquez, E.; Martins-de-Souza, D.; Velásquez, I.; Carneiro, G.R.; Schmitt, A.; Falkai, P.; Domont, G.B.; Nogueira, F.C.S. Quantitative subcellular proteomics of the orbitofrontal cortex of schizophrenia patients. J. Proteome Res. 2019, 18, 4240–4253. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Cropley, V.L.; Klauser, P.; Lenroot, R.K.; Bruggemann, J.; Sundram, S.; Bousman, C.; Pereira, A.; Di Biase, M.A.; Weickert, T.W.; Weickert, C.S.; et al. Accelerated gray and white matter deterioration with age in schizophrenia. Am. J. Psychiatry 2017, 174, 286–295. [Google Scholar] [CrossRef] [PubMed]
- O’Hanlon, E.; Howley, S.; Prasad, S.; McGrath, J.; Leemans, A.; McDonald, C.; Garavan, H.; Murphy, K.C. Multimodal MRI reveals structural connectivity differences in 22q11 deletion syndrome related to impaired spatial working memory. Hum. Brain Mapp. 2016, 37, 4689–4705. [Google Scholar] [CrossRef] [PubMed]
- Altamura, A.C.; Delvecchio, G.; Marotta, G.; Oldani, L.; Pigoni, A.; Ciappolino, V.; Caletti, E.; Rovera, C.; Dobrea, C.; Arici, C.; et al. Structural and metabolic differentiation between bipolar disorder with psychosis and substance-induced psychosis: An integrated MRI/PET study. Eur. Psychiatry 2017, 41, 85–94. [Google Scholar] [CrossRef]
- Shan, X.X.; Ou, Y.P.; Pan, P.; Ding, Y.D.; Zhao, J.; Liu, F.; Chen, J.D.; Guo, W.B.; Zhao, J.P. Increased frontal gray matter volume in individuals with prodromal psychosis. CNS Neurosci. 2019, 25, 987–994. [Google Scholar] [CrossRef]
- Mechelli, A.; Riecher-Rössler, A.; Meisenzahl, E.M. Neuroanatomical abnormalities that predate the onset of psychosis: A multicenter study. Arch. Gen. Psychiatry 2011, 68, 489–495. [Google Scholar] [CrossRef]
- Pollak, T.A.; Lennox, B.R.; Müller, S.; Benros, M.E.; Prüss, H.; van Elst, L.T.; Klein, H.; Steiner, J.; Frodl, T.; Bogerts, B.; et al. Autoimmune psychosis: An international consensus on an approach to the diagnosis and management of psychosis of suspected autoimmune origin. Lancet Psychiatry 2019, 7, 93–108. [Google Scholar] [CrossRef]
- Fernandez, A.; Meechan, D.W.; Karpinski, B.A.; Paronett, E.M.; Bryan, C.A.; Rutz, H.L.; Radin, E.A.; Lubin, N.; Bonner, E.R.; Popratiloff, A.; et al. Mitochondrial dysfunction leads to cortical under-connectivity and cognitive impairment. Neuron 2019, 102, 1127–1142. [Google Scholar] [CrossRef] [PubMed]
- Mayo, D.; Bolden, K.A.; Simon, T.J.; Niendam, T.A. Bullying and psychosis: The impact of chronic traumatic stress on psychosis risk in 22q11.2 deletion syndrome - a uniquely vulnerable population. J. Psychiatr. Res. 2019, 114, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Núñez, C.; Stephan-Otto, C.; Usall, J.; Bioque, M.; Lobo, A.; González-Pinto, A.; Pina-Camacho, L.; Vieta, E.; Castro-Fornieles, J.; Rodriguez-Jimenez, R.; et al. Neutrophil count is associated with reduced gray matter and enlarged ventricles in first-episode psychosis. Schizophr. Bull. 2019, 45, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Smucny, J.; Lesh, T.A.; Zarubin, V.C.; Niendam, T.A.; Ragland, J.D.; Tully, L.M.; Carter, C.S. One-year stability of frontoparietal cognitive control network connectivity in recent onset schizophrenia: A task-related 3T fMRI study. Schizophr. Bull. 2020. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Farooq, S.; Edwards, J.; Chew-Graham, C.A.; Shiers, D.; Frisher, M.; Hayward, R.; Sumathipala, A.; Jordan, K.P. Patterns of symptoms before a diagnosis of first episode psychosis: A latent class analysis of UK primary care electronic health records. BMC Med. 2019, 17, 227. [Google Scholar] [CrossRef] [PubMed]
- Anda, L.; Brønnick, K.K.; Johannessen, J.O.; Joa, I.; Kroken, R.A.; Johnsen, E.; Rettenbacher, M.; Fathian, F.; Løberg, E.M. Cognitive profile in ultra high risk for psychosis and schizophrenia: A comparison using coordinated norms. Front. Psychiatry 2019, 10, 695. [Google Scholar] [CrossRef]
- Schulmann, A.; Ryu, E.; Goncalves, V.; Rollins, B.; Christiansen, M.; Frye, M.A.; Biernacka, J.; Vawter, M.P. Novel complex interactions between mitochondrial and nuclear DNA in schizophrenia and bipolar disorder. Mol. Neuropsychiatry 2019, 5, 13–27. [Google Scholar] [CrossRef]
- Llorca, P.M.; Pereira, B.; Jardri, R.; Chereau-Boudet, I.; Brousse, G.; Misdrahi, D.; de Chazeron, I. Hallucinations in schizophrenia and Parkinson’s disease: An analysis of sensory modalities involved and the repercussion on patients. Sci. Rep. 2016, 6, 38152. [Google Scholar] [CrossRef]
- Häfner, H. From onset and prodromal stage to a life-long course of schizophrenia and its symptom dimensions: How sex, age, and other risk factors influence incidence and course of illness. Psychiatry J. 2019, 2019, 9804836. [Google Scholar] [CrossRef]
- Chang, W.C.; Westbrook, A.; Strauss, G.P.; Chu, A.O.K.; Chong, C.S.Y.; Siu, C.M.W.; Chan, S.K.W.; Lee, E.H.M.; Hui, C.L.M.; Suen, Y.M.; et al. Abnormal cognitive effort allocation and its association with amotivation in first-episode psychosis. Psychol. Med. 2019. [Google Scholar] [CrossRef]
- Avery, S.N.; McHugo, M.; Armstrong, K.; Blackford, J.U.; Woodward, N.D.; Heckers, S. Disrupted habituation in the early stage of psychosis. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2019, 4, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- Fusar-Poli, P.; Broome, M.R.; Woolley, J.B. Altered brain function directly related to structural abnormalities in people at ultra high risk of psychosis: Longitudinal VBM-fMRI study. J. Psychiatr. Res. 2011, 45, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, I.; Kumar, V.; Sharma, S.; Dhingra, D.; Rana, B.; Agarwal, M.; Kumar, N. Identification of brain regions associated with working memory deficit in schizophrenia. F1000Research 2019, 8, 124. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liang, S.; Pu, W.; Song, Y.; Mwansisya, T.E.; Yang, Q.; Liu, H.; Liu, Z.; Shan, B.; Xue, Z. Reduced cortical thickness in right Heschl’s gyrus associated with auditory verbal hallucinations severity in first-episode schizophrenia. BMC Psychiatry 2015, 15, 152. [Google Scholar] [CrossRef]
- Kasai, K.; Shenton, M.E.; Salisbury, D.F. Progressive decrease of left Heschl gyrus and planum temporale gray matter volume in first-episode schizophrenia: A longitudinal magnetic resonance imaging study. Arch. Gen. Psychiatry 2003, 60, 766–775. [Google Scholar] [CrossRef]
- Li, J.; Wu, C.; Zheng, Y.; Li, R.; Li, X.; She, S.; Wu, H.; Peng, H.; Ning, Y.; Li, L. Schizophrenia affects speech-induced functional connectivity of the superior temporal gyrus under cocktail-party listening conditions. Neuroscience 2017, 17, 248–257. [Google Scholar] [CrossRef]
- Bandeira, I.D.; Barouh, J.L.; Bandeira, I.D.; Quarantini, L. Analysis of the superior temporal gyrus as a possible biomarker in schizophrenia using voxel-based morphometry of the brain magnetic resonance imaging: A comprehensive review. CNS Spectr. 2020, 10, 1–7. [Google Scholar] [CrossRef]
- Gao, J.; Tang, X.; Wang, C.; Yu, M.; Sha, W.; Wang, X.; Zhang, H.; Zhang, X.; Zhang, X. Aberrant cerebellar neural activity and cerebro-cerebellar functional connectivity involving executive dysfunction in schizophrenia with primary negative symptoms. Brain Imaging Behav. 2019. [Google Scholar] [CrossRef]
- Zhang, R.; Wei, Q.; Kang, Z.; Zalesky, A.; Li, M.; Xu, Y.; Li, L.; Wang, J.; Zheng, L.; Wang, B.; et al. Disrupted brain anatomical connectivity in medication-naïve patients with first-episode schizophrenia brain. Struct. Funct. 2015, 220, 1145–1159. [Google Scholar] [CrossRef]
- Gil-Berrozpe, G.J.; Sánchez-Torres, A.; García de Jalón, E.; Moreno-Izco, L.; Fañanás, L.; Peralta, V.; Cuesta, M.J.; SEGPEPs group. Utility of the MoCA for cognitive impairment screening in long-term psychosis patients. Schizophr. Res. 2019. [Google Scholar] [CrossRef]
- Zanelli, J.; Mollon, J.; Sandin, S.; Morgan, C.; Dazzan, P.; Pilecka, I.; Reis Marques, T.; David, A.S.; Morgan, K.; Fearon, P.; et al. Cognitive change in schizophrenia and other psychoses in the decade following the first episode. Am. J. Psychiatry 2019, 176, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Goulding, S.M.; Holtzman, C.W.; Trotman, H.D.; Ryan, T.; MacDonald, A.N.; Shapiro, D.I.; Brasfield, J.L.; Walker, E.F. The prodrome and clinical risk for psychotic disorders. Child Adolesc. Psychiatr. Clin. N. Am. 2013, 22, 557–567. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Oliver, D.; Davies, C.; Crossland, G.; Lim, S.; Gifford, G.; McGuire, P.; Fusar-Poli, P. Can we reduce the duration of untreated psychosis? A systematic review and meta-analysis of controlled interventional studies. Schizophr. Bull. 2018, 44, 1362–1372. [Google Scholar] [CrossRef] [PubMed]
- Romanov, D.V.; Lepping, P.; Bewley, A.; Huber, M.; Freudenmann, R.W.; Lvov, A.; Squire, S.B.; Noorthoorn, E.O. Longer duration of untreated psychosis is associated with poorer outcomes for patients with delusional infestation. Acta Derm. Venereol. 2018, 10, 848–854. [Google Scholar] [CrossRef]
- Penttilä, M.; Jääskeläinen, E.; Hirvonen, N.; Isohanni, M.; Miettunen, J. Duration of untreated psychosis as predictor of long-term outcome in schizophrenia: Systematic review and meta-analysis. Br. J. Psychiatry 2014, 205, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Ordak, M.; Nasierowski, T.; Muszyńska, E. The pharmacological basis of drug interactions: An aspect overlooked in psychiatry. Lancet Psychiatry 2019, 6, 984. [Google Scholar] [CrossRef]
- Shimomura, Y.; Kikuchi, Y.; Suzuki, T.; Uchida, H.; Mimura, M.; Takeuchi, H. Antipsychotic treatment in the maintenance phase of schizophrenia: An updated systematic review of the guidelines and algorithms. Schizophr. Res. 2019. [Google Scholar] [CrossRef]
- Correll, C.U.; Lencz, T.; Malhotra, A.K. Antipsychotic drugs and obesity. Trends Mol. Med. 2011, 17, 97–107. [Google Scholar] [CrossRef]
- Correll, C.U.; Robinson, D.G.; Schooler, N.R.; Brunette, M.F.; Mueser, K.T.; Rosenheck, R.A.; Marcy, P.; Addington, J.; Estroff, S.E.; Robinson, J.; et al. Cardiometabolic risk in patients with first-episode schizophrenia spectrum disorders baseline results from the RAISE-ETP study. JAMA Psychiatry 2014, 71, 1350–1363. [Google Scholar] [CrossRef]
- Yang, N.; Yu, L.; Deng, Y.; Han, Q.; Wang, J.; Yu, L.; Zhai, Z.; Li, W. Identification and characterization of proteins that are differentially expressed in adipose tissue of olanzapine-induced insulin resistance rat by iTRAQ quantitative proteomics. J. Proteom. 2019, 212, 103570. [Google Scholar] [CrossRef]
- Xu, P.T.; Song, Z.; Zhang, W.C.; Jiao, B.; Yu, Z.B. Impaired translocation of GLUT4 results in insulin resistance of atrophic soleus muscle. BioMed Res. Int. 2015, 2015, 291987. [Google Scholar] [CrossRef]
- Greenhalgh, A.M.; Gonzalez-Blanco, L.; Garcia-Rizo, C.; Fernandez-Egea, E.; Miller, B.; Arroyo, M.B.; Kirkpatrick, B. Meta-analysis of glucose tolerance, insulin, and insulin resistance in antipsychotic-naïve patients with nonaffective psychosis. Schizophr. Res. 2017, 179, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Grover, S.; Chakrabarti, S.; Kulhara, P.; Avasthi, A. Clinical practice guidelines for management of schizophrenia. Indian J. Psychiatr. 2017, 59, 19. [Google Scholar] [CrossRef]
