
Density Functional Theory (DFT) Study on the Ternary Interaction System of the Fluorinated Ethylene Carbonate, Li+ and Graphene Model


Abstract
:1. Introduction
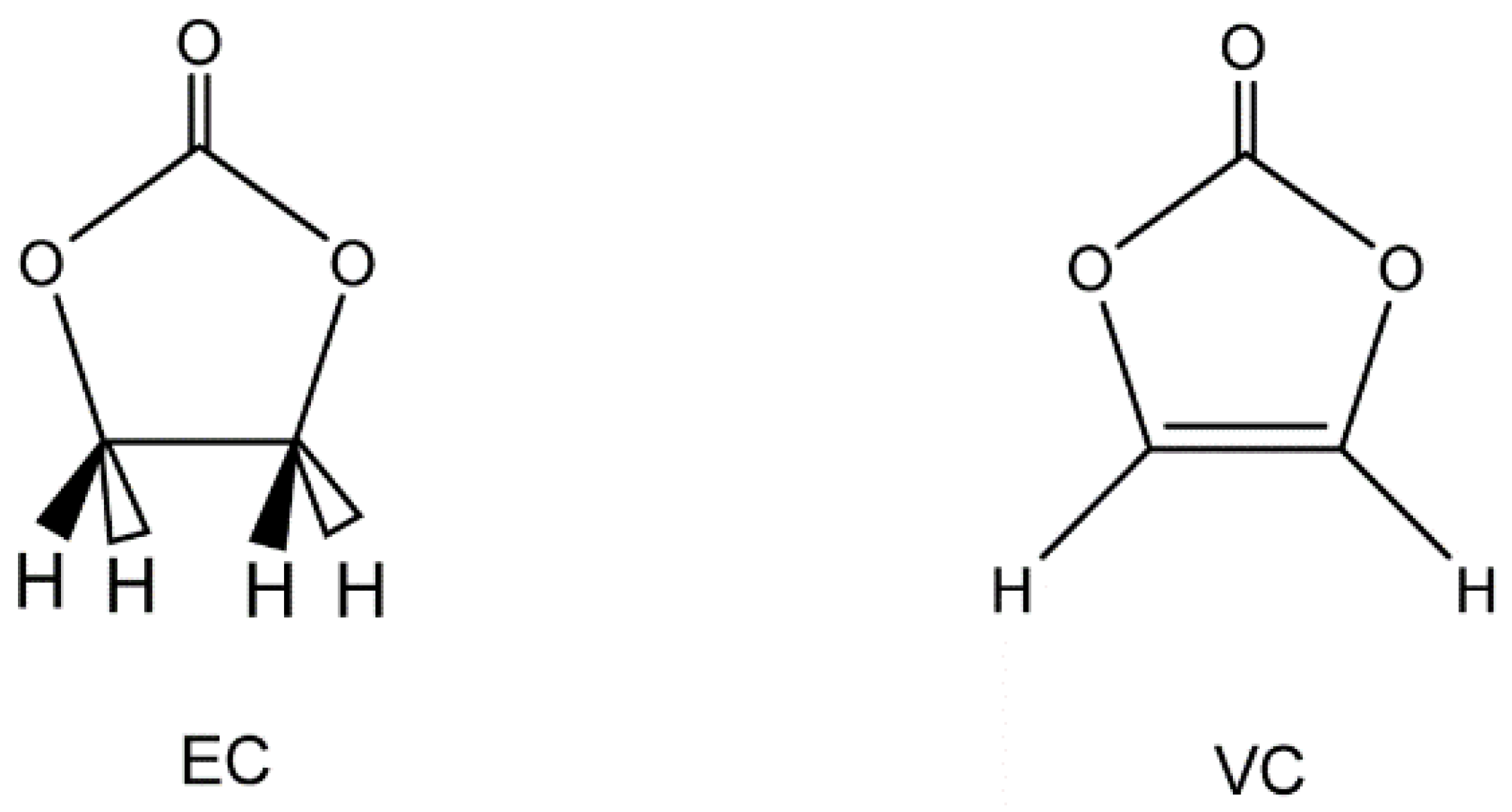
2. Method of Calculations
3. Results
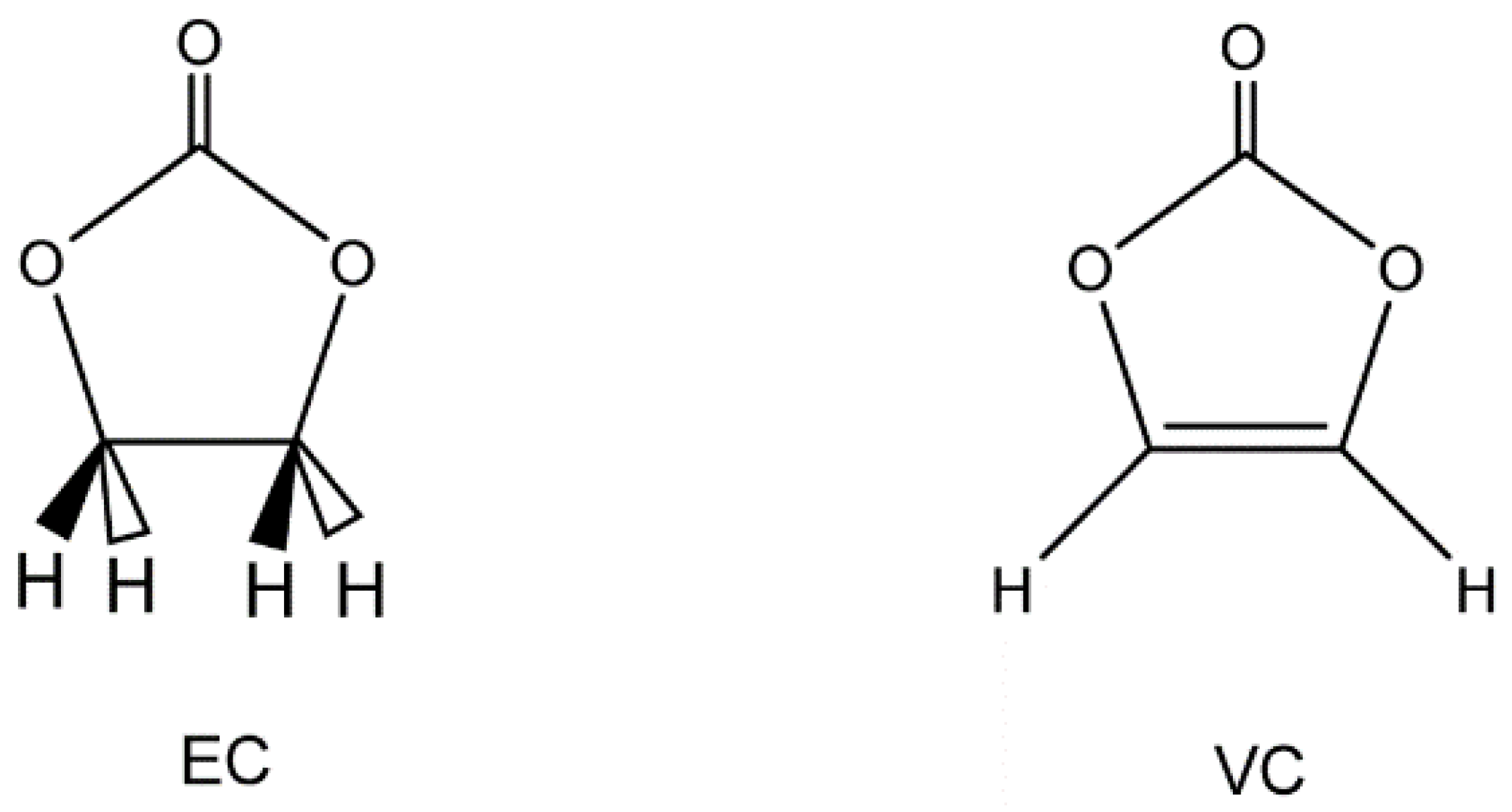
3.1. Structures of EC(F) and VC(F)
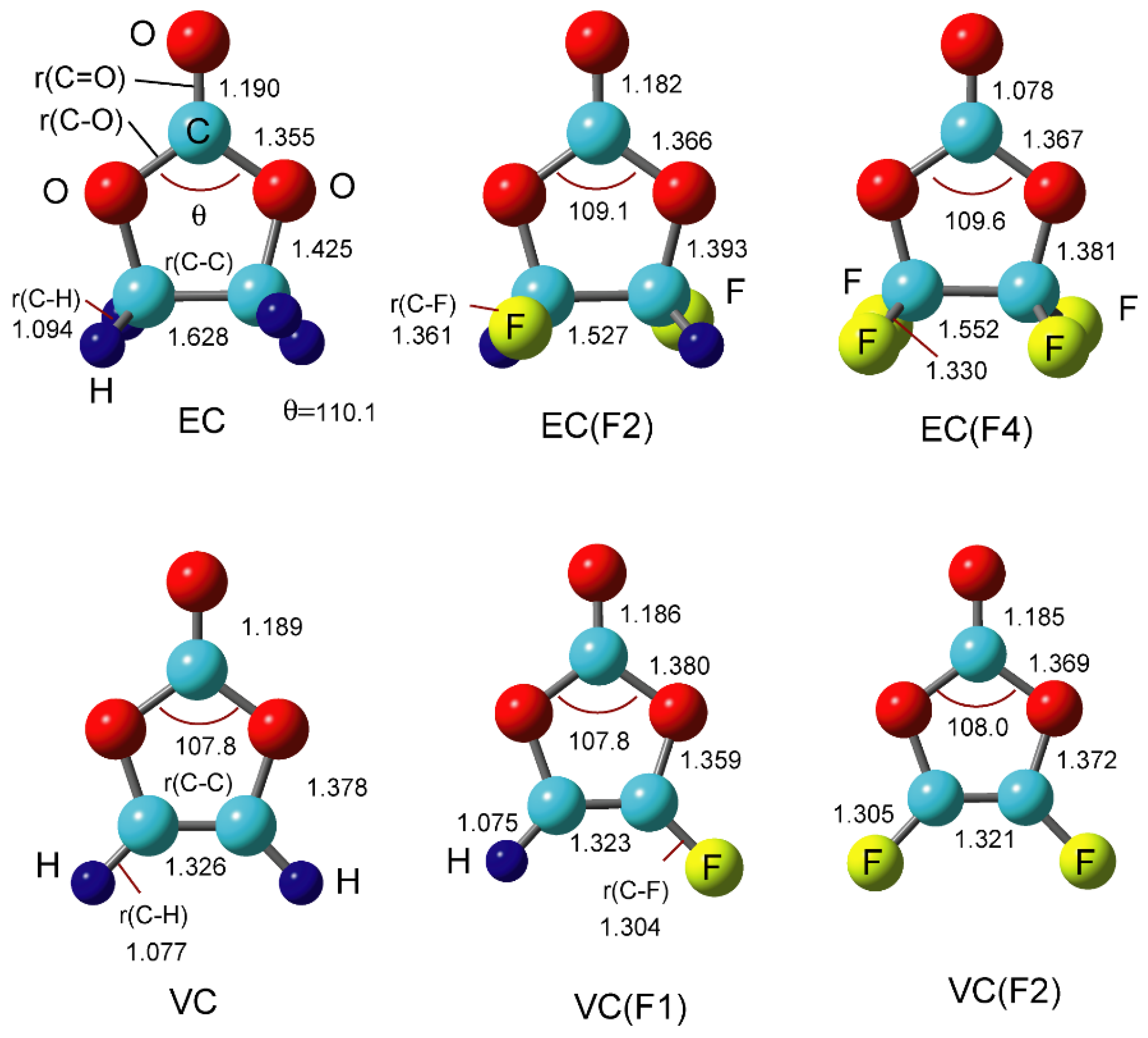

{kind=link}
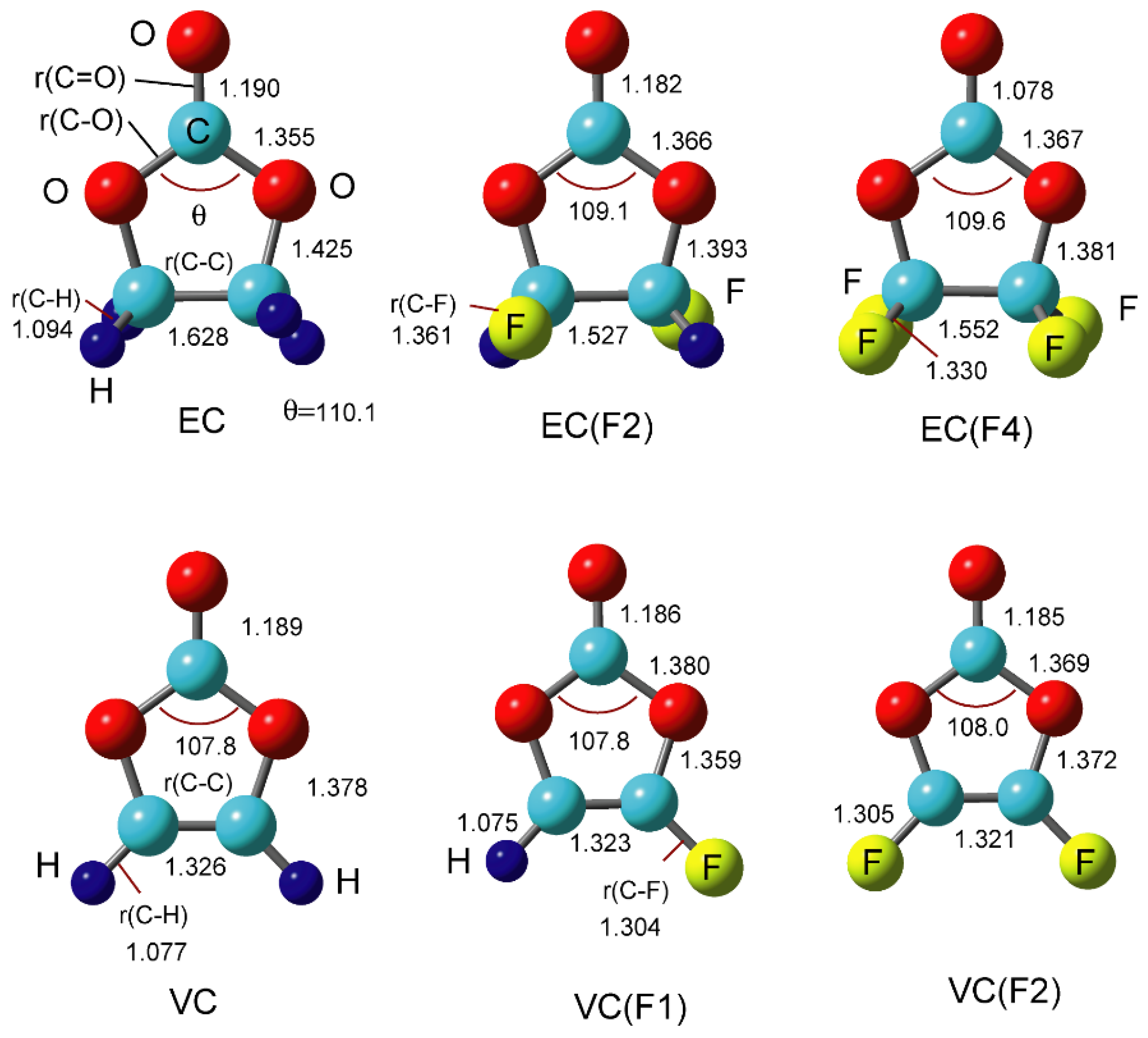{kind=link}
{kind=link}
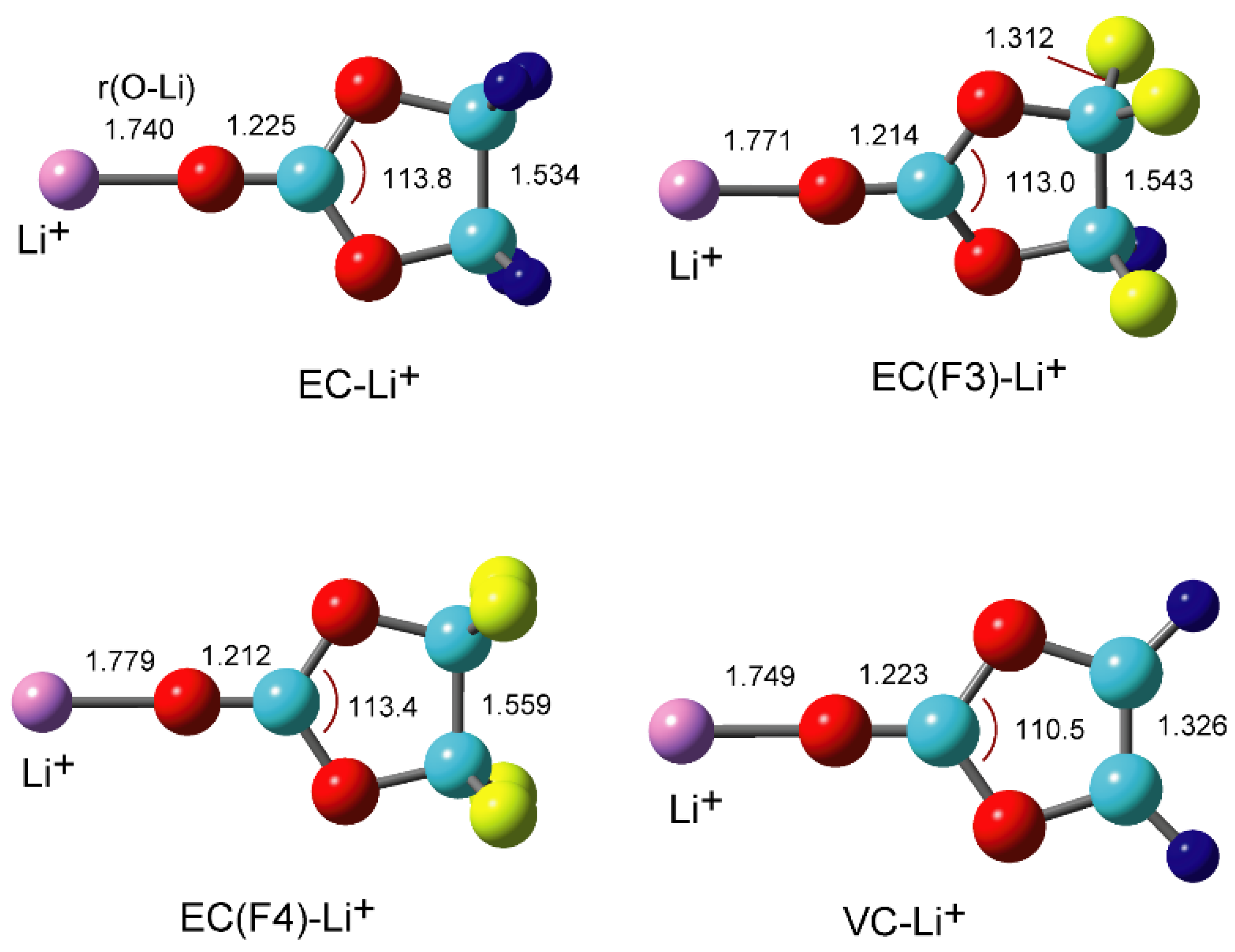
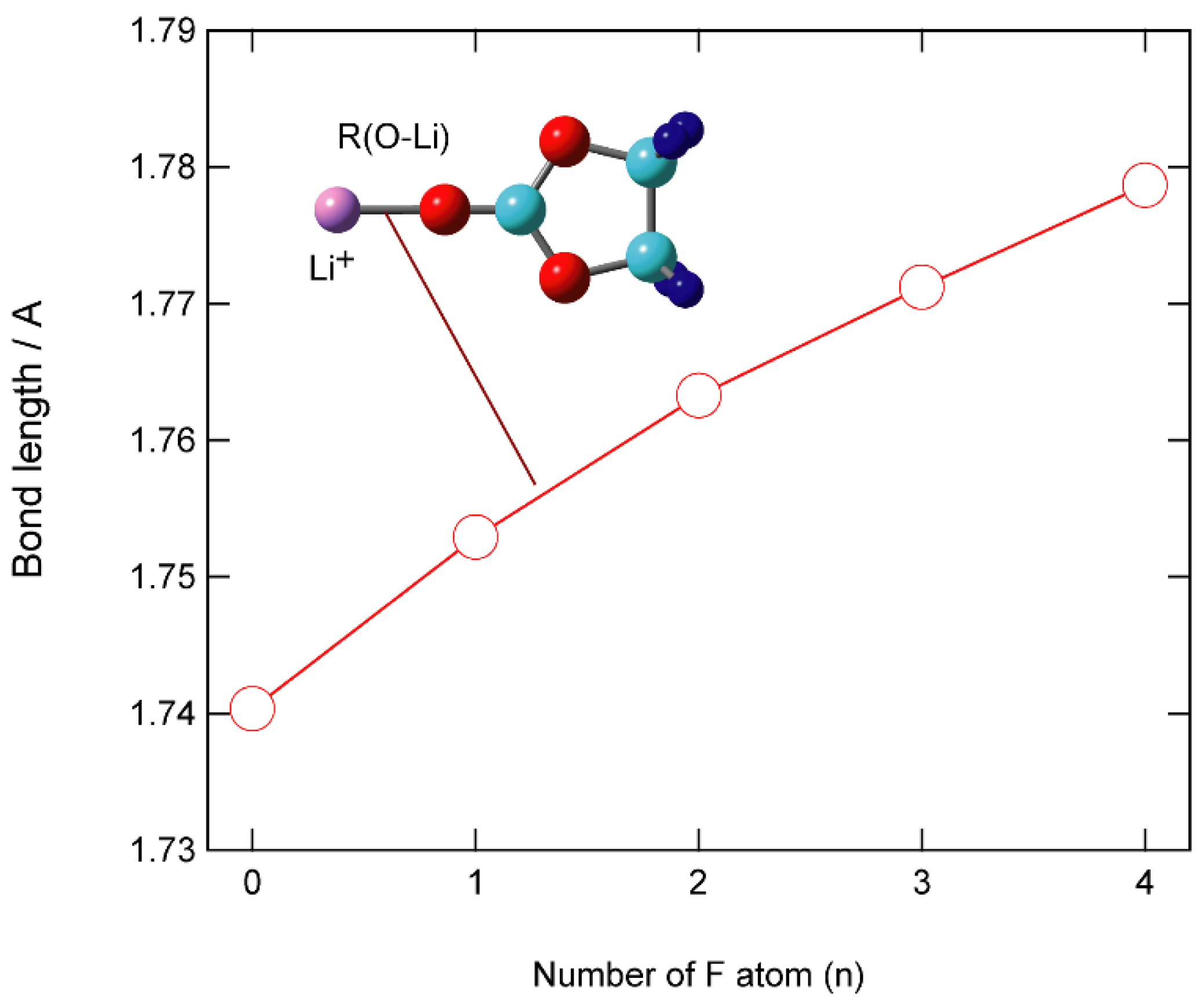{kind=link}
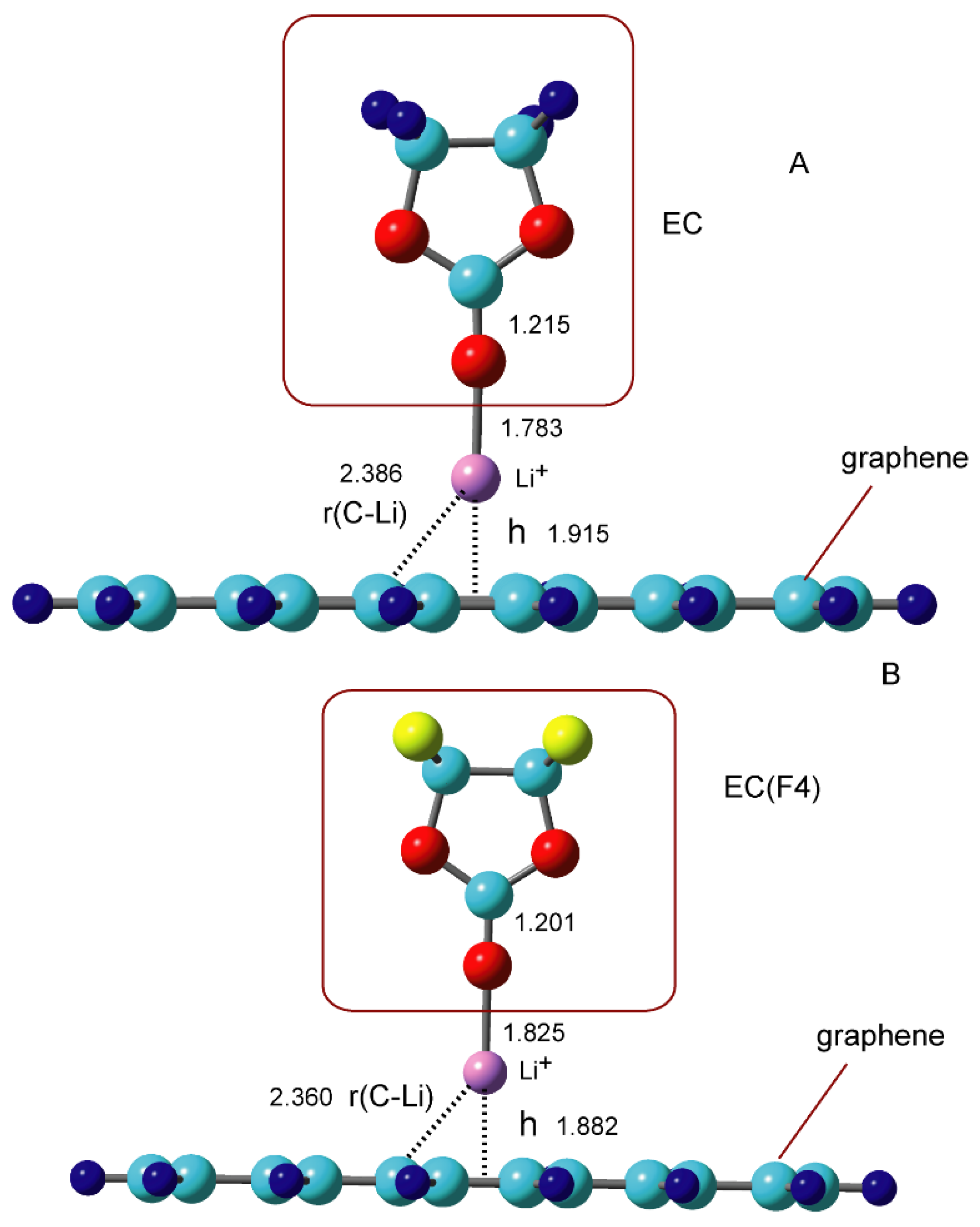{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dipole Moment | ||
|---|---|---|
| n | CAM-B3LYP/6-31G(d) | CAM-B3LYP/6-311G(dp) |
| EC0 | 5.36 (4.9) | 5.42 |
| EC(F1)1 | 4.76 | 4.80 |
| EC(F2)2 | 3.61 | 3.50 |
| EC(F3)3 | 2.87 | 2.75 |
| EC(F4)4 | 1.67 | 1.39 |
| VC0 | 4.71 | 4.69 |
