An Integrative Multi-Omics Workflow to Address Multifactorial Toxicology Experiments

 , , , ,
, , , , 
Abstract
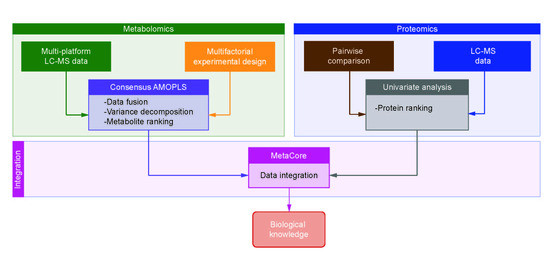
1. Introduction
2. Materials and Methods
2.1. Solvents and Additives
2.2. Culture of 3D Rat Brain Cell Cultures and TMT Exposure
2.3. Sample Preparation for Metabolomic Analyses
2.4. LC-MS Analyses for Metabolomics
2.5. Metabolomics Data Pretreatment and Metabolite Identification
2.6. Sample Preparation for Proteomic Analysis
2.7. MS-Data Acquisition for Proteomics Analyses
2.8. Protein Identification and Quantification
2.9. Data Processing and Analysis
3. Results and Discussion
3.1. Metabolomic Analyses
3.1.1. Sample Analysis, Data Acquisition and Preprocessing
3.1.2. Multivariate Analysis Using the AMOPLS Algorithm
3.2. Complementary Proteomic Analyses
3.3. Joint Multi-Omics Analyses
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wilkins, J.M.; Trushina, E. Application of Metabolomics in Alzheimer’s Disease. Front. Neurol. 2018, 8, 719. [Google Scholar] [CrossRef] [PubMed]
- Bridges, J.; Sauer, U.G.; Buesen, R.; Deferme, L.; Tollefsen, K.E.; Tralau, T.; van Ravenzwaay, B.; Poole, A.; Pemberton, M. Framework for the quantitative weight-of-evidence analysis of ‘omics data for regulatory purposes. Regul. Toxicol. Pharmacol. 2017, 91, S46–S60. [Google Scholar] [CrossRef] [PubMed]
- Kauffmann, H.-M.; Kamp, H.; Fuchs, R.; Chorley, B.N.; Deferme, L.; Ebbels, T.; Hackermüller, J.; Perdichizzi, S.; Poole, A.; Sauer, U.G.; et al. Framework for the quality assurance of ‘omics technologies considering GLP requirements. Regul. Toxicol. Pharmacol. 2017, 91, S27–S35. [Google Scholar] [CrossRef]
- Viant, M.R.; Ebbels, T.M.D.; Beger, R.D.; Ekman, D.R.; Epps, D.J.T.; Kamp, H.; Leonards, P.E.G.; Loizou, G.; MacRae, J.I.; van Ravenzwaay, B.; et al. Best Practice and Reporting Standards for Applications of Metabolomics in Regulatory Toxicology.
- Choudhuri, S.; Patton, G.W.; Chanderbhan, R.F.; Mattia, A.; Klaassen, C.D. From Classical Toxicology to Tox21: Some Critical Conceptual and Technological Advances in the Molecular Understanding of the Toxic Response Beginning From the Last Quarter of the 20th Century. Toxicol. Sci. 2017, 161, 5–22. [Google Scholar] [CrossRef]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef]
- Rabilloud, T.; Lescuyer, P. Proteomics in mechanistic toxicology: History, concepts, achievements, caveats, and potential. Proteomics 2014, 15, 1051–1074. [Google Scholar] [CrossRef] [PubMed]
- Miller, I.; Serchi, T.; Murk, A.J.; Gutleb, A.C. The Added Value of Proteomics for Toxicological Studies. J. Toxicol. Environ. Health 2014, 17, 225–246. [Google Scholar] [CrossRef]
- Jeanneret, F.; Tonoli, D.; Hochstrasser, D.; Saurat, J.-H.; Sorg, O.; Boccard, J.; Rudaz, S. Evaluation and identification of dioxin exposure biomarkers in human urine by high-resolution metabolomics, multivariate analysis and in vitro synthesis. Toxicol. Lett. 2016, 240, 22–31. [Google Scholar] [CrossRef]
- Li, R.-S.; Takeda, T.; Ohshima, T.; Yamada, H.; Ishii, Y. Metabolomic profiling of brain tissues of mice chronically exposed to heroin. Drug Metab. Pharmacokinet. 2017, 32, 108–111. [Google Scholar] [CrossRef]