- Pillinger, T.; Beck, K.; Gobjila, C.; Donocik, J.G.; Jauhar, S.; Howes, O.D. Impaired glucose homeostasis in first-episode schizophrenia. JAMA Psychiatry 2017, 74, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Kapogiannis, D.; Dobrowolny, H.; Tran, J.; Mustapic, M.; Frodl, T.; Meyer-Lotz, G.; Schiltz, K.; Schanze, D.; Rietschel, M.; Bernstein, H.G.; et al. Insulin-signaling abnormalities in drug-naïve first-episode schizophrenia: Transduction protein analyses in extracellular vesicles of putative neuronal origin. Eur. Psychiatry 2019, 62, 124–129. [Google Scholar] [CrossRef]
- Jurcovicova, J. Glucose transport in brain—Effect of inflammation. Endocr. Regul. 2014, 48, 35–48. [Google Scholar] [CrossRef] [PubMed]
- De Silva, P.N. Does the association with diabetes say more about schizophrenia and its treatment?—The GLUT hypothesis. Med. Hypotheses 2011, 77, 529–531. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.M.; Caravaggio, F.; Costa-Dookhan, K.A.; Castellani, L.; Kowalchuk, C.; Asgariroozbehani, R.; Graff-Guerrero, A.; Hahn, M. Brain insulin action in schizophrenia: Something borrowed and something new. Neuropharmacology 2020, 163, 107633. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, M.; Du, X.; Liao, W.; Chen, D.; Fan, F.; Xiu, M.; Jia, Q.; Ning, Y.; Huang, X.; et al. Glucose disturbances, cognitive deficits and white matter abnormalities in first-episode drug-naive schizophrenia. Mol. Psychiatry 2019. [Google Scholar] [CrossRef]
- Fernandez-Egea, E.; Walker, R.; Ziauddeen, H.; Cardinal, R.N.; Bullmore, E.T. Birth weight, family history of diabetes and diabetes onset in schizophrenia. BMJ Open Diabetes Res. Care 2020, 8, e001036. [Google Scholar] [CrossRef]
- Laurens, K.R.; Luo, L.; Matheson, S.L.; Carr, V.J.; Raudino, A.; Harris, F.; Green, M.J. Common or distinct pathways to psychosis? A systematic review of evidence from prospective studies for developmental risk factors and antecedents of the schizophrenia spectrum disorders and affective psychoses. BMC Psychiatry 2015, 15, 205. [Google Scholar] [CrossRef] [PubMed]
- Sesti, G. Pathophysiology of insulin resistance. Best Pract. Res. Clin. Endocrinol. Metab. 2006, 20, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Wijtenburg, S.A.; Kapogiannis, D.; Korenic, S.A.; Mullins, R.J.; Tran, J.; Gaston, F.E.; Chen, S.; Mustapic, M.; Hong, L.E.; Rowland, L.M. Brain insulin resistance and altered brain glucose are related to memory impairments in schizophrenia. Schizophr. Res. 2019, 208, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, A.C.; Bourbonnais, A.; Frisch, F.; Giacca, A.; Lewis, G.F. Plasma nonesterified fatty acid intolerance and hyperglycemia are associated with intravenous lipid-induced impairment of insulin sensitivity and disposition index. J. Clin. Endocrinol. Metab. 2010, 95, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Norris, D.M.; Geddes, T.A.; James, D.E.; Fazakerley, D.J.; Burchfield, J.G. Glucose transport: Methods for interrogating GLUT4 trafficking in adipocytes. Methods Mol. Biol. 2018, 1713, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Mokáň, M.; Galajda, P. Primary and secondary insulin resistance. Vnitr. Lek. 2019, 65, 264–272. [Google Scholar]
- Rojo, L.E.; Gaspar, P.A.; Silva, H.; Risco, L.; Arena, P.; Cubillos-Robles, K.; Jara, B. Metabolic syndrome and obesity among users of second generation antipsychotics: A global challenge for modern psychopharmacology. Pharm. Res. 2015, 101, 74–85. [Google Scholar] [CrossRef]
- Groenewoud, M.J.; Zwartkruis, F.J. Rheb and rags come together at the lysosome to activate mTORC1. Biochem. Soc. Trans. 2013, 41, 951–955. [Google Scholar] [CrossRef]
- Zhu, M.; Wang, X.Q. Regulation of mTORC1 by Small GTPases in response to nutrients. J. Nutr. 2020, 21. [Google Scholar] [CrossRef]
- Noda, T. Regulation of autophagy through TORC1 and mTORC1. Biomolecules 2017, 7, 52. [Google Scholar] [CrossRef]
- Sengupta, S.; Peterson, T.R.; Sabatini, D.M. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol. Cell 2010, 40, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Ryskalin, L.; Limanaqi, F.; Frati, A.; Busceti, C.L.; Fornai, F. mTOR-related brain dysfunctions in neuropsychiatric disorders. Int. J. Mol. Sci. 2018, 19, 2226. [Google Scholar] [CrossRef] [PubMed]
- Lamming, D.W.; Sabatini, D.M. A central role for mTOR in lipid homeostasis. Cell Metab. 2013, 18, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Kuma, A.; Komatsu, M.; Mizushima, N. Autophagy-monitoring and autophagy-deficient mice. Autophagy 2017, 13, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Heras-Sandoval, D.; Pérez-Rojas, J.M.; Hernández-Damián, J.; Pedraza-Chaverri, J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal. 2014, 26, 2694–2701. [Google Scholar] [CrossRef]
- Olsen, J.M.; Sato, M.; Dallner, O.S. Glucose uptake in brown fat cells is dependent on mTOR complex 2-promoted GLUT1 translocation. J. Cell Biol. 2014, 207, 365–374. [Google Scholar] [CrossRef]
- Kumar, A.; Lawrence, J.C., Jr.; Jung, D.Y.; Ko, H.J.; Keller, S.R.; Kim, J.K.; Magnuson, M.A.; Harris, T.E. Fat cell-specific ablation of rictor in mice impairs insulin-regulated fat cell and whole-body glucose and lipid metabolism. Diabetes 2010, 59, 1397–1406. [Google Scholar] [CrossRef]
- Jensen, K.G.; Correll, C.U.; Rudå, D.; Klauber, D.G.; Decara, M.S.; Fagerlund, B.; Jepsen, J.R.M.; Eriksson, F.; Fink-Jensen, A.; Pagsberg, A.K. Cardiometabolic adverse effects and its predictors in children and adolescents with first-episode psychosis during treatment with quetiapine-extended release versus aripiprazole: 12-week results from the tolerance and effect of antipsychotics in children and adolescents with psychosis (TEA) trial. J. Am. Acad. Child Adolesc. Psychiatry 2019, 58, 1062–1078. [Google Scholar] [CrossRef]
- Das, A.; Reis, F.; Mishra, P.K. mTOR signaling in cardiometabolic disease, cancer, and aging 2018. Oxid. Med. Cell. Longev. 2019, 2019, 9692528. [Google Scholar] [CrossRef]
- Uchinaka, A.; Yoneda, M.; Yamada, Y.; Murohara, T.; Nagata, K. Effects of mTOR inhibition on cardiac and adipose tissue pathology and glucose metabolism in rats with metabolic syndrome. Pharmacol. Res. Perspect. 2017, 5, e00331. [Google Scholar] [CrossRef] [PubMed]
- Alsabban, A.H.; Morikawa, M.; Tanaka, Y.; Takei, Y.; Hirokawa, N. Kinesin Kif3b mutation reduces NMDAR subunit NR2A trafficking and causes schizophrenia-like phenotypes in mice. EMBO J. 2020, 39, e101090. [Google Scholar] [CrossRef] [PubMed]
- Karki, S.; Maksimainen, M.M.; Lehtiö, L.; Kajander, T. Inhibitor screening assay for neurexin-LRRTM adhesion protein interaction involved in synaptic maintenance and neurological disorders. Anal. Biochem. 2019, 587, 113463. [Google Scholar] [CrossRef] [PubMed]
- Steiner, J.; Bernstein, H.G.; Schiltz, K.; Müller, U.J.; Westphal, S.; Drexhage, H.A.; Bogerts, B. Immune system and glucose metabolism interaction in schizophrenia: A chicken-egg dilemma. Prog Neuro-psychopharmacol. Biol. Psychiatry 2014, 48, 287–294. [Google Scholar] [CrossRef]
- Wedervang-Resell, K.; Friis, S.; Lonning, V.; Smelror, R.E.; Johannessen, C.; Reponen, E.J.; Lyngstad, S.H.; Lekva, T.; Aukrust, P.; Ueland, T.; et al. Increased interleukin 18 activity in adolescents with early-onset psychosis is associated with cortisol and depressive symptoms. Psychoneuroendocrinology 2020, 112, 104513. [Google Scholar] [CrossRef]
- Pedersen, C.B.; Mors, O.; Bertelsen, A.; Waltoft, B.L.; Agerbo, E.; McGrath, J.J.; Mortensen, P.B.; Eaton, W.W. A comprehensive nationwide study of the incidence rate and lifetime risk for treated mental disorders. JAMA Psychiatry 2014, 71, 573–581. [Google Scholar] [CrossRef]
- Gerasimou, C.; Tsoporis, J.N.; Siafakas, N.; Hatziagelaki, E.; Kallergi, M.; Chatziioannou, S.N.; Parker, T.G.; Parissis, J.; Salpeas, V.; Papageorgiou, C.; et al. A longitudinal study of alterations of S100B, sRAGE and fas ligand in association to olanzapine medication in a sample of first episode patients with schizophrenia. CNS Neurol. Disord. Drug Targets 2018, 17, 383–388. [Google Scholar] [CrossRef]
- Ramírez-Jirano, L.J.; Velasco-Ramírez, S.F.; Pérez-Carranza, G.A.; Domínguez-Díaz, C.; Bitzer-Quintero, O.K. Cytokines and nervous system: Relationship with schizophrenia. Rev. Med. Inst. Mex. Seguro Soc. 2019, 57, 107–112. [Google Scholar]
- Balaji, R.; Subbanna, M.; Shivakumar, V.; Abdul, F.; Venkatasubramanian, G.; Debnath, M. Pattern of expression of toll like receptor (TLR)-3 and -4 genes in drug-naïve and antipsychotic treated patients diagnosed with schizophrenia. Psychiatry Res. 2019. [Google Scholar] [CrossRef]
- Okun, E.; Barak, B.; Saada-Madar, R.; Rothman, S.M.; Griffioen, K.J.; Roberts, N.; Castro, K.; Mughal, M.R.; Pita, M.A.; Stranahan, A.M.; et al. Evidence for a developmental role for TLR4 in learning and memory. PLoS ONE 2012, 7, e47522. [Google Scholar] [CrossRef]
- Okun, E.; Griffioen, K.; Barak, B.; Roberts, N.J.; Castro, K.; Pita, M.A.; Cheng, A.; Mughal, M.R.; Wan, R.; Ashery, U.; et al. Toll-like receptor 3 inhibits memory retention and constrains adult hippocampal neurogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 15625–15630. [Google Scholar] [CrossRef] [PubMed]
- Okun, E.; Griffioen, K.J.; Mattson, M.P. Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci. 2011, 34, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Momtazmanesh, S.; Zare-Shahabadi, A.; Rezaei, N. Cytokine alterations in schizophrenia: An updated review. Front. Psychiatry 2019, 10, 892. [Google Scholar] [CrossRef] [PubMed]
- McOmish, C.E.; Demireva, E.Y.; Gingrich, J.A. Developmental expression of mGlu2 and mGlu3 in the mouse brain. Gene. Expr. Patterns 2016, 22, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Matrisciano, F.; Dong, E.; Nicoletti, F.; Guidotti, A. Epigenetic alterations in prenatal stress mice as an endophenotype model for schizophrenia: Role of metabotropic glutamate 2/3 receptors. Front. Mol. Neurosci. 2018, 11, 423. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, A.; Venkatasubramanian, G.; Berk, M.; Debnath, M. Mitochondrial dysfunction in schizophrenia: Pathways, mechanisms and implications. Neurosci. Biobehav. Rev. 2015, 48, 10–21. [Google Scholar] [CrossRef]
- Sullivan, C.R.; Koene, R.H.; Hasselfeld, K.; O’Donovan, S.M.; Ramsey, A.; McCullumsmith, R.E. Neuron-specific deficits of bioenergetic processes in the dorsolateral prefrontal cortex in schizophrenia. Mol. Psychiatry 2019, 24, 1319–1328. [Google Scholar] [CrossRef]
- Jiang, J.; Peng, C.; Sun, L.; Li, J.; Qing, Y.; Hu, X.; Yang, X.; Li, Y.; Xu, C.; Zhang, J.; et al. Leukocyte proteomic profiling in first-episode schizophrenia patients: Does oxidative stress play central roles in the pathophysiology network of schizophrenia? Antioxid. Redox Signal. 2019, 31, 579–588. [Google Scholar] [CrossRef]
- Ide, M.; Ohnishi, T.; Toyoshima, M.; Balan, S.; Maekawa, M.; Shimamoto-Mitsuyama, C.; Iwayama, Y.; Ohba, H.; Watanabe, A.; Ishii, T.; et al. Excess hydrogen sulfide and polysulfides production underlies a schizophrenia pathophysiology. EMBO Mol. Med. 2019, 11, e10695. [Google Scholar] [CrossRef]
- Rowland, L.M.; Pradhan, S.; Korenic, S.; Wijtenburg, S.A.; Hong, L.E.; Edden, R.A.; Barker, P.B. Elevated brain lactate in schizophrenia: A 7 T magnetic resonance spectroscopy study. Transl. Psychiatry 2016, 6, e967. [Google Scholar] [CrossRef]
- Roberts, R.C. Postmortem studies on mitochondria in schizophrenia. Schizophr. Res. 2017, 187, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.L.; Li, X.S.; Chen, G.Y.; Du, Y.; Wei, Z.X.; Chen, X.; Zheng, G.E.; Deng, W.; Cheng, Y. Serum oxidative stress marker levels in unmedicated and medicated patients with schizophrenia. J. Mol. Neurosci. 2018, 66, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, S.; Noda, Y.; Tarumi, R.; Mimura, Y.; Yoshida, K.; Iwata, Y.; Elsalhy, M.; Kuromiya, M.; Kurose, S.; Masuda, F.; et al. Glutathione levels and activities of glutathione metabolism enzymes in patients with schizophrenia: A systematic review and meta-analysis. J. Psychopharmacol. 2019, 33, 1199–1214. [Google Scholar] [CrossRef] [PubMed]