| VC(F1)1 | 3.70 | 3.59 |
| VC(F2)2 | 2.61 | 2.40 |
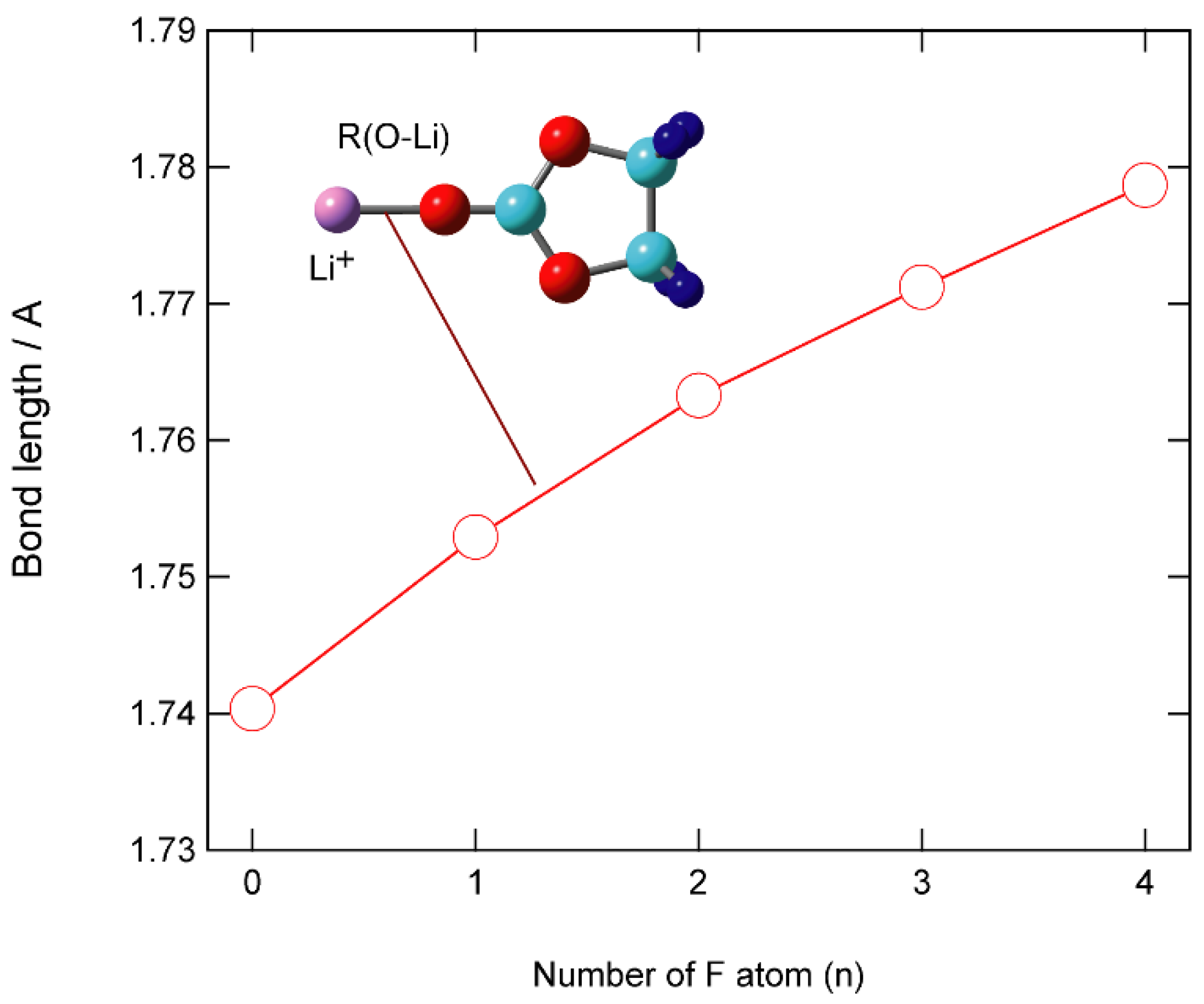
3.2. Structures of Li+-EC and Li+-VC Complexes
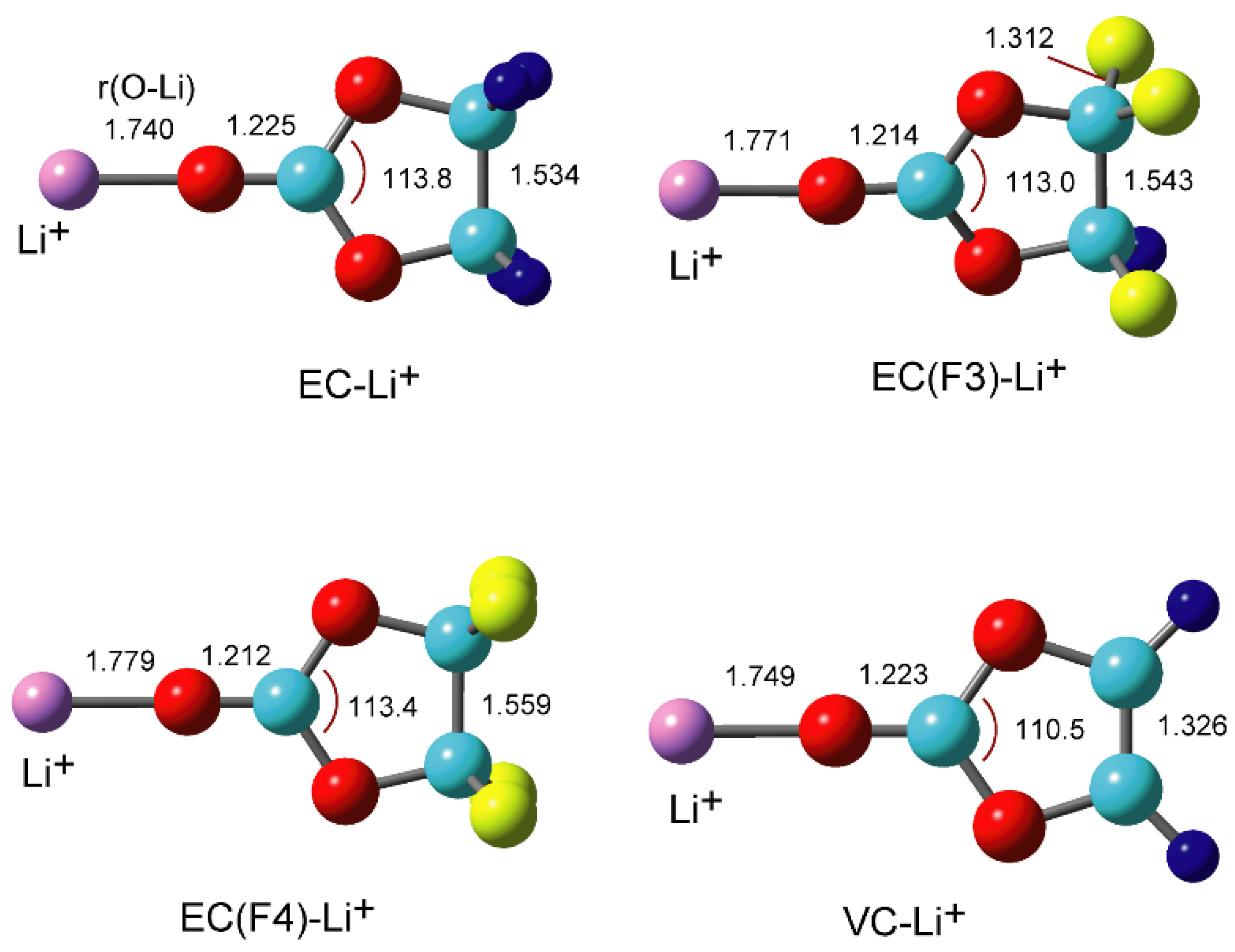
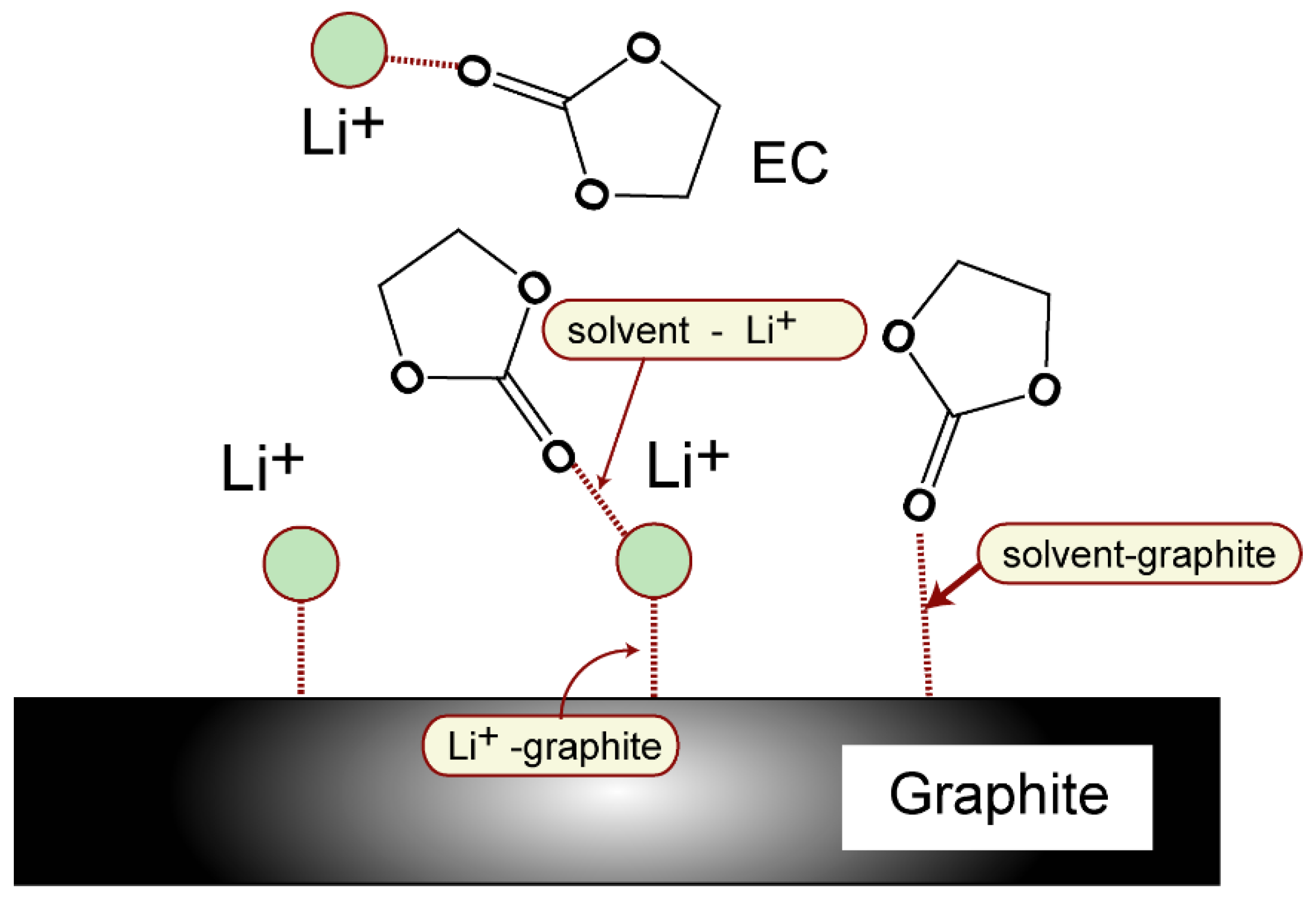
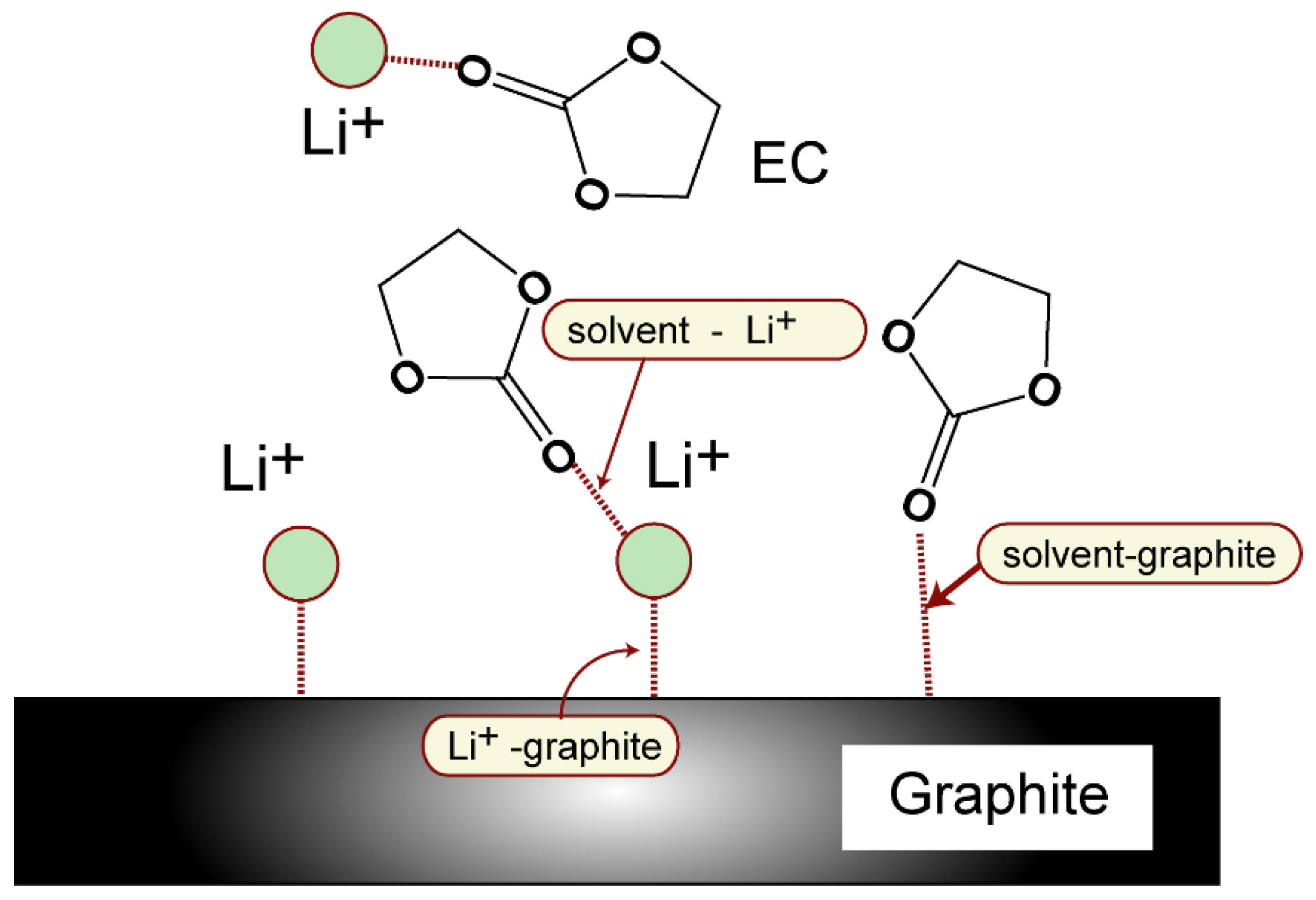
3.3. Binding Structures of Li+-EC Complexes on Graphene
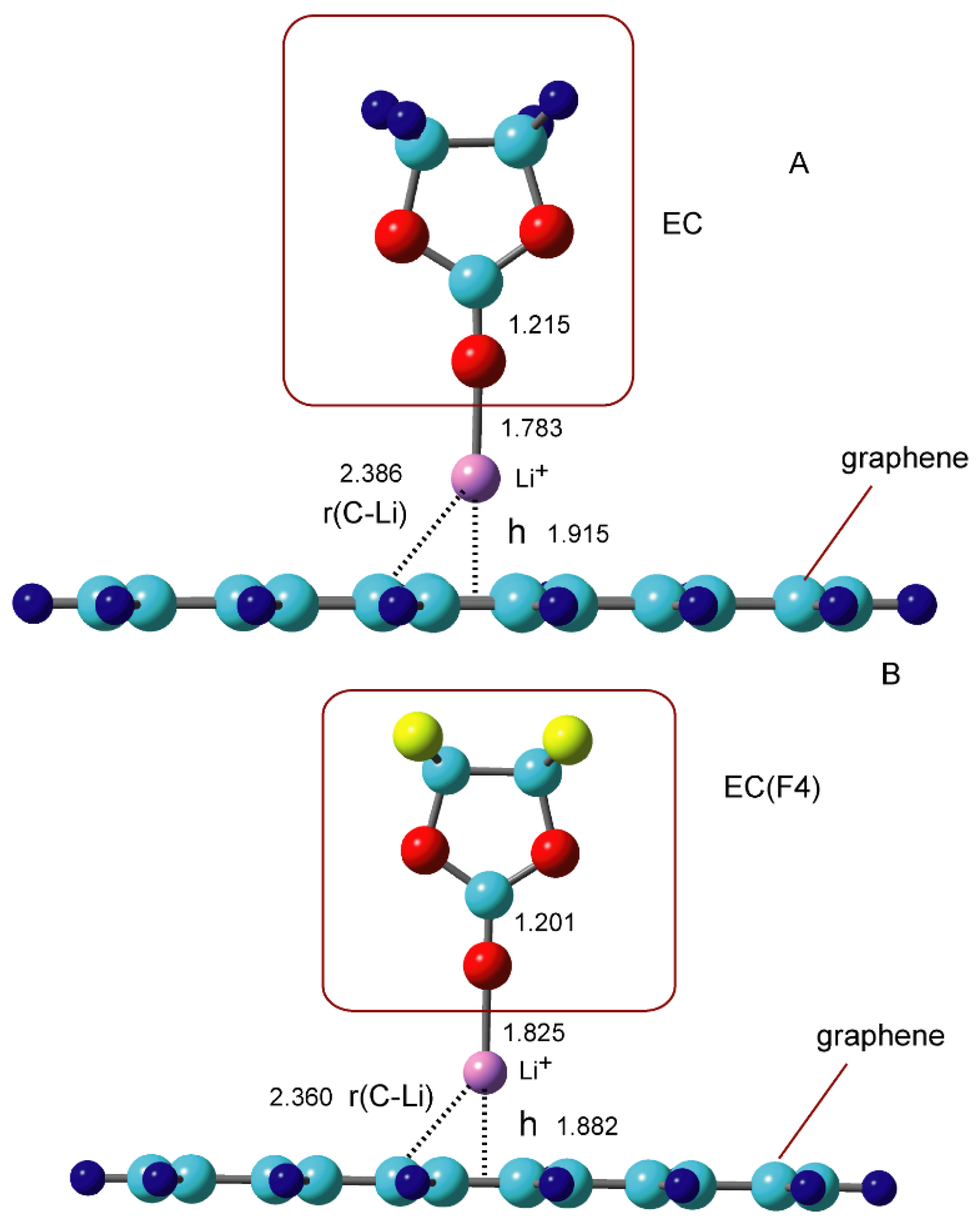
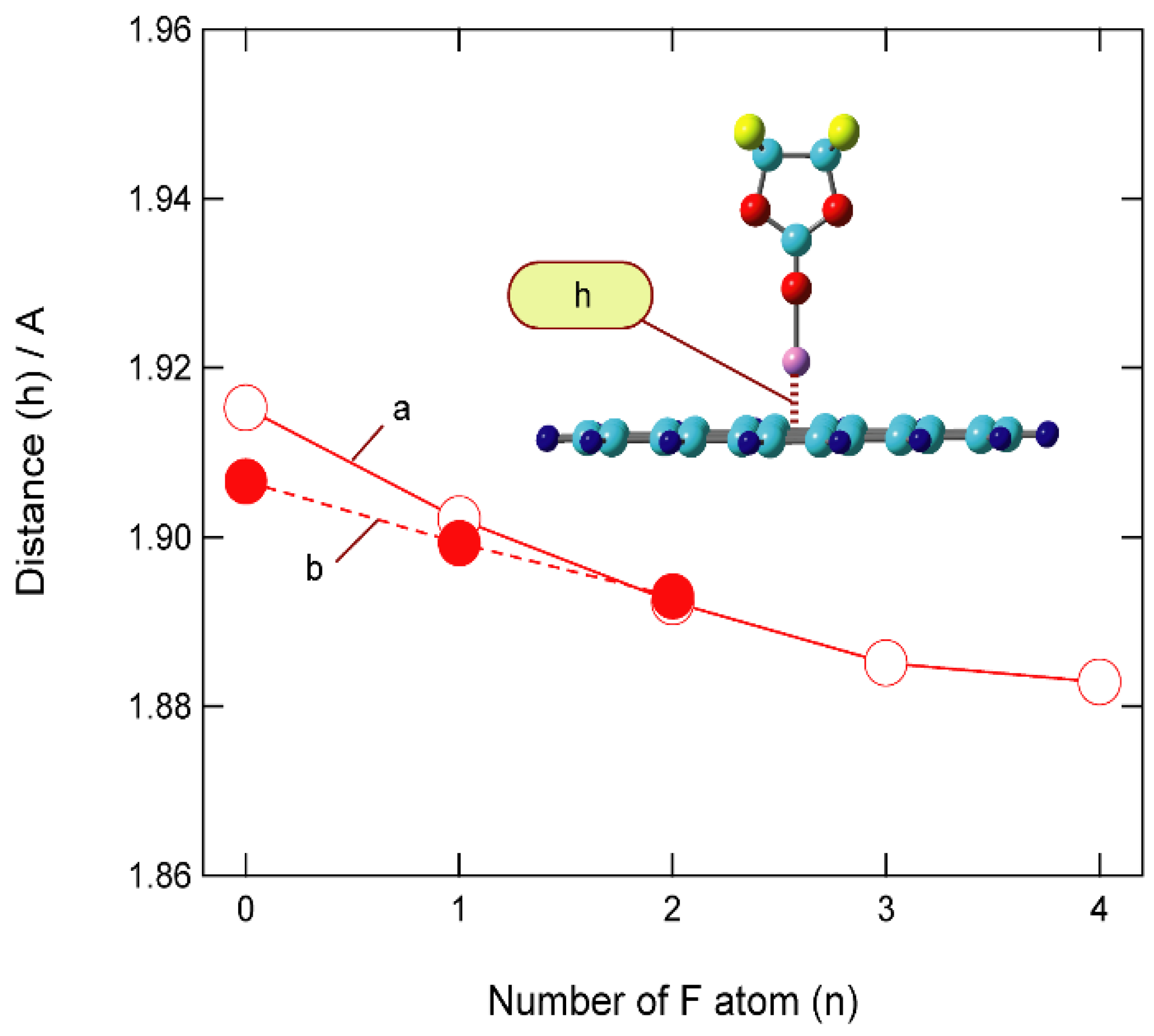
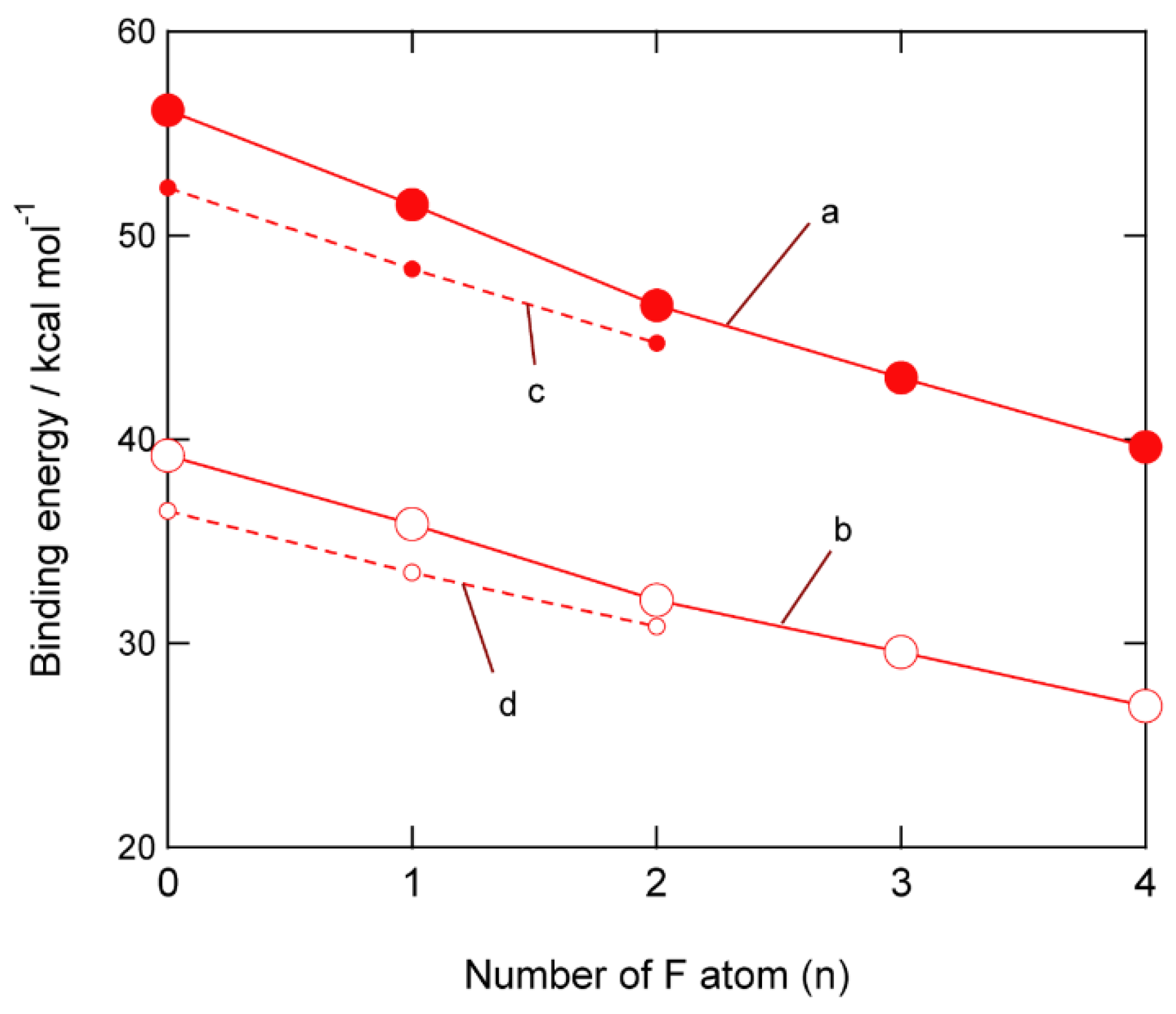
3.4. Binding Energies of Li+-EC Complexes on Graphene
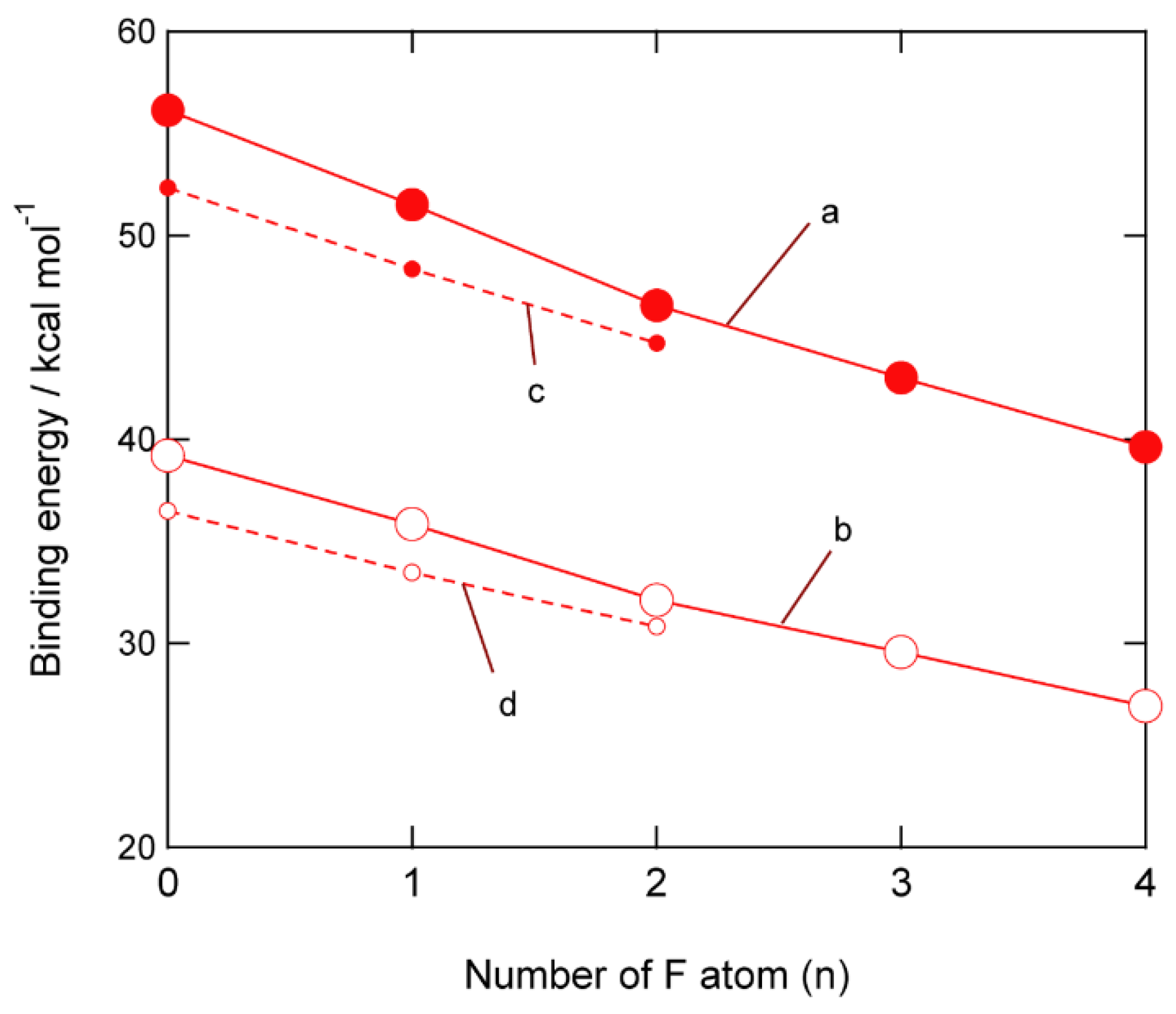
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bhatta, M.D.; Cho, M.; Cho, K. Interaction of Li+ ions with ethylene carbonate (EC): Density functional theory calculations. Appl. Surf. Sci. 2010, 257, 1463–1468. [Google Scholar] [CrossRef]