- Tolstikov, V. Metabolomics: Bridging the Gap between Pharmaceutical Development and Population Health. Metabolites 2016, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Ankley, G.T.; Bennett, R.S.; Erickson, R.J.; Hoff, D.J.; Hornung, M.W.; Johnson, R.D.; Mount, D.R.; Nichols, J.W.; Russom, C.L.; Schmieder, P.K.; et al. Adverse outcome pathways: A conceptual framework to support ecotoxicology research and risk assessment. Environ. Toxicol. Chem. 2010, 29, 730–741. [Google Scholar] [CrossRef]
- Boccard, J.; Rudaz, S. Harnessing the complexity of metabolomic data with chemometrics. J. Chemom. 2013, 28, 1–9. [Google Scholar] [CrossRef]
- Boccard, J.; Rudaz, S. Exploring Omics data from designed experiments using analysis of variance multiblock Orthogonal Partial Least Squares. Analytica Chimica Acta 2016, 920, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Little, A.R.; Miller, D.B.; Li, S.; Kashon, M.L.; O’Callaghan, J.P. Trimethyltin-induced neurotoxicity: Gene expression pathway analysis, q-RT-PCR and immunoblotting reveal early effects associated with hippocampal damage and gliosis. Neurotoxicol. Teratol. 2012, 34, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yang, M.; Son, Y.; Jang, H.; Kim, D.; Kim, J.-C.; Kim, S.-H.; Kang, M.-J.; Im, H.-I.; Shin, T.; et al. Glial activation with concurrent up-regulation of inflammatory mediators in trimethyltin-induced neurotoxicity in mice. Acta Histochemica 2014, 116, 1490–1500. [Google Scholar] [CrossRef]
- Imam, S.Z.; He, Z.; Cuevas, E.; Rosas-Hernandez, H.; Lantz, S.M.; Sarkar, S.; Raymick, J.; Robinson, B.; Hanig, J.P.; Herr, D.; et al. Changes in the metabolome and microRNA levels in biological fluids might represent biomarkers of neurotoxicity: A trimethyltin study. Exp. Biol. Med. 2018, 243, 228–236. [Google Scholar] [CrossRef]
- Lee, S.; Yang, M.; Kim, J.; Kang, S.; Kim, J.; Kim, J.-C.; Jung, C.; Shin, T.; Kim, S.-H.; Moon, C. Trimethyltin-induced hippocampal neurodegeneration: A mechanism-based review. Brain Res. Bull. 2016, 125, 187–199. [Google Scholar] [CrossRef]
- Eskes, C.; Jeanneret, L.J.; Leuba, G.; Honegger, P.; Tschudi, F.M. Involvement of microglia–neuron interactions in the tumor necrosis factor-α release, microglial activation, and neurodegeneration induced by trimethyltin. J. Neurosci. Res. 2003, 71, 583–590. [Google Scholar] [CrossRef]
- Monnet-Tschudi, F.; Zurich, M.G.; Pithon, E.; van Melle, G.; Honegger, P. Microglial responsiveness as a sensitive marker for trimethyltin (TMT) neurotoxicity. Brain Res. 1995, 690, 8–14. [Google Scholar] [CrossRef]
- Honegger, P.; Defaux, A.; Monnet-Tschudi, F.; Zurich, M.-G. Preparation, maintenance, and use of serum-free aggregating brain cell cultures. Methods Mol. Biol. 2011, 758, 81–97. [Google Scholar]
- Pezzatti, J.; González-Ruiz, V.; Codesido, S.; Gagnebin, Y.; Joshi, A.; Guillarme, D.; Schappler, J.; Picard, D.; Boccard, J.; Rudaz, S. A scoring approach for multi-platform acquisition in metabolomics. J. Chromatogr. A 2019, 1592, 47–54. [Google Scholar] [CrossRef]
- González-Ruiz, V.; Pezzatti, J.; Roux, A.; Stoppini, L.; Boccard, J.; Rudaz, S. Unravelling the effects of multiple experimental factors in metabolomics, analysis of human neural cells with hydrophilic interaction liquid chromatography hyphenated to high resolution mass spectrometry. J. Chromatogr. A 2017, 1527, 53–60. [Google Scholar] [CrossRef]