- Herberth, M.; Koethe, D.; Cheng, T.M.K. Impaired glycolytic response in peripheral blood mononuclear cells of first-onset antipsychotic-naive schizophrenia patients. Mol. Psychiatry 2011, 16, 848–859. [Google Scholar] [CrossRef] [PubMed]
- Ebertowska, A.; Ludkiewicz, B.; Klejbor, I.; Melka, N.; Moryś, J. Pyruvate dehydrogenase deficiency—Morphological and metabolic effects, creation of animal model to study and research for treatment therapy. Folia Morphol. (Warsz) 2020, 19. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, M.A.; Kontek, B. Lipid peroxidation in patients with schizophrenia. Psychiatry Clin. Neurosci. 2010, 64, 469–475. [Google Scholar] [CrossRef]
- Joshi, Y.B.; Praticò, D. Lipid peroxidation in psychiatric illness: Overview of clinical evidence. Oxidative medicine and cellular longevity. Oxid. Med. Cell. Longev. 2014, 2014, 828702. [Google Scholar] [CrossRef]
- Sharma, P.; Sampath, H. Mitochondrial DNA Integrity: Role in health and disease. Cells 2019, 8, 100. [Google Scholar] [CrossRef]
- Paglialunga, S.; Ludzki, A.; Root-McCaig, J.; Holloway, G.P. In adipose tissue, increased mitochondrial emission of reactive oxygen species is important for short-term high-fat diet-induced insulin resistance in mice. Diabetologia 2015, 58, 1071–1080. [Google Scholar] [CrossRef]
- Fazakerley, D.J.; Minard, A.Y.; Krycer, J.R.; Thomas, K.C.; Stöckli, J.; Harney, D.J.; Burchfield, J.G.; Maghzal, G.J.; Caldwell, S.T.; Hartley, R.C.; et al. Mitochondrial oxidative stress causes insulin resistance without disrupting oxidative phosphorylation. J. Biol. Chem. 2018, 293, 7315–7328. [Google Scholar] [CrossRef]
- Ijaz, S.; Bolea, B.; Davies, S.; Savović, J.; Richards, A.; Sullivan, S.; Moran, P. Antipsychotic polypharmacy and metabolic syndrome in schizophrenia: A review of systematic reviews. BMC Psychiatry 2018, 18, 275. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.W.; Kim, Y.K. Unresolved issues for utilization of atypical antipsychotics in schizophrenia: Antipsychotic polypharmacy and metabolic syndrome. Int. J. Mol. Sci. 2017, 18, 2174. [Google Scholar] [CrossRef] [PubMed]
- Mueser, K.T.; DeTore, N.R.; Kredlow, M.A.; Bourgeois, M.L.; Penn, D.L.; Hintz, K. Clinical and demographic correlates of stigma in first-episode psychosis: The impact of duration of untreated psychosis. Acta Psychiatr. Scand. 2020, 141, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Hambsch, B. Altered glyoxalase 1 expression in psychiatric disorders: Cause or consequence? Semin. Cell Dev. Biol. 2011, 22, 302–308. [Google Scholar] [CrossRef]
- Koike, S.; Kayama, T.; Arai, M.; Horiuchi, Y.; Kobori, A.; Miyashita, M.; Itokawa, M.; Ogasawara, Y. Characterization of modified proteins in plasma from a subtype of schizophrenia based on carbonyl stress: Protein carbonyl is a possible biomarker of psychiatric disorders. Biochem. Biophys. Res. Commun. 2015, 67, 361–366. [Google Scholar] [CrossRef]
- Koike, S.; Nishimoto, S.; Ogasawara, Y. Cysteine persulfides and polysulfides produced by exchange reactions with H2S protect SH-SY5Y cells from methylglyoxal-induced toxicity through Nrf2 activation. Redox Biol. 2017, 12, 530–539. [Google Scholar] [CrossRef]
- Itokawa, M.; Miyashita, M.; Arai, M.; Dan, T.; Takahashi, K.; Tokunaga, T.; Ishimoto, K.; Toriumi, K.; Ichikawa, T.; Horiuchi, Y.; et al. Pyridoxamine: A novel treatment for schizophrenia with enhanced carbonyl stress. Psychiatry Clin. Neurosci. 2018, 72, 35–44. [Google Scholar] [CrossRef]
- Kouidrat, Y.; Amad, A.; Arai, M.; Miyashita, M.; Lalau, J.D.; Loas, G.; Itokawa, M. Advanced glycation end products and schizophrenia: A systematic review. J. Psychiatr. Res. 2015, 66–67, 112–117. [Google Scholar] [CrossRef]
- Takeda, M.; Ohnuma, T.; Takeuchi, M.; Katsuta, N.; Maeshima, H.; Takebayashi, Y.; Arai, H. Altered serum glyceraldehyde-derived advanced glycation end product (AGE) and soluble AGE receptor levels indicate carbonyl stress in patients with schizophrenia. Neuroscience Letters. 2015, 593, 51–55. [Google Scholar] [CrossRef]
- Du Yan, S.; Zhu, H.; Fu, J.; Yan, S.F.; Roher, A.; Tourtellotte, W.W.; Rajavashisth, T.; Chen, X.; Godman, G.C.; Stern, D.; et al. Amyloid-beta peptide-receptor for advanced glycation end product interaction elicits neuronal expression of macrophage-colony stimulating factor: A proinflammatory pathway in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1997, 94, 5296–5301. [Google Scholar] [CrossRef]
- Hofmann, M.A.; Drury, S.; Fu, C.; Qu, W.; Taguchi, A.; Lu, Y.; Avila, C.; Kambham, N.; Bierhaus, A.; Nawroth, P.; et al. RAGE mediates a novel proinflammatory axis: A central cell surface receptor for S100/calgranulin polypeptides. Cell 1999, 97, 889–901. [Google Scholar] [CrossRef]
- Hori, O.; Brett, J.; Slattery, T.; Cao, R.; Zhang, J.; Chen, J.X.; Nagashima, M.; Lundh, E.R.; Vijay, S.; Nitecki, D. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J. Biol. Chem. 1995, 270, 25752–25761. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Huang, Y.; Wu, X.; Lin, X.; Xu, J.; Chen, X.; Bai, X.; Li, Q. Endogenous secretory receptor for advanced glycation end products protects endothelial cells from AGEs induced apoptosis. BioMed Res. Int. 2018, 2018, 8216578. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, M.; Watanabe, T.; Ichikawa, T.; Toriumi, K.; Horiuchi, Y.; Kobori, A.; Kushima, I.; Hashimoto, R.; Fukumoto, M.; Koike, S.; et al. The regulation of soluble receptor for AGEs contributes to carbonyl stress in schizophrenia. BioMed Res. Int. 2018, 2018, 8216578. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, M.; Jiang, X.; Ogawa, T.; Ohnishi, T.; Yoshihara, S.; Balan, S.; Yoshikawa, T.; Hirokawa, N. Enhanced carbonyl stress induces irreversible multimerization of CRMP2 in schizophrenia pathogenesis. Life Sci. Alliance 2019, 2, e201900478. [Google Scholar] [CrossRef] [PubMed]
- Brekk, O.R.; Makridakis, M.; Mavroeidi, P.; Vlahou, A.; Xilouri, M.; Stefanis, L. Impairment of chaperone-mediated autophagy affects neuronal homeostasis through altered expression of DJ-1 and CRMP-2 proteins. Mol. Cell. Neurosci. 2019, 95, 1–12. [Google Scholar] [CrossRef]
- Fang, W.; Gao, G.; Zhao, H.; Xia, Y.; Guo, X.; Li, N.; Li, Y.; Yang, Y.; Chen, L.; Wang, Q.; et al. Role of the Akt/GSK-3β/CRMP-2 pathway in axon degeneration of dopaminergic neurons resulting from MPP+ toxicity. Brain Res. 2015, 30, 9–19. [Google Scholar] [CrossRef]
- Hensley, K.; Venkova, K.; Christov, A.; Gunning, W.; Park, J. Collapsin response mediator protein-2: An emerging pathologic feature and therapeutic target for neurodisease indications. Mol. Neurobiol. 2011, 43, 180–191. [Google Scholar] [CrossRef]
- Scheijen, J.L.; van de Waarenburg, M.P.; Stehouwer, C.D.; Schalkwijk, C.G. Measurement of pentosidine in human plasma protein by a single-column high-performance liquid chromatography method with fluorescence detection. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 610–614. [Google Scholar] [CrossRef]
- Ohnuma, T.; Nishimon, S.; Takeda, M.; Sannohe, T.; Katsuta, N.; Arai, H. Carbonyl stress and microinflammation-related molecules as potential biomarkers in schizophrenia. Front. Psychiatry 2018, 9, 82. [Google Scholar] [CrossRef]
- Ohnishi, T.; Balan, S.; Toyoshima, M.; Maekawa, M.; Ohba, H.; Watanabe, A.; Iwayama, Y.; Fujita, Y.; Tan, Y.; Hisano, Y.; et al. Investigation of betaine as a novel psychotherapeutic for schizophrenia. EBioMedicine 2019, 45, 432–446. [Google Scholar] [CrossRef]
- Park, S.Y.; Hwang, I.S.; Lee, H.J.; Song, C.E. Biomimetic catalytic transformation of toxic α-oxoaldehydes to high-value chiral α-hydroxythioesters using artificial glyoxalase I. Nat. Commun. 2017, 4, 14877. [Google Scholar] [CrossRef]
- Distler, M.G.; Palmer, A.A. Role of Glyoxalase 1 (Glo1) and methylglyoxal (MG) in behavior: Recent advances and mechanistic insights. Front. Genet. 2012, 3, 250. [Google Scholar] [CrossRef]
- Emendato, A.; Milordini, G.; Zacco, E.; Sicorello, A.; Dal Piaz, F.; Guerrini, R.; Thorogate, R.; Picone, D.; Pastore, A. Glycation affects fibril formation of Aβ peptides. J. Biol. Chem. 2018, 293, 13100–13111. [Google Scholar] [CrossRef]
- Cai, H.Q.; Catts, V.S.; Webster, M.J.; Galletly, C.; Liu, D.; O’Donnell, M.; Weickert, T.W.; Weickert, C.S. Increased macrophages and changed brain endothelial cell gene expression in the frontal cortex of people with schizophrenia displaying inflammation. Mol. Psychiatry 2018. [Google Scholar] [CrossRef]
- López-Díez, R.; Shekhtman, A.; Ramasamy, R.; Schmidt, A.M. Cellular mechanisms and consequences of glycation in atherosclerosis and obesity. Biochim. Biophys. Acta 2016, 1862, 2244–2252. [Google Scholar] [CrossRef]
- van der Flier, W.M.; Skoog, I.; Schneider, J.A.; Pantoni, L.; Mok, V.; Chen, C.L.H.; Scheltens, P. Vascular cognitive impairment. Nat. Rev. Dis. Primers 2018, 4, 18003. [Google Scholar] [CrossRef]
- Kim, E.; Keskey, Z.; Kang, M.; Kitchen, C.; Bentley, W.E.; Chen, S.; Kelly, D.L.; Payne, G.F. Validation of oxidative stress assay for schizophrenia. Schizophr. Res. 2019, 212, 126–133. [Google Scholar] [CrossRef]
- Boll, K.M.; Noto, C.; Bonifácio, K.L.; Bortolasci, C.C.; Gadelha, A.; Bressan, R.A.; Barbosa, D.S.; Maes, M.; Moreira, E.G. Oxidative and nitrosative stress biomarkers in chronic schizophrenia. Psychiatry Res. 2017, 253, 43–48. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef]
- Solberg, D.K.; Refsum, H.; Andreassen, O.A.; Bentsen, H. A five-year follow-up study of antioxidants, oxidative stress and polyunsaturated fatty acids in schizophrenia. Acta Neuropsychiatr. 2019, 31, 202–212. [Google Scholar] [CrossRef]
- Byron, K.; Bitanihirwe, M.; Tsung-Ung, W.; Woo, M. Oxidative stress in schizophrenia: An integrated approach. Neurosci. Biobehav. Rev. 2011, 35, 878–893. [Google Scholar] [CrossRef]
- Wass, C.; Klamer, D.; Fejgin, K.; Pålsson, E. The importance of nitric oxide in social dysfunction. Behav. Brain Res. 2009, 200, 113–116. [Google Scholar] [CrossRef]
- Marrocco, I.; Altieri, F.; Peluso, I. Measurement and clinical significance of biomarkers of oxidative stress in humans. Oxid. Med. Cell. Longev. 2017. [Google Scholar] [CrossRef]
- Brieger, K.; Schiavone, S.; Miller, F.J.; Krause, K.H. Reactive oxygen species: From health to disease. Swiss Med. Wkly. 2012, 142, 1–14. [Google Scholar] [CrossRef]
- Korotkova, E.I.; Misini, B.; Dorozhko, E.V.; Bukkel, M.V.; Plotnikov, E.V.; Linert, W. Study of OH radicals in human serum blood of healthy individuals and those with pathological schizophrenia. Int. J. Mol. Sci. 2011, 12, 401–410. [Google Scholar] [CrossRef]
- Adibhatla, R.M.; Hatcher, J.F. Altered lipid metabolism in brain injury and disorders. Subcell. Biochem. 2008, 49, 241–268. [Google Scholar] [CrossRef]
- Śmierciak, N.; Krzyściak, W.; Szwajca, M.; Szczęsny-Małysiak, E.; Kij, A.; Gawęda, P.; Chłopicki, S.; Pilecki, M. Short-term improvement in clinical symptoms of first episode psychosis is associated with a fall in plasma nitrite concentration, worsering lipid profile and systemic inflammation—A pilot study. Psychiatr. Q. Manuscript PSAQ-D-20-00033 status; added to the editors on 12 Feb 2020.