- Nakagawa, H.; Fujino, Y.; Kozono, S.; Katayama, Y.; Nukuda, T.; Sakaebe, H.; Matsumoto, H.; Tatsumi, K. Application of nonflammable electrolyte with room temperature ionic liquids (RTILs) for lithium-ion cells. J. Power Sources 2007, 174, 1021–1026. [Google Scholar] [CrossRef]
- Xing, L.; Li, W.; Wang, C.; Gu, F.; Xu, M.; Tan, C.; Yi, J. Theoretical Investigations on Oxidative Stability of Solvents and Oxidative Decomposition Mechanism of Ethylene Carbonate for Lithium Ion Battery Use. J. Phys. Chem. B 2009, 113, 16596–16602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Sun, C.H.; Li, F.; Liu, C.; Tan, J.H.; Cheng, M. New insight into the interaction between propylene carbonate-based electrolytes and graphite anode material for lithium ion batteries. J. Phys. Chem. C 2007, 111, 4740–4748. [Google Scholar] [CrossRef]
- Zhu, Y.; Casselman, M.D.; Li, Y.; Wei, A.; Abrahama, D.P. Perfluoroalkyl-substituted ethylene carbonates: Novel electrolyte additives for high-voltage lithium-ion batteries. J. Power Sources 2014, 246, 184–191. [Google Scholar] [CrossRef]