- León, Z.; García-Cañaveras, J.C.; Donato, M.T.; Lahoz, A. Mammalian cell metabolomics: Experimental design and sample preparation. Electrophoresis 2013, 34, 2762–2775. [Google Scholar] [CrossRef]
- Lu, W.; Su, X.; Klein, M.S.; Lewis, I.A.; Fiehn, O.; Rabinowitz, J.D. Metabolite Measurement: Pitfalls to Avoid and Practices to Follow. Annu. Rev. Biochem. 2017, 86, 277–304. [Google Scholar] [CrossRef]
- Teng, Q.; Huang, W.; Collette, T.W.; Ekman, D.R.; Tan, C. A direct cell quenching method for cell-culture based metabolomics. Metabolomics 2008, 5, 199–208. [Google Scholar] [CrossRef]
- Ser, Z.; Liu, X.; Tang, N.N.; Locasale, J.W. Extraction parameters for metabolomics from cultured cells. Anal. Biochem. 2015, 475, 22–28. [Google Scholar] [CrossRef]
- Randazzo, G.M.; Tonoli, D.; Strajhar, P.; Xenarios, I.; Odermatt, A.; Boccard, J.; Rudaz, S. Enhanced metabolite annotation via dynamic retention time prediction: Steroidogenesis alterations as a case study. J. Chromatogr. B 2017, 1071, 11–18. [Google Scholar] [CrossRef]
- Dunn, W.B.; Erban, A.; Weber, R.J.M.; Creek, D.J.; Brown, M.; Breitling, R.; Hankemeier, T.; Goodacre, R.; Neumann, S.; Kopka, J.; et al. Mass appeal: Metabolite identification in mass spectrometry-focused untargeted metabolomics. Metabolomics 2012, 9, 44–66. [Google Scholar] [CrossRef]
- Azzollini, A.; Boggia, L.; Boccard, J.; Sgorbini, B.; Lecoultre, N.; Allard, P.-M.; Rubiolo, P.; Rudaz, S.; Gindro, K.; Bicchi, C.; et al. Dynamics of Metabolite Induction in Fungal Co-cultures by Metabolomics at Both Volatile and Non-volatile Levels. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef]
- Boccard, J.; Tonoli, D.; Strajhar, P.; Jeanneret, F.; Odermatt, A.; Rudaz, S. Removal of batch effects using stratified subsampling of metabolomic data for in vitro endocrine disruptors screening. Talanta 2019, 195, 77–86. [Google Scholar] [CrossRef]
- Snoeij, N.J.; van Rooijen, H.J.M.; Penninks, A.; Seinen, W. Effects of various inhibitors of oxidative phosphorylation on energy metabolism, macromolecular synthesis and cyclic AMP production in isolated rat thymocytes. A regulating role for the cellular energy state in macromolecular synthesis and cyclic AMP production. Biochimica et Biophysica Acta 1986, 852, 244–253. [Google Scholar]
- Moffett, J.; Ross, B.; Arun, P.; Madhavarao, C.; NAMBOODIRI, A. N-Acetylaspartate in the CNS: From neurodiagnostics to neurobiology. Prog. Neurobiol. 2007, 81, 89–131. [Google Scholar] [CrossRef]
- Aschner, M.; Aschner, J.L. Cellular and molecular effects of trimethyltin and triethyltin: Relevance to organotin neurotoxicity. Neurosci. Biobehav. Rev. 1992, 16, 427–435. [Google Scholar] [CrossRef]
- Costa, L.G. Interactions of neurotoxicants with neurotransmitter systems. Toxicology 1988, 49, 359–366. [Google Scholar] [CrossRef]
- Ferraz da Silva, I.; Freitas-Lima, L.C.; Graceli, J.B.; Rodrigues, L.C. Organotins in Neuronal Damage, Brain Function, and Behavior: A Short Review. Front. Endocrinol. 2018, 8, 366. [Google Scholar] [CrossRef]
- Schvartz, D.; González-Ruiz, V.; Walter, N.; Antinori, P.; Jeanneret, F.; Tonoli, D.; Boccard, J.; Zurich, M.-G.; Rudaz, S.; Tschudi-Monnet, F.; et al. Protein pathway analysis to study development-dependent effects of acute and repeated Trimethyltin (TMT) treatments in 3D rat brain cell cultures. Toxicology in Vitro. submitted.