- Olson, K.R. Hydrogen sulfide, reactive sulfur species and coping with reactive oxygen species. Free Radic. Biol. Med. 2019, 140, 74–83. [Google Scholar] [CrossRef]
- Koike, S.; Kayama, T.; Yamamoto, S.; Komine, D.; Tanaka, R.; Nishimoto, S.; Suzuki, T.; Kishida, A.; Ogasawara, Y. Polysulfides protect SH-SY5Y cells from methylglyoxal-induced toxicity by suppressing protein carbonylation: A possible physiological scavenger for carbonyl stress in the brain. NeuroToxicology 2016, 55, 13–19. [Google Scholar] [CrossRef]
- Giles, G.I.; Nasim, M.J.; Ali, W.; Jacob, C. The reactive sulfur species concept: 15 years on. Antioxidants 2017, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Wei, F.Y.; Watanabe, S.; Hirayama, M.; Ohuchi, Y.; Fujimura, A.; Kaitsuka, T.; Ishii, I.; Sawa, T.; Nakayama, H.; et al. Reactive sulfur species regulate tRNA methylthiolation and contribute to insulin secretion. Nucleic Acids Res. 2017, 45, 435–445. [Google Scholar] [CrossRef]
- Berry, T.; Abohamza, E.; Moustafa, A.A. Treatment-resistant schizophrenia: Focus on the transsulfuration pathway. Rev. Neurosci. 2019, 31, 219–232. [Google Scholar] [CrossRef]
- Pontes-Neto, J.G.; Fontes, D.A.F.; de Lyra, M.A.M.; Brito, M.D.R.M.; Chaves, L.L.; Rolim-Neto, P.J.; De La Roca Soares, M.F.; Quintans Júnior, L.J.; de Freitas, R.M.; Soares-Sobrinho, J.L. Evaluation of antioxidant potencial of novel CaAl and NiAl layered double hydroxides loaded with olanzapine. Life Sci. 2018, 207, 246–252. [Google Scholar] [CrossRef]
- Chan, S.T.; McCarthy, M.J.; Vawter, M.P. Psychiatric drugs impact mitochondrial function in brain and other tissues. Schizophr. Res. 2019. [Google Scholar] [CrossRef]
- Leza, J.C.; Bueno, B.; Bioque, M.; Arango, C.; Parellada, M.; Do, K.; O’Donnell, P.; Bernardo, M. Inflammation in schizophrenia: A question of balance. Neurosci. Biobehav. Rev. 2015, 55, 612–626. [Google Scholar] [CrossRef]
- Frustaci, A.; Neri, M.; Cesario, A.; Adams, J.B.; Domenici, E.; Dalla Bernardina, B.; Bonassi, S. Oxidative stress-related biomarkers in autism: Systematic review and meta-analyses. Free Radic. Biol. Med. 2012, 52, 2128–2141. [Google Scholar] [CrossRef]
- Flatow, J.; Buckley, P.; Miller, B.J. Meta-analysis of oxidative stress in schizophrenia. Biol. Psychiatry 2013, 74, 400–409. [Google Scholar] [CrossRef]
- Liochev, S.I. Reactive oxygen species and the free radical theory of aging. Free Radic. Biol. Med. 2013, 60, 1–4. [Google Scholar] [CrossRef]
- Campolo, N.; Issoglio, F.M.; Estrin, D.A.; Bartesaghi, S.R.R. 3-Nitrotyrosine and related derivatives in proteins: Precursors, radical intermediates and impact in function. Essays Biochem. 2020, 64, 111–133. [Google Scholar] [CrossRef]
- Hashimoto, K. Recent advances in the early intervention in schizophrenia: Future direction from preclinical findings. Curr. Psychiatry Rep. 2019, 21. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.; Cotter, D.R. Special issue: Psychosis from early intervention to treatment resistance. Ir. J. Psychol. Med. 2019, 36, 239–241. [Google Scholar] [CrossRef] [PubMed]
- Potkin, S.G.; Kane, J.M.; Correll, C.; Lindenmayer, J.-P.; Agid, O.; Marder, S.R.; Olfson, M.; Howes, O.D. The neurobiology of treatment-resistant schizophrenia: Paths to antipsychotic resistance and a roadmap for future research. npj Schizophr. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Dainin, K.; Ide, R.; Maeda, A.; Suyama, K.; Akagawa, M. Pyridoxamine scavenges protein carbonyls and inhibits protein aggregation in oxidative stress-induced human HepG2 hepatocytes. Biochem. Biophys. Res. Commun. 2017, 486, 845–851. [Google Scholar] [CrossRef]
- Fedorova, M.; Bollineni, R.C.; Hoffmann, R. Protein carbonylation as a major hallmark of oxidative damage: Update of analytical strategies. Mass Spectrom. Rev. 2014, 33, 79–97. [Google Scholar] [CrossRef]
- Vázquez, G.; Caballero, A.B.; Kokinda, J.; Hijano, A.; Sabaté, R.; Gamez, P. Copper, dityrosine cross-links and amyloid-β aggregation. J. Biol. Inorg. Chem. 2019, 24, 1217–1229. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Li, B.; Ge, Y.; Xu, Y.; Lu, Y.; Yang, Y.; Han, L.; Jiang, Y.; Shi, Y.; Le, G. Spatial learning and memory impairment in growing mice induced by major oxidized tyrosine product dityrosine. J. Agric. Food Chem. 2019, 67, 9039–9049. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, H.; Yan, B.; Zhang, T.; Gao, Y.; Shi, Y.; Le, G. Health effects of dietary oxidized tyrosine and dityrosine administration in mice with nutrimetabolomic strategies. J. Agric. Food Chem. 2017, 65, 6957–6971. [Google Scholar] [CrossRef]
- Levine, R.L.; Moskovitz, J.; Stadtman, E.R. Oxidation of methionine in proteins: Roles in antioxidant defense and cellular regulation. IUBMB Life 2000, 50, 301–307. [Google Scholar] [CrossRef]
- Reiterer, M.; Schmidt-Kastner, R.; Milton, S.L. Methionine sulfoxide reductase (Msr) dysfunction in human brain disease. Free Radic. Res. 2019, 53, 1144–1154. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Davies, M.J. Evidence for the formation of adducts and S-(carboxymethyl)cysteine on reaction of α-dicarbonyl compounds with thiol groups on amino acids, peptides, and proteins. Chem. Res. Toxicol. 2005, 18, 1232–1241. [Google Scholar] [CrossRef] [PubMed]
- Miglio, G.; Sabatino, A.D.; Veglia, E.; Giraudo, M.T.; Beccuti, M.; Cordero, F. A computational analysis of S-(2-succino)cysteine sites in proteins. Biochim. Biophys. Acta Proteins Proteom. 2016, 1864, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Blatnik, M.; Frizzell, N.; Thorpe, S.R.; Bayes, J.W. Inactivation of glyceraldehyde-3-phosphate dehydrogenase by fumarate in diabetes: Formation of S-(2-succinyl)cysteine, a novel chemical modification of protein and possible biomarker of mitochondrial stress. Diabetes 2008, 57, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Andrade, C. Carboxymethyl-lysine: Thirty years of investigation in the field of AGE formation. Food Funct. 2016, 7, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, D.W. Homocysteine and vitamins in cardiovascular disease. Clin. Chem. 1998, 44, 1833–1843. [Google Scholar] [PubMed]
- Sung, C.C.; Hsu, Y.C.; Chen, C.C.; Lin, Y.F.; Wu, C.C. Oxidative stress and nucleic acid oxidation in patients with chronic kidney disease. Oxid. Med. Cell. Longev. 2013, 2013, 301982. [Google Scholar] [CrossRef]
- de Jager, C.A. Critical levels of brain atrophy associated with homocysteine and cognitive decline. Neurobiol. Aging 2014, 35, S35–S39. [Google Scholar] [CrossRef]
- Wang, L.J.; Lin, P.Y.; Lee, Y.; Huang, Y.C.; Wu, C.C.; Hsu, S.T.; Chen, C.C.; Chong, M.Y.; Lin, C.H.; Hung, C.F. Increased serum levels of cysteine in patients with schizophrenia: A potential marker of cognitive function preservation. Schizophr. Res. 2018, 192, 391–397. [Google Scholar] [CrossRef]
- Gaschler, M.; Stockwell, B. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Yao, J.K.; Keshavan, M.S. Antioxidants, redox signaling, and pathophysiology in Schizophrenia: An integrative view. Antioxid. Redox Signal. 2011, 15, 2011–2035. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.L.; Hwu, H.G.; Hwang, T.J.; Hsieh, M.H.; Liu, C.C.; Lin-Shiau, S.Y.; Liu, C.M. Clinical implications of oxidative stress in schizophrenia: Acute relapse and chronic stable phase. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2020, 99, 109868. [Google Scholar] [CrossRef] [PubMed]
- Petronijević, N.D.; Mićić, D.; Duričić, B.; Marinković, D.; Paunović, V.R. Substrate kinetics of erythrocyte membrane Na,K-ATPase and lipid peroxides in schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2003, 27, 431–440. [Google Scholar] [CrossRef]
- del Rio, D.; Stewart, A.J.; Pellegrini, N. A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr. Metab. Cardiovasc. Dis. 2005, 15, 316–328. [Google Scholar] [CrossRef]
- Romano, A.; Serviddio, G.; Calcagnini, S.; Villani, R.; Giudetti, A.; Cassano, T.; Gaetani, S. Linking lipid peroxidation and neuropsychiatric disorders: Focus on 4-hydroxy-2-nonenal. Free Radic. Biol. Med. 2017, 111, 281–293. [Google Scholar] [CrossRef]
- Mertsch, K.; Blasig, I.; Grune, T. 4-Hydroxynonenal impairs the permeability of an in vitro rat blood-brain barrier. Neurosci. Lett. 2001, 314, 135–138. [Google Scholar] [CrossRef]
- Proudfoot, J.; Barden, A.; Mori, T.A.; Burke, V.; Croft, K.D.; Beilin, L.J.; Puddey, I.B. Measurement of urinary F2-isoprostanes as markers of in Vivo lipid peroxidation—A comparison of enzyme immunoassay with gas chromatography/mass spectrometry. Anal. Biochem. 1999, 272, 209–215. [Google Scholar] [CrossRef]
- Montuschi, P.; Barnes, P.J.; Roberts, L.J. Isoprostanes: Markers and mediators of oxidative stress. FASEB J. 2004, 18, 1791–1800. [Google Scholar] [CrossRef]
- Davies, S.S.; Amarnath, V.; Montine, K.S.; Bernoud-Hubac, N.; Boutaud, O.; Montine, T.J.; Roberts, L.J., II. Effects of reactive gamma-ketoaldehydes formed by the isoprostane pathway (isoketals) and cyclooxygenase pathway (levuglandins) on proteasome function. FASEB J. 2002, 16, 715–717. [Google Scholar] [CrossRef]
- Kohen, R.; Nyska, A. Oxidation of biological systeme. Toxicol. Pathol. 2002, 30, 620–650. [Google Scholar] [CrossRef]