- Werber, T.; Walther, J.H.; Jaffe, R.L.; Halicioglu, T.; Koumoutsakos, P. On the water-carbon interaction for use in molecular dynamics simulations of graphite and carbon nanotubes. J. Phys. Chem. B 2003, 107, 1345–1352. [Google Scholar]
- Rana, M.; Chandra, A. Filled and empty states of carbon nanotubes in water: Dependence on nanotube diameter, wall thickness and dispersion interactions. J. Chem. Sci. 2007, 119, 367–376. [Google Scholar] [CrossRef]
- Castriota, M.; Cazzanelli, E.; Nicotera, I.; Coppola, L.; Oliviero, C.; Ranieri, G.A. Temperature dependence of lithium ion solvation in ethylene carbonate-LiClO4 solutions. J. Chem. Phys. 2003, 118, 5537–5541. [Google Scholar] [CrossRef]
- Tachikawa, H. A direct molecular orbital-molecular dynamics study on the diffusion of the Li ion on a fluorinated graphene surface. J. Phys. Chem. C 2008, 112, 10193–10199. [Google Scholar] [CrossRef]
- Abe, S.; Nagoya, Y.; Watari, F.; Tachikawa, H. Interaction of Water Molecules with Graphene: A Density Functional Theory and Molecular Dynamics Study. Jpn. J. Apl. Phys. 2010, 49, AH071–AH074. [Google Scholar] [CrossRef]
- Abe, S.; Nagoya, Y.; Watari, F.; Tachikawa, H. Structures and Electronic States of Water Molecules on Graphene Surface: A Density Functional Theory Study. Jpn. J. Apl. Phys. 2010, 49, GJ131–GJ133. [Google Scholar] [CrossRef]
- Amft, M.; Sanyal, B.; Eriksson, O.; Skorodumova, N.V. Small gold clusters on graphene, their mobility and clustering: A DFT study. J. Phys. Condens. Matter 2011, 23, 205301. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.; Lu, X.Q.; Wu, C.M.L. Nitrated tyrosine adsorption on metal-doped graphene: A DFT study. Comput. Mater. Sci. 2012, 51, 141–145. [Google Scholar] [CrossRef]