- Boccard, J.; Rudaz, S. Integration of Metabolomic Data From Multiple Analytical Platforms: Towards Extensive Coverage of the Metabolome; Comprehensive Analytical Chemistry, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 1–28. [Google Scholar]
- Rajasundaram, D.; Selbig, J. More Effort—More Results: Recent Advances in Integrative “Omics” Data Analysis. Curr. Opin. Plant. Biol. 2016, 30, 57–61. [Google Scholar] [CrossRef]
- Rohart, F.; Gautier, B.; Singh, A.; Cao, K.-A.L. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef]
- Lê Cao, K.-A.; González, I.; Déjean, S. integrOmics: An R package to unravel relationships between two omics datasets. Bioinformatics 2009, 25, 2855–2856. [Google Scholar] [CrossRef]
- Wu, C.; Sun, D. GABA receptors in brain development, function, and injury. Metab. Brain Dis. 2014, 30, 367–379. [Google Scholar] [CrossRef]
- Andersson, H.; Radesäter, A.C.; Luthman, J. Trimethyltin-induced loss of NMDA and kainate receptors in the rat brain. Amino Acids 1995, 8, 23–35. [Google Scholar] [CrossRef]
- Baciak, L.; Gasparova, Z.; Liptaj, T.; Juranek, I. In vivo magnetic resonance approach to trimethyltin induced neurodegeneration in rats. Brain Res. 2017, 1673, 111–116. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Maturation State | Exposure Duration | TMT Conc. | Maturation × Exposure | Maturation × Conc. | Exposure × Conc. | Residuals | |
|---|---|---|---|---|---|---|---|
| Contribution | 22.3% | 15.3% | 6.3% | 13.5% | 2.1% | 3.8% | 36.6% |
| RSR | 2.42 | 1.77 | 1.37 | 1.49 | 1.01 | 1.07 | 1.00 |
| p-value | 0.01 | 0.01 | 0.01 | 0.01 | 83.53 | 29.10 | - |
| Proteomics | Metabolomics | |||||
|---|---|---|---|---|---|---|
| Maps | p-value | FDR | Found Elements | p-value | FDR | Found Elements |
| Neurophysiological process: GABA-A receptor life cycle | 3.721 × 10−9 | 9.208 × 10−7 | 7/27 | 1.316 × 10−1 | 2.466 × 10−1 | 1/27 |
| Neurophysiological process: Role of CDK5 in presynaptic signaling | 4.924 × 10−9 | 9.208 × 10−7 | 7/28 | 9.239 × 10−3 | 4.070 × 10−2 | 2/28 |
| Neurophysiological process: HTR2A signaling in the nervous system | 1.177 × 10−8 | 1.761 × 10−6 | 8/48 | 2.221 × 10−1 | 2.784 × 10−1 | 1/48 |
| Stem cells: Schema: Adult neuron differentiation in the Subventricular and Subgranular Zones | 4.166 × 10−2 | 1.484 × 10−1 | 2/35 | 2.227 × 10−8 | 1.815 × 10−6 | 6/35 |
| Neurophysiological process: Activity-dependent synaptic AMPA receptor removal | 1.221 × 10−7 | 1.142 × 10−5 | 8/64 | 2.847 × 10−1 | 2.994 × 10−1 | 1/64 |
| Nicotine signaling in dopaminergic neurons, Pt. 1 - cell body | 9.946 × 10−3 | 6.526 × 10−2 | 3/48 | 1.598 × 10−7 | 7.598 × 10−6 | 6/48 |
| Gamma-aminobutyrate (GABA) biosynthesis and metabolism | 8.397 × 10−2 | 2.073 × 10−1 | 2/52 | 2.608 × 10−7 | 8.503 × 10−6 | 6/52 |
| Histidine-glutamate-glutamine metabolism | 2.240 × 10−1 | 3.870 × 10−1 | 2/96 | 6.016 × 10−7 | 1.613 × 10−5 | 7/96 |
| Neurophysiological process: Constitutive and regulated NMDA receptor trafficking | 2.185 × 10−6 | 1.634 × 10−4 | 7/65 | 4.498 × 10−2 | 1.094 × 10−1 | 2/65 |
| Neurophysiological process: GABAergic neurotransmission | 1.051 × 10−4 | 3.574 × 10−3 | 5/51 | 6.315 × 10−6 | 1.287 × 10−4 | 5/51 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Ruiz, V.; Schvartz, D.; Sandström, J.; Pezzatti, J.; Jeanneret, F.; Tonoli, D.; Boccard, J.; Monnet-Tschudi, F.; Sanchez, J.-C.; Rudaz, S. An Integrative Multi-Omics Workflow to Address Multifactorial Toxicology Experiments. Metabolites 2019, 9, 79. https://doi.org/10.3390/metabo9040079
González-Ruiz V, Schvartz D, Sandström J, Pezzatti J, Jeanneret F, Tonoli D, Boccard J, Monnet-Tschudi F, Sanchez J-C, Rudaz S. An Integrative Multi-Omics Workflow to Address Multifactorial Toxicology Experiments. Metabolites. 2019; 9(4):79. https://doi.org/10.3390/metabo9040079
Chicago/Turabian StyleGonzález-Ruiz, Víctor, Domitille Schvartz, Jenny Sandström, Julian Pezzatti, Fabienne Jeanneret, David Tonoli, Julien Boccard, Florianne Monnet-Tschudi, Jean-Charles Sanchez, and Serge Rudaz. 2019. "An Integrative Multi-Omics Workflow to Address Multifactorial Toxicology Experiments" Metabolites 9, no. 4: 79. https://doi.org/10.3390/metabo9040079
APA StyleGonzález-Ruiz, V., Schvartz, D., Sandström, J., Pezzatti, J., Jeanneret, F., Tonoli, D., Boccard, J., Monnet-Tschudi, F., Sanchez, J.-C., & Rudaz, S. (2019). An Integrative Multi-Omics Workflow to Address Multifactorial Toxicology Experiments. Metabolites, 9(4), 79. https://doi.org/10.3390/metabo9040079