- Fenga, C.; Gangemi, S.; Teodoro, M.; Rapisarda, V.; Golokhvast, K.; Docea, A.; Tsatsakis, A.M.; Costa, C. 8-hydroxydeoxyguanosine as a biomarker of oxidative DNA damage in workers exposed to low-dose benzene. Toxicol. Rep. 2017, 4, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Dedon, P.C.; Tannenbaum, S.R. Reactive nitrogen species in the chemical biology of inflammation. Arch. Biochem. Biophys. 2004, 423, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Shamanna, R.A.; Croteau, D.L.; Bohr, V.A. Base excision DNA repair levels in mitochondrial lysates of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Michel, T.M.; Sheldrick, A.J.; Camara, S.; Grnblatt, E.; Schneider, F.; Riederer, P. Alteration of the pro-oxidant xanthine oxidase (XO) in the thalamus and occipital cortex of patients with schizophrenia. World J. Biol. Psychiatry 2011, 12, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Asagoshi, K.; Terato, H.; Suzuki, T.; Ide, H. Assessment of the genotoxic potential of nitric oxide-induced guanine lesions by in vitro reactions with Escherichia coli DNA polymerase I. Mutagenesis 2005, 20, 209–216. [Google Scholar] [CrossRef]
- Lin, C.Y.; Lee, H.L.; Hwang, Y.T.; Su, T.C. The association between total serum isomers of per- and polyfluoroalkyl substances, lipid profiles, and the DNA oxidative/nitrative stress biomarkers in middle-aged Taiwanese adults. Environ. Res. 2020, 182, 109064. [Google Scholar] [CrossRef]
- Horiike, S.; Kawanishi, S.; Kaito, M.; Ma, N.; Tanaka, H.; Fujita, N.; Iwasa, M.; Kobayashi, Y.; Hiraku, Y.; Oikawa, S.; et al. Accumulation of 8-nitroguanine in the liver of patients with chronic hepatitis C. J. Hepatol. 2005, 43, 403–410. [Google Scholar] [CrossRef]
- Murata, M. Inflammation and cancer. Environ. Health Prev. Med. 2018, 23, 1–8. [Google Scholar] [CrossRef]
- Hoki, Y.; Murata, M.; Hiraku, Y.; Ma, N.; Matsumine, A.; Uchida, A.; Kawanishi, S. 8-nitroguanine as a potential biomarker for progression of malignant fibrous histiocytoma, a model of inflammation-related cancer. Oncol. Rep. 2007, 18, 1165–1169. [Google Scholar] [CrossRef][Green Version]
- Sawa, T.; Tatemichi, M.; Akaike, T.; Barbin, A.; Ohshima, H. Analysis of urinary 8-nitroguanine, a marker of nitrative nucleic acid damage, by high-performance liquid chromatography-electrochemical detection coupled with immunoaffinity purification: Association with cigarette smoking. Free Radic. Biol. Med. 2006, 40, 711–720. [Google Scholar] [CrossRef]
- Fedeles, B.I.; Freudenthal, B.D.; Yau, E.; Singh, V.; Chang, S.; Li, D.; Delaney, J.C.; Wilson, S.H.; Essigmann, J. Intrinsic mutagenic properties of 5-chlorocytosine: A mechanistic connection between chronic inflammation and cancer. Proc. Natl. Acad. Sci. USA 2015, 112, E4571–E4580. [Google Scholar] [CrossRef]
- McGregor, D. The genetic toxicology of N-nitrosodiphenylamine. Mutat. Res. Rev. Genet. Toxicol. 1994, 317, 195–211. [Google Scholar] [CrossRef]
- Knaapen, A.M.; Güngör, N.; Schins, R.P.F.; Borm, P.J.A.; van Schooten, F.J. Neutrophils and respiratory tract DNA damage and mutagenesis: A review. Mutagenesis 2006, 21, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.L.; Kavli, B.; Slupphaug, G.; Dianov, G.L. NEIL1 is the major DNA glycosylase that processes 5-hydroxyuracil in the proximity of a DNA single-strand break. Biochemistry 2007, 46, 4158–4163. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Markesbery, W.R. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Res. 2007, 35, 7497–7504. [Google Scholar] [CrossRef]
- Liebert, M.A.; Markesbery, W.R.; Lovell, M.A. DNA oxidation in Alzheimer’s disease clinical manifestations of Alzheimer’s disease. Antioxid. Redox Signal. 2006, 8, 2039–2045. [Google Scholar] [CrossRef]
- Rozalski, R.; Gackowski, D.; Siomek-Gorecka, A.; Starczak, M.; Modrzejewska, M.; Banaszkiewicz, Z.; Olinski, R. Urinary 5-hydroxymethyluracil and 8-oxo-7, 8-dihydroguanine as potential biomarkers in patients with colorectal cancer. Biomarkers 2015, 5804, 287–291. [Google Scholar] [CrossRef]
- Olinski, R.; Starczak, M.; Gackowski, D. Enigmatic 5-hydroxymethyluracil: Oxidatively modified base, epigenetic mark or both? Mutat. Res. Rev. Mutat. Res. 2016, 767, 59–66. [Google Scholar] [CrossRef]
- Mueller, S.; Peccerella, T.; Qin, H.; Glassen, K.; Waldherr, R.; Flechtenmacher, C.; Straub, B.K.; Millonig, G.; Stickel, F.; Bruckner, T.; et al. Carcinogenic etheno DNA adducts in alcoholic liver disease: Correlation with cytochrome P-4502E1 and fibrosis. Alcohol. Clin. Exp. Res. 2018, 42, 252–259. [Google Scholar] [CrossRef]
- Liu, T.; Cai, J.P.; Zhang, L.Q.; Sun, N.; Cui, J.; Wang, H.; Yang, J.F. The mechanism of RNA oxidation involved in the development of heart failure. Free Radic. Res. 2019, 53, 910–921. [Google Scholar] [CrossRef]
- Guo, C.; Chen, Q.; Chen, J.; Yu, J.; Hu, Y.; Zhang, S.; Zheng, S. 8-hydroxyguanosine as a possible RNA oxidative modification marker in urine from colorectal cancer patients: Evaluation by ultra performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2020, 1136, 121931. [Google Scholar] [CrossRef]
- Linhart, K.B.; Glassen, K.; Peccerella, T.; Waldherr, R.; Linhart, H.; Bartsch, H.; Seitz, H. The generation of carcinogenic etheno-DNA adducts in the liver of patients with nonalcoholic fatty liver disease. Hepatobiliary Surg. Nutr. 2015, 4, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.D.; Snyder, S.H. Therapeutic applications of cysteamine and cystamine in neurodegenerative and neuropsychiatric diseases. Front. Neurol. 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Vojdani, A.; Geffard, M.; Moreira, E.G.; Barbosa, D.S.; Michelin, A.P.; Semeão, L.O.; Sirivichayakul, S.; Kanchanatawan, B. Schizophrenia phenomenology comprises a bifactorial general severity and a single-group factor, which are differently associated with neurotoxic immune and immune-regulatory pathways. Biomol. Concepts 2019, 10, 209–225. [Google Scholar] [CrossRef]
- Andreazza, A.C.; Wang, J.F.; Salmasi, F.; Shao, L.; Young, L.T. Specific subcellular changes in oxidative stress in prefrontal cortex from patients with bipolar disorder. J. Neurochem. 2013, 127, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.W.; Sun, Y.; Chen, N.; Chen, S.; Xiu, M.H.; Zhang, X.Y. Interaction of oxidative stress and BDNF on executive dysfunction in patients with chronic schizophrenia. Psychoneuroendocrinology 2020, 111, 104473. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, M.; Arai, M.; Kobori, A.; Ichikawa, T.; Toriumi, K.; Niizato, K.; Oshima, K.; Okazaki, Y.; Yoshikawa, T.; Amano, N.; et al. Clinical features of schizophrenia with enhanced carbonyl stress. Schizophr. Bull. 2014, 40, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.; Kalani, A.; Rai, S.; Swarnkar, S.; Tota, S.; Nath, C.; Tyagi, N. Mechanism of oxidative stress and synapse dysfunction in the pathogenesis of Alzheimer’s disease: Understanding the therapeutics strategies. Mol. Neurobiol. 2016, 53, 648–661. [Google Scholar] [CrossRef]
- Ogawa, F.; Malavasi, E.L.V.; Crummie, D.K.; Eykelenboom, J.E.; Soares, D.C.; Shaun, M.; Porteous, D.J.; Millar, J.K. DISC1 complexes with TRAK1 and Miro1 to modulate anterograde axonal mitochondrial trafficking. Hum. Mol. Genet. 2014, 23, 906–919. [Google Scholar] [CrossRef]
- Xu, Y.; Ren, J.; Ye, H. Association between variations in the disrupted in schizophrenia 1 gene and schizophrenia: A meta-analysis. Gene 2018, 651, 94–99. [Google Scholar] [CrossRef]
- Mattson, M.P.; LaFerla, F.M.; Chan, S.L.; Leissring, M.A.; Shepel, P.N.; Geiger, J.D. Calcium signaling in the ER: Its role in neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2000, 23, 222–229. [Google Scholar] [CrossRef]
- Johnson, A.W.; Jaaro-Peled, H.; Shahani, N.; Sedlak, T.W.; Zoubovsky, S.; Burruss, D.; Emiliani, F.; Sawa, A.; Gallagher, M. Cognitive and motivational deficits together with prefrontal oxidative stress in a mouse model for neuropsychiatric illness. Proc. Natl. Acad. Sci. USA 2013, 110, 12462–12467. [Google Scholar] [CrossRef] [PubMed]
- Carless, M.A.; Glahn, D.C.; Winkler, A.M.; Blangero, J. Dissecting the functions of DISC1. Mol. Psychiatry 2011, 16, 1063. [Google Scholar] [CrossRef][Green Version]
- Forsyth, J.K.; Lewis, D.A. Mapping the consequences of impaired synaptic plasticity in schizophrenia through development: An integrative model for diverse clinical features. Trends Cogn. Sci. 2017, 21, 760–778. [Google Scholar] [CrossRef] [PubMed]
- Abboud, R.; Noronha, C.; Diwadkar, V.A. Motor system dysfunction in the schizophrenia diathesis: Neural systems to neurotransmitters. Eur. Psychiatry 2017, 44, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Elustondo, P.A.; Nichols, M.; Robertson, G.S.; Pavlov, E.V. Mitochondrial Ca2+ uptake pathways. J. Bioenerg. Biomembr. 2017, 49, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Hroudová, J.; Fišar, Z. Control mechanisms in mitochondrial oxidative phosphorylation. Neural Regen. Res. 2013, 8, 363–375. [Google Scholar] [CrossRef]
- Li, J.; Ryan, S.K.; Deboer, E.; Cook, K.; Fitzgerald, S.; Lachman, H.; Wallace, D.C.; Goldberg, E.; Anderson, S. Mitochondrial deficits in human iPSC-derived neurons from patients with 22q11.2 deletion syndrome and schizophrenia. Transl. Psychiatry 2019, 9. [Google Scholar] [CrossRef]
- Xia, J.; Wang, H.; Zhang, Q.; Han, Z. Modulation of P2X purinoceptor 3 (P2x3) in pentylenetetrazole-induced kindling epilepsy in rats. Med. Sci. Monit. 2018, 24, 6165–6177. [Google Scholar] [CrossRef]
- Papa, S.; Sardanelli, A.M.; Scacco, S.; Technikova-Dobrova, Z. cAMP-dependent protein kinase and phosphoproteins in mammalian mitochondria. An extension of the cAMP-mediated intracellular signal transduction. FEBS Lett. 1999, 444, 245–249. [Google Scholar] [CrossRef]