- Lim, D.H.; Wilcox, J. DFT-Based Study on Oxygen Adsorption on Defective Graphene-Supported Pt Nanoparticles. J. Phys. Chem. 2011, 115, 22742–22747. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.; Handy, N. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision B.04; Gaussian, Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Payne, R.; Theodorou, I.E. Dielectric properties and relaxation in ethylene carbonate and propylene carbonate. J. Phys. Chem. 1972, 76, 2892–2900. [Google Scholar] [CrossRef]
- Mirzaei, M.; Ahangari, R.S. Formations of CNT modified 5-(halogen)uracil hybrids: DFT studies. Superlattice Microstruct. 2014, 65, 375–379. [Google Scholar] [CrossRef]
- Peyghan, A.A.; Noei, M.; Yourdkhani, S. Al-doped graphene-like BN nanosheet as a sensor for para-nitrophenol: DFT study. Superlattice Microstruct. 2013, 59, 115–122. [Google Scholar] [CrossRef]
- Jenness, G.R.; Karalti, O.; Jordan, K.D. Benchmark calculations of water-acene interaction energies: Extrapolation to the water-graphene limit and assessment of dispersion-corrected DFT methods. Phys. Chem. Chem. Phys. 2010, 12, 6375–6381. [Google Scholar] [CrossRef] [PubMed]
- Pisula, W.; Feng, X.; Mullen, K. Charge-Carrier Transporting Graphene-Type Molecules. Chem. Mater. 2011, 23, 554–567. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mutoh, M.; Abe, S.; Kusaka, T.; Nakamura, M.; Yoshida, Y.; Iida, J.; Tachikawa, H. Density Functional Theory (DFT) Study on the Ternary Interaction System of the Fluorinated Ethylene Carbonate, Li+ and Graphene Model. Atoms 2016, 4, 4. https://doi.org/10.3390/atoms4010004
Mutoh M, Abe S, Kusaka T, Nakamura M, Yoshida Y, Iida J, Tachikawa H. Density Functional Theory (DFT) Study on the Ternary Interaction System of the Fluorinated Ethylene Carbonate, Li+ and Graphene Model. Atoms. 2016; 4(1):4. https://doi.org/10.3390/atoms4010004
Chicago/Turabian StyleMutoh, Mami, Shigeaki Abe, Teruo Kusaka, Mariko Nakamura, Yasuhiro Yoshida, Junichiro Iida, and Hiroto Tachikawa. 2016. "Density Functional Theory (DFT) Study on the Ternary Interaction System of the Fluorinated Ethylene Carbonate, Li+ and Graphene Model" Atoms 4, no. 1: 4. https://doi.org/10.3390/atoms4010004
APA StyleMutoh, M., Abe, S., Kusaka, T., Nakamura, M., Yoshida, Y., Iida, J., & Tachikawa, H. (2016). Density Functional Theory (DFT) Study on the Ternary Interaction System of the Fluorinated Ethylene Carbonate, Li+ and Graphene Model. Atoms, 4(1), 4. https://doi.org/10.3390/atoms4010004