- Signorile, A.; Sardanelli, A.M.; Nuzzi, R.; Papa, S. Serine (threonine) phosphatase(s) acting on cAMP-dependent phosphoproteins in mammalian mitochondria. FEBS Lett. 2002, 512, 91–94. [Google Scholar] [CrossRef]
- Lee, I.; Bender, E.; Kadenbach, B. Control of mitochondrial membrane potential and ROS formation by reversible phosphorylation of cytochrome c oxidase. Mol. Cell. Biochem. 2002, 234–235, 63–70. [Google Scholar] [CrossRef]
- Klaunig, J.E. Oxidative stress and cancer. Curr. Pharm. Des. 2019, 24, 4771–4778. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.; Lin, C.l.G. Quantification of oxidized RNAs in Alzheimer’s disease. Neurobiol. Aging 2006, 27, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Katerji, M.; Filippova, M.; Duerksen-Hughes, P. Approaches and methods to measure oxidative stress in clinical samples: Research applications in the cancer field. Oxid. Med. Cell. Longev. 2019, 2019, 1279250. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Alessio, H.M.; Cutler, R.G. Oxygen-radical absorbance capacity assay for antioxidants. Free Radic. Biol. Med. 1993, 14, 303–311. [Google Scholar] [CrossRef]
- Miller, N.J.; Rice-Evans, C.; Davies, M.J.; Gopinathan, V.; Milner, A. A novel method for measuring antioxidant capacity and its application to monitoring the antioxidant status in premature neonates. Clin. Sci. 1993, 84, 407–412. [Google Scholar] [CrossRef]
- Ialongo, C. Preanalytic of total antioxidant capacity assays performed in serum, plasma, urine and saliva. Clin. Biochem. 2017, 50, 356–363. [Google Scholar] [CrossRef]
- Lissi, E.; Salim-Hanna, M.; Pascual, C.; del Castillo, M.D. Evaluation of total antioxidant potential (TRAP) and total antioxidant reactivity from luminol-enhanced chemiluminescence measurements. Free Radic. Biol. Med. 1995, 18, 153–158. [Google Scholar] [CrossRef]
- Benzie, I.F.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: The FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef]
- Erel, O. A novel automated direct measurement method for total antioxidant capacity using a new generation, more stable ABTS radical cation. Clin. Biochem. 2004, 37, 277–285. [Google Scholar] [CrossRef]
- Lissi, E.; Pascual, C.; del Castillo, M.D. Luminol luminescence induced by 2,2′-Azo-Bis(2-Amidinopropane) thermolysis. Free Radic. Res. Commun. 1992, 17, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Güçlü, K.; Kıbrıslıoğlu, G.; Özyürek, M.; Apak, R. Development of a fluorescent probe for measurement of peroxyl radical scavenging activity in biological samples. J. Agric. Food Chem. 2014, 26, 1839–1845. [Google Scholar] [CrossRef] [PubMed]
- Chapple, I.L.C.; Mason, G.I.; Garner, I.; Matthews, J.B.; Thorpe, G.H.; Maxwell, S.R.J.; Whitehead, T.P. Enhanced chemiluminescent assay for measuring the total antioxidant capacity of serum, saliva and crevicular fluid. Ann. Clin. Biochem. 1997, 34, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959, 82, 70–77. [Google Scholar] [CrossRef]
- Wayner, D.D.M.; Burton, G.W.; Ingold, K.U.; Locke, S. Quantitative measurement of the total, peroxyl radical-trapping antioxidant capability of human blood plasma by controlled peroxidation. The important contribution made by plasma proteins. FEBS Lett. 1985, 187, 33–37. [Google Scholar] [CrossRef]
- Lorente, L.; Martín, M.M.; Pérez-Cejas, A.; González-Rivero, A.F.; Abreu-González, P.; Ramos, L.; Argueso, M.; Solé-Violán, J.; Cáceres, J.J.; Jiménez, A.; et al. Traumatic brain injury patients mortality and serum total antioxidant capacity. Brain Sci. 2020, 18, 110. [Google Scholar] [CrossRef]
- Chen, B.; Shen, Z.; Wu, D.; Xie, X.; Xu, X.; Lv, L.; Dai, H.; Chen, J.; Gan, X. Glutathione peroxidase 1 promotes NSCLC resistance to cisplatin via ROS-induced activation of PI3K/AKT pathway. BioMed Res. Int. 2019, 27, 7640547. [Google Scholar] [CrossRef]
- Sinan, K.I.; Chiavaroli, A.; Orlando, G.; Bene, K.; Zengin, G.; Cziáky, Z.; Jekő, J.; Mahomoodally, M.F.; Picot-Allain, M.C.N.; Menghini, L.; et al. Biopotential of bersama abyssinica fresen stem bark extracts: UHPLC profiles, antioxidant, enzyme inhibitory, and antiproliferative propensities. Antioxidant 2020, 17, 163. [Google Scholar] [CrossRef]
- Morera-Fumero, A.L.; Díaz-Mesa, E.; Abreu-Gonzalez, P.; Fernandez-Lopez, L.; Cejas-Mendez, M. Low levels of serum total antioxidant capacity and presence at admission and absence at discharge of a day/night change as a marker of acute paranoid schizophrenia relapse. Psychiatry Res. 2017, 249, 200–205. [Google Scholar] [CrossRef]
- Benzie, I.F.F.; Strain, J.J. Ferric reducing/antioxidant power assay: Direct measure of total antioxidant activity of biological fluids and modified version for simultaneous measurement of total antioxidant power and ascorbic acid concentration. Methods Enzymol. 1999, 299, 15–27. [Google Scholar] [CrossRef]
- Pedrini, M.; Massuda, R.; Fries, G.R.; de Bittencourt Pasquali, M.A.; Schnorr, C.E.; Moreira, J.C.F.; Gama, C.S. Similarities in serum oxidative stress markers and inflammatory cytokines in patients with overt schizophrenia at early and late stages of chronicity. J. Psychiatr. Res. 2012, 46, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Dresch, M.T.K.; Rossato, S.B.; Kappel, V.D.; Biegelmeyer, R.; Hoff, M.L.M.; Mayorga, P.; Zuanazzi, J.A.S.; Henriques, A.T.; Moreira, J.C.F. Optimization and validation of an alternative method to evaluate total reactive antioxidant potential. Anal. Biochem. 2009, 385, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Koracevic, D.; Koracevic, G.; Djordjevic, V.; Andrejevic, S.; Cosic, V. Method for the measurement of antioxidant activity in human fluids. J. Clin. Pathol. 2001, 54, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Krawczuk-Rybak, M.; Panasiuk, A.; Czygier, M.; Muszynska-Roslan, K.; Wysocka, J.; Szmitkowski, M. Total antioxidant status (TAS) in childhood cancer survivors. Folia Histochem. Cytobiol. 2012, 50, 468–472. [Google Scholar] [CrossRef]
- Pinchuk, I.; Shoval, H.; Dotan, Y.; Lichtenberg, D. Evaluation of antioxidants: Scope, limitations and relevance of assays. Chem. Phys. Lipids 2012, 165, 638–647. [Google Scholar] [CrossRef]
- Ghiselli, A.; Serafini, M.; Maiani, G.; Azzini, E.; Ferro-Luzzi, A. A fluorescence-based method for measuring total plasma antioxidant capability. Free Radic. Biol. Med. 1995, 18, 29–36. [Google Scholar] [CrossRef]
- Kim, E.; Winkler, T.E.; Kitchen, C.; Mijeong, K.; Banis, G.; Bentley, W.E.; Kelly, D.L.; Ghodssi, R.; Payne, G.F. Redox probing for chemical information of oxidative stress. Anal. Chem. 2017, 89, 1583–1592. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Tan, Y.L.; Cao, L.Y.; Wu, G.Y.; Xu, Q.; Shen, Y.; Zhou, D.F. Antioxidant enzymes and lipid peroxidation in different forms of schizophrenia treated with typical and atypical antipsychotics. Schizophr. Res. 2006, 81, 291–300. [Google Scholar] [CrossRef]
- Czerska, M.; Mikołajewska, K.; Zieliński, M.; Gromadzińska, J.; Wąsowicz, W. Today’s oxidative stress markers. Med. Pract. 2015, 66, 393–405. [Google Scholar] [CrossRef]
- Peluso, I.; Raguzzini, A. Salivary and urinary total antioxidant capacity as biomarkers of oxidative stress in humans. Pathol. Res. Int. 2016, 2016, 5480267. [Google Scholar] [CrossRef]
- Avval, F.Z.; Mahmoudi, N.; Tirkani, A.N.; Jarahi, L.; Alamdari, D.H.; Sadjadi, S.A. Determining pro-oxidant antioxidant balance (PAB) and total antioxidant capacity (TAC) in patients with schizophrenia. Iran. J. Psychiatry 2018, 13, 223–227. [Google Scholar]
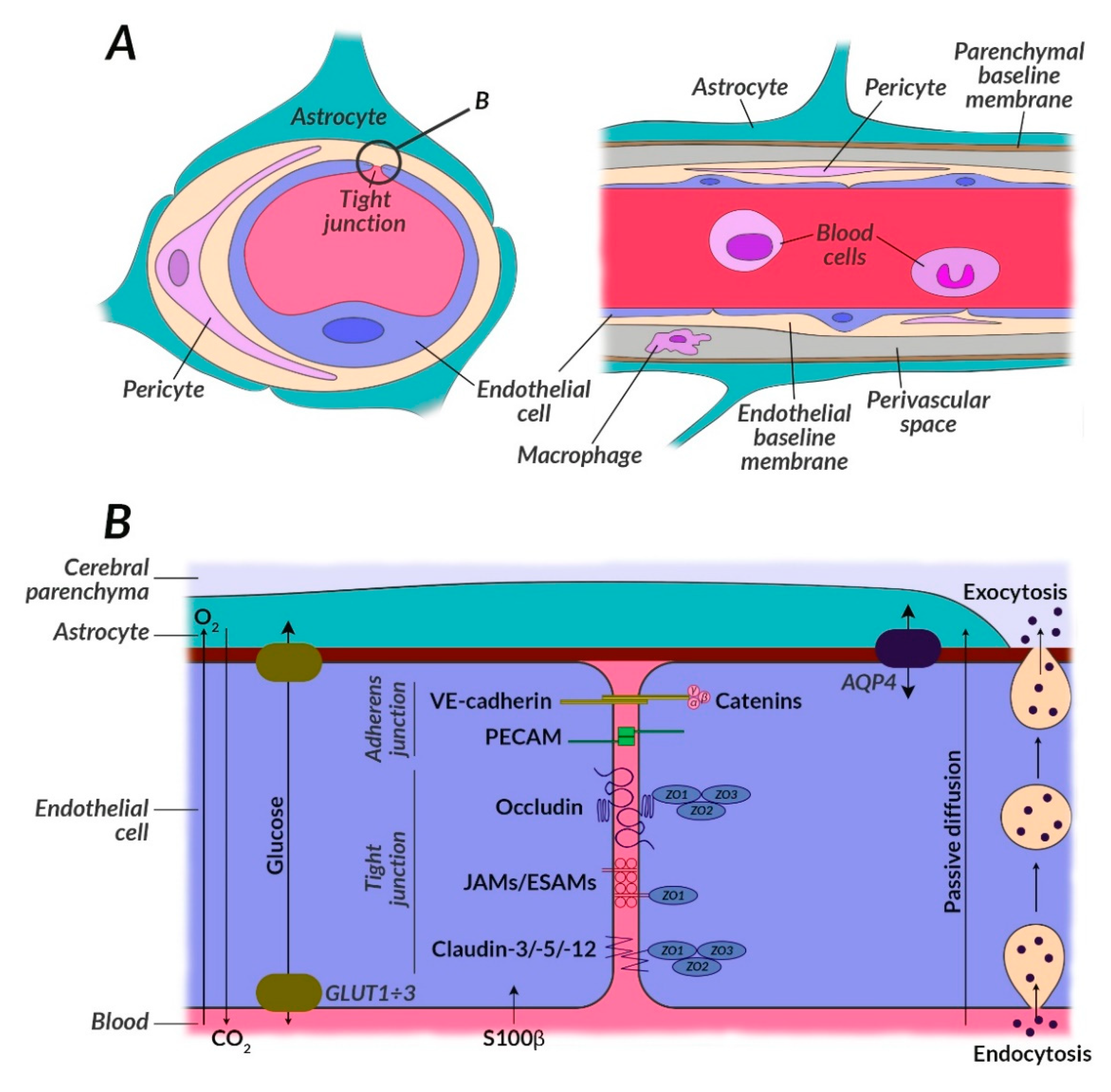
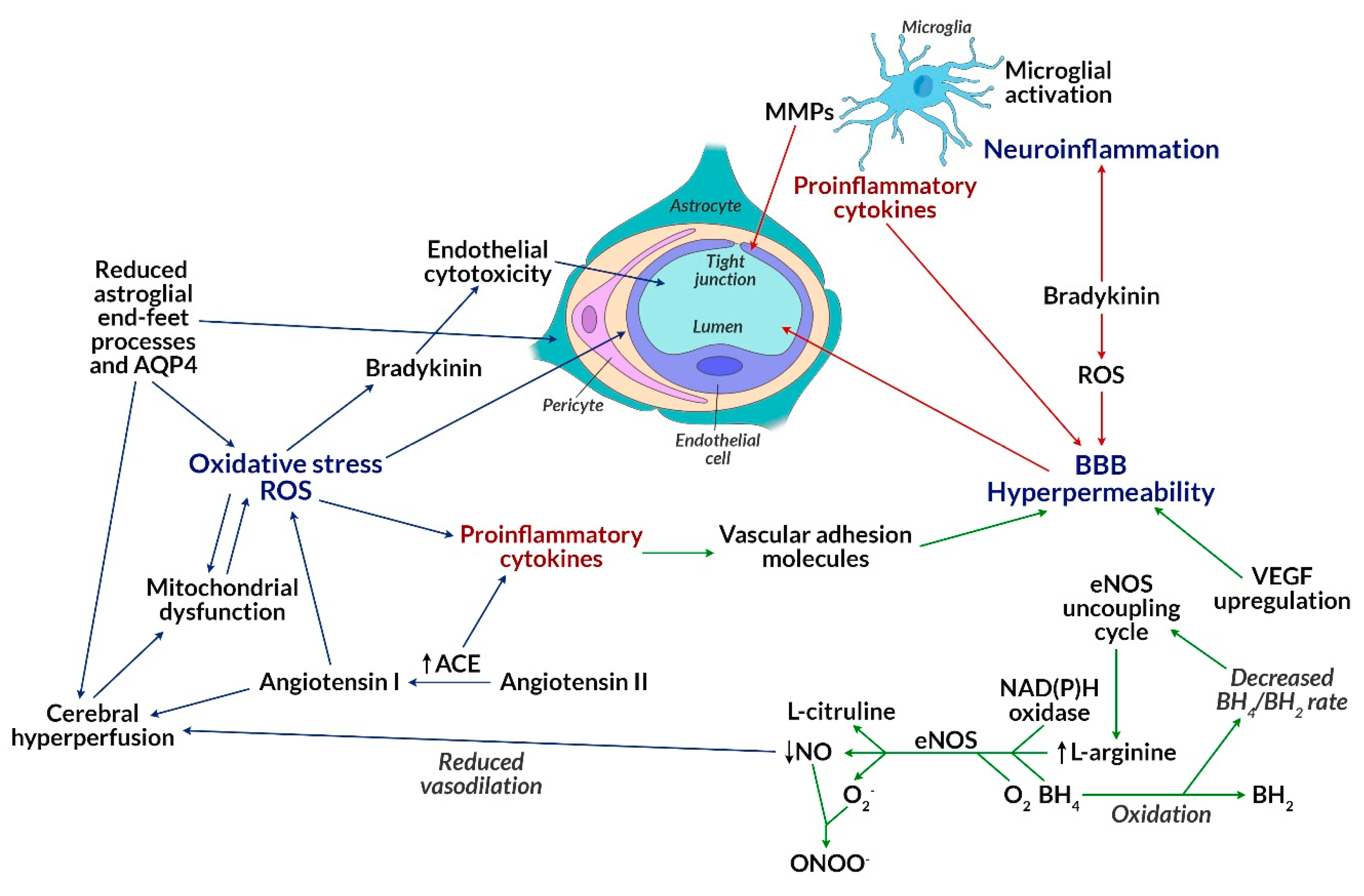
- Farrell, M.; Aherne, S.; O’Riordan, S.; O’Keeffe, E.; Greene, C.; Campbell, M. Blood-brain barrier dysfunction in a boxer with chronic traumatic encephalopathy and schizophrenia. Clin. Neuropathol. 2019, 38, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Kealy, J.; Greene, C.; Campbell, M. Blood-brain barrier regulation in psychiatric disorders. Neurosci. Lett. 2018. [Google Scholar] [CrossRef] [PubMed]
- Costea, L.; Mészáros, Á.; Bauer, H.; Bauer, H.; Traweger, A.; Wilhelm, I.; Farkas, A.E.; Krizbai, I. The blood-brain barrier and its intercellular junctions in age-related brain disorders. Int. J. Mol. Sci. 2019, 20, 5472. [Google Scholar] [CrossRef]
- Deng, H.; Kahlon, R.S.; Mohite, S.; Amin, P.; Zunta-Soares, G.; Colpo, G.; Stertz, L.; Fries, G.R.; Walss-Boss, C.; Soures, J.C.; et al. Elevated plasma S100B, psychotic symptoms, and cognition in schizophrenia. Psychiatr. Q. 2018, 89, 53–60. [Google Scholar] [CrossRef]
- Lara, D.R.; Gama, C.S.; Belmonte-De-Abreu, P.; Portela, L.; Gonçalves, C.; Fonseca, M.; Hauck, S.; Souza, D. Increased serum S100B protein in schizophrenia: A study in medication-free patients. J. Psychiatr. Res. 2001, 35, 11–14. [Google Scholar] [CrossRef]
- Agosta, J.B.; Mari, Y.M.; Rodriquez, N.R.; Herrera, D.G.d.B.; Ojalvo, A.G.; Mayola, M.F.; Nielo, G.G.; Sosa, P.V. Burn injury insulin resistance and central nervous system complications: A review. Burn. Open 2020. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Wands, J.R. Alzheimer’s disease is type 3 diabetes—Evidence reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef]
- Fung, A.; Vizcaychipi, M.; Lloyd, D.; Wan, Y.; Ma, D. Central nervous system inflammation in disease related conditions: Mechanistic prospects. Brain Res. 2012, 1446, 144–155. [Google Scholar] [CrossRef]
- De Felice, F.G.; Lourenco, M.V.; Ferreira, S.T. How does brain insulin resistance develop in Alzheimer’s disease? Alzheimer’s Dement. 2014, 10, S26–S32. [Google Scholar] [CrossRef]
- Yang, H.; Shan, W.; Zhu, F.; Wu, J.; Wang, Q. Ketone bodies in neurological diseases: Focus on neuroprotection and underlying mechanisms. Front. Neurol. 2019, 10, 585. [Google Scholar] [CrossRef] [PubMed]
- Knezovic, A.; Osmanovic-Barilar, J.; Curlin, M.; Hof, P.R.; Simic, G.; Riederer, P.; Salkovic-Petrisic, M. Staging of cognitive deficits and neuropathological and ultrastructural changes in streptozotocininduced rat model of Alzheimer’s disease. J. Neural Transm. 2015, 122, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Adilijiang, A.; Wang, W.; You, P.; Lin, D.; Li, X.; He, J. Arecoline attenuates memory impairment and demyelination in a cuprizone-induced mouse model of schizophrenia. Neuroreport 2019, 16, 134–138. [Google Scholar] [CrossRef] [PubMed]
- McNay, E.C.; Pearson-Leary, J. GluT4: A central player in hippocampal memory and brain insulin resistance. Exp. Neurol. 2020, 323, 113076. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Qiao, S.; Shi, C.; Wang, S.; Ji, G. Metabolomics window into diabetic complications. J. Diabetes Investig. 2018, 9, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Antenor-Dorsey, J.A.; Meyer, E.; Rutlin, J.; Perantie, D.C.; White, N.H.; Arbelaez, A.M.; Shimony, J.S.; Hershey, T. White matter microstructural integrity in youth with type 1 diabetes. Diabetes 2013, 62, 581–589. [Google Scholar] [CrossRef]
- Ooi, S.L.; Green, R.; Pak, S.C. N-acetylcysteine for the treatment of psychiatric disorders: A review of current evidence. BioMed Res. Int. 2018, 2018, 2469486. [Google Scholar] [CrossRef]
- Górny, M.; Wnuk, A.; Kamińska, A.; Kamińska, K.; Chwatko, G.; Bilska-Wilkosz, A.; Iciek, M.; Kajta, M.; Rogóż, Z.; Lorenc-Koci, E. Glutathione deficiency and alterations in the sulfur amino acid homeostasis during early postnatal development as potential triggering factors for schizophrenia-like behavior in adult rats. Molecules 2019, 24, 4253. [Google Scholar] [CrossRef]
- Mullier, E.; Roine, T.; Griffa, A.; Xin, L.; Baumann, P.S.; Klauser, P.; Cleusix, M.; Jenni, R.; Alemàn-Gómez, Y.; Gruetter, R.; et al. N-acetyl-cysteine supplementation improves functional connectivity within the cingulate cortex in early psychosis: A pilot study. Int. J. Neuropsychopharmacol. 2019, 22, 478–487. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| ROS | RNS | RSS | |
|---|---|---|---|
| Reactive independently | Hydroxyl radical (OH•) | Nitric oxide (II)/(IV) – nitric oxide and nitrogen dioxide (NO• and NOO) | Sulfhydryl radical (HS•) |
| Thiyl radical (RS•) | |||
| Perhydroxyl radical, the protonated form of superoxide radical (HOO·) | Peroxynitrite (ONOO-) | Persulfide radical anion, ‘supersulfide’ (HS2•−) | |
| Superoxide radical (O2•−) | Protonated form of peroxynitrite (peroxyacid) (ONOOH) | Sulfenic acids (RSOH), e.g., sulfinic acid (–SO2H) and sulfonic acid (–SO3H) | |
| Ozone (O3) | Sulfans, np.: Hydrogen disulfide (H2S2), trisulfan (H2S3) | ||
| Singlet oxygen (½ O2) | Polysulfides (H2Sn) | ||
| Not reactive independently, reactive with other radicals | Hydrogen peroxide (H2O2) | Nitroxyl (HNO) | Cysteine hydropersulfide (CysSSH) |
| Hypochlorous acid (HOCl) | Nitrosyl cation (NO+) | Thiols (RSH) | |
| Organic peroxides (ROOH) | Nitrosyl anion (NO-) | Hydrogen sulfide (H2S) |
| ROS Target | Directly Measured Product of Oxidative Damage | Mechanisms | Effects |
|---|---|---|---|
| Proteins | 3-nitrotyrosine | Produced during radical–radical reaction between: Superoxide radical (O2•-) with nitric oxide (NO•) to form peroxynitrite (ONOO−), which leads to nitration of Tyr residues in proteins; between the tyrosyl radical and nitrogen dioxide (•NO2). Moreover, occurs during reactions catalyzed by peroxidases, including myeloperoxidase (MPO) or eosinophilic peroxidase. 3-Nitrotyrosine is also formed in a mechanism catalyzed by modified superoxide dismutase (Cu, Zn-SOD), which has a greater ability to catalyze the nitration reaction of tyrosine residues caused by ONOO−. This occurs in motor neurons [149]. | Affects the structure and function of proteins in vitro and in vivo; present in inflammation associated with vascular endothelial dysfunction and cardiovascular complications in schizophrenia [150]. Correlates with the severity of atherosclerotic lesions; serves as an independent indicator of the risk of coronary artery disease in patients with FEP or schizophrenia. Biomarker, whose level depends on the pharmacotherapy, e.g., clozapine induces oxidative and nitrosative stress (in the caspase-3, NF-κB p65 or Nrf2 mechanism) depending on the dose in the cardiovascular system [151,152,153]. |
| Protein carbonyls | Carbonylated proteins are formed in the course of the following mechanisms: 1. The formation of aldehydes due to the cleavage of the peptide chain resulting in the formation of free radicals that convert into aldehydes; 2. Formation of oxidized amino acid side chains (oxo-histidine); 3. Combining proteins with lipid peroxidation products; 4. Creating advanced glycation end-products (AGEs). | Protein carbonyls increase: Carbonyl stress; production of conformationally altered polypeptide chains, which contributes to cellular dysfunction; excessive aggregation of proteins by promoting unfolding and formation of noncovalent, as well as covalent bonds between proteins; increased toxicity; may lead to apoptotic cell death [154,155]. | |
| Dityrosine | Can be formed as a result of metal-catalyzed bond formation between two tyrosine residues. The reaction proceeds to form a tyrosine radical that isomerizes the entire aromatic ring. As a result of the combination of two radicals in the ortho–ortho position, an unsaturated bis-ketone is formed, which leads to the formation of dityrosine [156]. The change is irreversible [157]. | Affects the redox state disorder; elevates the level of inflammatory factors, causing oxidative damage to the hippocampus; contributes to the deterioration of learning and memory skills [158]. It may contribute to the amino acid metabolism disorder and inhibit energy production [159]. | |
| Methionine sulfoxide | Hydrogen peroxide reacts with methionine residues at pH = 5 to form methionine sulfoxide. Under these conditions, cysteine residues are resistant to methylation. Cysteine thiol group must first be ionized in order to be oxidized. At acidic pH, methionine is oxidized by dimethyl sulfoxide to methionine sulfoxide. At neutral or alkaline pH it is oxidized under the influence of hypochlorous acid, oxygen, ozone, peroxynitrite, superoxide radical [160]. Under the influence of further oxidation, methionine sulfoxide can be converted to methionine sulfone [157]. | Methionine sulfoxide reductase is present in many organs, including the brain. It has a protective function against the effects of oxidative stress. Disturbances in the functioning of methionine sulfoxide reductase may have a significant impact on the development of many diseases, including schizophrenia. Various genetic variations of methionine sulfoxide reductase have been demonstrated in schizophrenic patients. They may be associated with dopamine disorders and affect the effects of treatment. These genetic variations affect various traits related to brain function. Chronic stress is associated with overexpression of methionine sulfoxide reductase in the hippocampus [161]. | |
| S-carboxymethyl-cysteine S-2-succinyl-cysteine | Under the influence of glyoxal or glucose on cysteine residues, protective amino acids, peptides containing thiol groups, and proteins form S-carboxymethyl-cysteine, which has been recognized as AGE. Studies show that thiohemiacetal that was initially formed undergoes an intramolecular Cannizzarro reaction [162]. S-succinyl-cysteine is formed as a result of a nonenzymatic Michael reaction under the influence of fumaric acid on the free thiol groups of cysteine residues [157,163]. | Fumarate, leading to the formation of S-(2-succinyl-cysteine, causes irreversible inhibition of many sulfhydryl enzymes. One of them is 3-phosphoglyceroldehyde dehydrogenase, which belongs to the glycolytic pathway, which is impaired in the course of schizophrenia [164]. | |
| Carboxymethyllysine | Formed as a result of various reaction mechanisms, i.e., oxidation of fructosyl-lysine (reaction chain leading to AGE), direct reaction of glyoxal with the ε-amino group of lysine (reaction chain leading to ALE) [162,165], oxidation of lysine, proline residues, arginine and threonine [130]. | Important AGE-epitope and RAGE ligand. It causes systemic glycoxidant load and increased body’s susceptibility to stress [165]. | |
| Cysteine/cystine Homocysteine/homocystine | Homocysteine and cysteine are reduced forms, homocystine and cystine are oxidized forms [166]. Homocysteine is a product of metabolism (demethylation) of methionine. It is sensitive to autoxidation and can be converted into cysteine [167]. | The accumulation of homocysteine and the lack of further metabolism of this compound causes a disorder of thymidine synthesis, DNA replication and neurogenesis, as well as the synthesis of neurotransmitters leading to a disorder of brain conductance [168]. Cysteine, which is a precursor of antioxidative glutathione, exhibits elevated levels in schizophrenia, compensating for the increase in oxidative stress [169]. | |
| Lipids | Lipid peroxides | Formed during oxidation of polyunsaturated long-chain fatty acids, e.g., linoleic, arachidonic, and docosahexaenoic acids. Hydrogen from the methyl group is removed first, resulting in the formation of two double bonds. Then, the fat is isomerized, a diene is formed, which reacts with molecular oxygen, leading to the formation of lipid peroxide. The second mechanism is based on oxidation catalyzed by metals (iron, copper). As a result of the Fenton reaction, radicals are formed that remove hydrogen from the methyl group, further reaction proceeds as described above [170]. | Inhibits glycolysis and synthesis of proteins and nucleic acids. Leads to the disorders in the transport of glucose and glutathione, damaging cholinergic neurons and accelerating apoptosis of neurons. Binds to thiol groups of proteins or to glutathione, reducing their level in the cell; binds to amino groups of proteins or DNA nitrogen bases, initiating mutagenesis and carcinogenesis processes. The concentration in schizophrenic patients is higher in both the brain and the periphery [171,172]. They probably inhibit Na, K-ATPase activity by lipid peroxidation, which leads to disruption of the phospholipid moiety [173]. |
| Malondialdehyde | MDA is formed during conversion of methyl linoleate to prostaglandin-like endoperoxide, which is considered a precursor of MDA under stress. Another mechanism is the production of hydrogen peroxide and β-cleavage of the fatty acid chain. Hydroperoxyaldehyde is then formed, from which MDA is generated as a result of β-scission or due to the reaction of the acrolein radical with a hydroxyl radical. MDA can also be formed during the enzymatic biosynthesis of thromboxane A2 [130,134,174]. | Inhibits glycolysis and synthesis of proteins and nucleic acids. Leads to the disorders in the transport of glucose and glutathione, damaging cholinergic neurons and accelerating apoptosis of neurons. Binds to thiol groups of proteins or to glutathione, reducing their level in the cell; binds to amino groups of proteins or DNA nitrogen bases, initiating mutagenesis and carcinogenesis processes. The concentration in schizophrenic patients is higher in both the brain and the periphery [171,172]. | |
| 4-hydroxynonenal | Oxidation of polyunsaturated fatty acids, i.e., linoleic, linolenic, arachidonic, and docosahexaenoic acids, by lipid peroxides [175]. Generally formed from omega-6 acids via β-fragmentation of 15-hydroperoxyarachidonic acid and 13-hydroperoxylolenic acid [130,134]. | HNE induces the formation of protein adducts, which then become the cause of a toxic neuronal disorder. Increases permeability of BBB and endothelium of blood vessels [175,176]. | |
| F2-isoprostanes | Eicosanoids result from the peroxidation of long-chain polyunsaturated fatty acids (mainly omega-3 and omega-6), e.g., arachidonic acid by OH• [130,134,177]. | Highly reactive products formed by the metabolism of isoprostanes (isoketals and their protein adducts). Inhibits the activity of simpleasomes, contributing to neurodegeneration [178,179]. | |
| DNA | 8-hydroxydeoxyguanosine | Formed during oxidation (hydroxylation of the C-8 position) of nucleotide guanine by OH•. | Leads to the destruction of DNA, which causes an increase in mutagenicity, cancer risk, and neurodegenerative diseases [177,180]. |
| Uracil, xanthine, oxanine | Exposure of DNA to RNS (N2O3—product of NO˙ auto-oxidation) causes deamination of bases and conversion of cytosine to uracil (2’-deoxyuridine), guanine to xanthine (2’-deoxyxanthosine), oxanine (2’-deoxyoxanine) (in the presence of HNO2 at acidic pH), and 8-nitroguanine [181,182]. | The correct level of uracil incision, abasic site cleavage, and dNTP incorporation activities in mitochondria originating from brains of patients with AD was examined [183]. Reduced activity of xanthine oxidase in the thalamus and occipital cortex of patients with chronic schizophrenia was observed. Studies show that elevated levels of xanthine oxidase also occur in the blood of schizophrenic patients [184]. | |
| 8-nitroguanine | ˙NO2 and NOO- react with guanine contained in DNA bases, nucleosides, and nucleotides, resulting in the formation of 8-nitroguanine [185,186]. | The presence of 8-nitroguanine has been shown in hepatocytes, Kuppfer’s cells, and inflammatory cells of patients with chronic hepatitis C [187]. 8-Nitroguanine can lead to the formation of tumors and is detected in cancer cells [188,189]. | |
| 5-chlorocytosine | Formed under the direct influence of HClO on cytosine contained in DNA [190]. | 5-Chlorocytosine may serve as a biomarker for chronic inflammation. The presence of 5-chlorothyrosine predisposes to mutagenesis [191]. | |
| 5-chlorouracil | Formed due to the enzymatic deamination of 5-chlorocytosine [190]. | Exhibits genotoxic and antimitotic effects [192]. | |
| 5-hydroxymethyluracil 5-hydroxyuracil | Formed under the influence of ROO• [191]. Oxidation of cytosine to unstable cytosine glycol which undergoes deamination leads to the formation of 5-hydroxyuracil [193]. 5-hydroxymethyluracil can be formed in two mechanisms:
| Elevated levels of 5-hydroxyuracil have been observed in vulnerable regions of the brains of patients in the late stages of Alzheimer’s disease [195]. Elevated levels of 5-hydroxyuracil have also been observed in other brain regions (such as temporal, parietal, and frontal lobes) with AD. Elevated levels of 5-hydroxyuracil in mtDNA were examined in the parietal and temporal lobes of AD brains, while neocortex showed an increased content of 5-hydroxyuracil in nuclear DNA [196]. A reduced level of incision and 5-hydroxyuracil ligase activity in mitochondria from brain postmortem investigations was demonstrated in patients with AD [183]. 5-Hydroxymethyluracil plays an important role in DNA demethylation. It can serve as a cancer biomarker [197]. | |
| Etheno-DNA-adducts | Reactive compounds formed in ROS-induced modification reactions, e.g., modified lipids, react with DNA either directly or through bi-functional intermediates, creating mutagenic etheno-DNA adducts, e.g., γ-linolenic acid peroxidation products as 4-hydroxynenenal react with adenine, cytosine, and guanine. This results in the formation of 1,N6-etheno-2’-deoxyadenosine (εdA), 3,N4-etheno-2’-deoxycytidine (εdC), 1,N2-etheno-2’-deoxyguanosine (1,N2εdG), and N2,3-etheno-2’-deoxyguanosine (N2,3εdG) [198]. | Can be used as cancer markers [199]. They are carcinogenic and play an important role in initiating the process of hepatocarcinogenesis [200]. They are detected in the livers of patients with nonalcoholic steatohepatitis (NASH) [201] and alcoholic liver disease (ALD) [200]. | |
| RNA | 8-hydroxyguanosine | Hydroxylation of nucleotide guanine by OH•, H2O2, O2•− [202]. | Increased serum 8-hydroxyguanosine levels have been demonstrated in patients with traumatic brain injury, which correlated with mortality [201]. Its occurrence is associated with many chronic diseases in old age, including Alzheimer’s disease [200], dementia and Parkinson’s disease [201]. |
| Oxidative Potential Found in Literature | Key Reaction | References |
|---|---|---|
| TAC (total antioxidant capacity) | R-PE: R-phycoerythrin | [233,237,238,239] |
| TAP/TAOP (total antioxidant power/potential) | Assessed via FRAP method: FeIIITPTZ + antioxidant → FeII-TPTZ | [230,240,241] |
| TRAP (total radical-trapping antioxidant parameter) | (ABAP) | [232,236,242] |
| TRAP (total reactive antioxidant potential) | Assessed via the method proposed by Lissi et al. | [232,243] |
| TAR (total antioxidant reactivity) | Assessed using the abovementioned method | [228] |
| TAR (total antioxidant response) | Based on the method proposed by Miller et al. ABTS - 2,2′-azinobis-(3-ethylbenzothiazoline)-6-sulphonic acid | [228,240] |
| TAA (total antioxidant activity) | [240,244] | |
| TAS (total antioxidant status) | ABTS - 2,2′-azinobis-(3-ethylbenzothiazoline)-6-sulphonic acid | [228,245,246] |
| TPAC (total plasma antioxidant capacity/capability) | R-PE: R-phycoerythrin | [237,238,247] |
| NEAC (nonenzymatic antioxidant capacity) | R-PE: R-phycoerythrin | [134,238] |
| Ir-reducing capacity (iridium-reducing capacity) | [128,248] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bryll, A.; Skrzypek, J.; Krzyściak, W.; Szelągowska, M.; Śmierciak, N.; Kozicz, T.; Popiela, T. Oxidative-Antioxidant Imbalance and Impaired Glucose Metabolism in Schizophrenia. Biomolecules 2020, 10, 384. https://doi.org/10.3390/biom10030384
Bryll A, Skrzypek J, Krzyściak W, Szelągowska M, Śmierciak N, Kozicz T, Popiela T. Oxidative-Antioxidant Imbalance and Impaired Glucose Metabolism in Schizophrenia. Biomolecules. 2020; 10(3):384. https://doi.org/10.3390/biom10030384
Chicago/Turabian StyleBryll, Amira, Justyna Skrzypek, Wirginia Krzyściak, Maja Szelągowska, Natalia Śmierciak, Tamas Kozicz, and Tadeusz Popiela. 2020. "Oxidative-Antioxidant Imbalance and Impaired Glucose Metabolism in Schizophrenia" Biomolecules 10, no. 3: 384. https://doi.org/10.3390/biom10030384
APA StyleBryll, A., Skrzypek, J., Krzyściak, W., Szelągowska, M., Śmierciak, N., Kozicz, T., & Popiela, T. (2020). Oxidative-Antioxidant Imbalance and Impaired Glucose Metabolism in Schizophrenia. Biomolecules, 10(3), 384. https://doi.org/10.3390/biom10030384