Software Tools and Approaches for Compound Identification of LC-MS/MS Data in Metabolomics




Abstract
1. Introduction
2. Compound Databases and Chemical Space
3. Mass Spectral Database Search for Fast Annotations
4. In Silico Generation of Mass Spectra and MS/MS Spectra
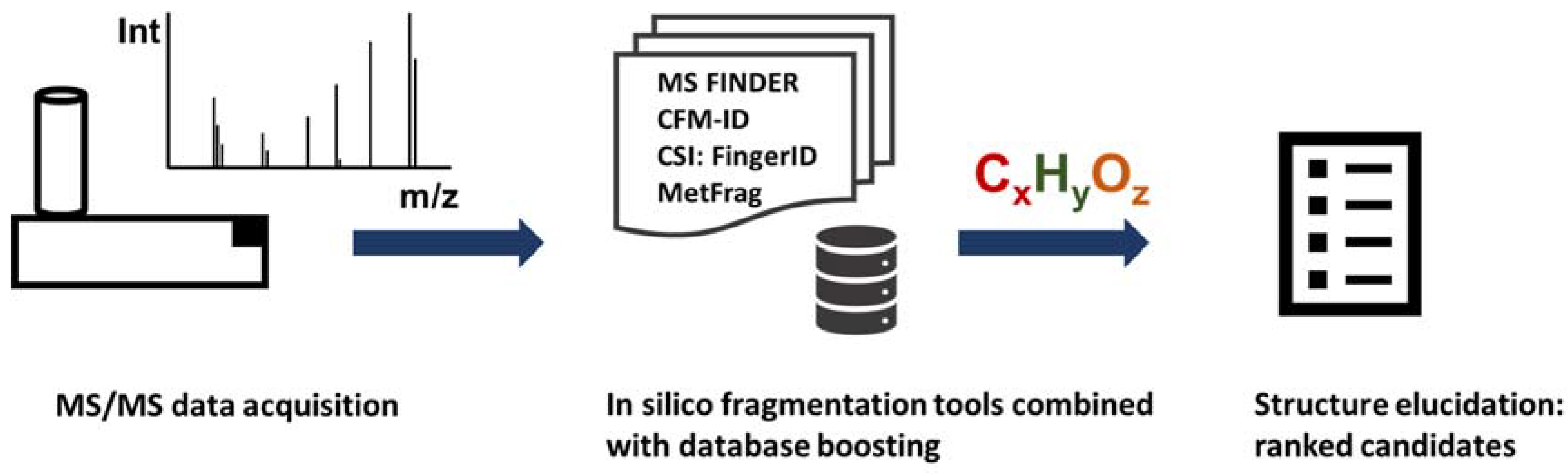
5. In Silico Fragmentation Software
6. Retention Time Prediction
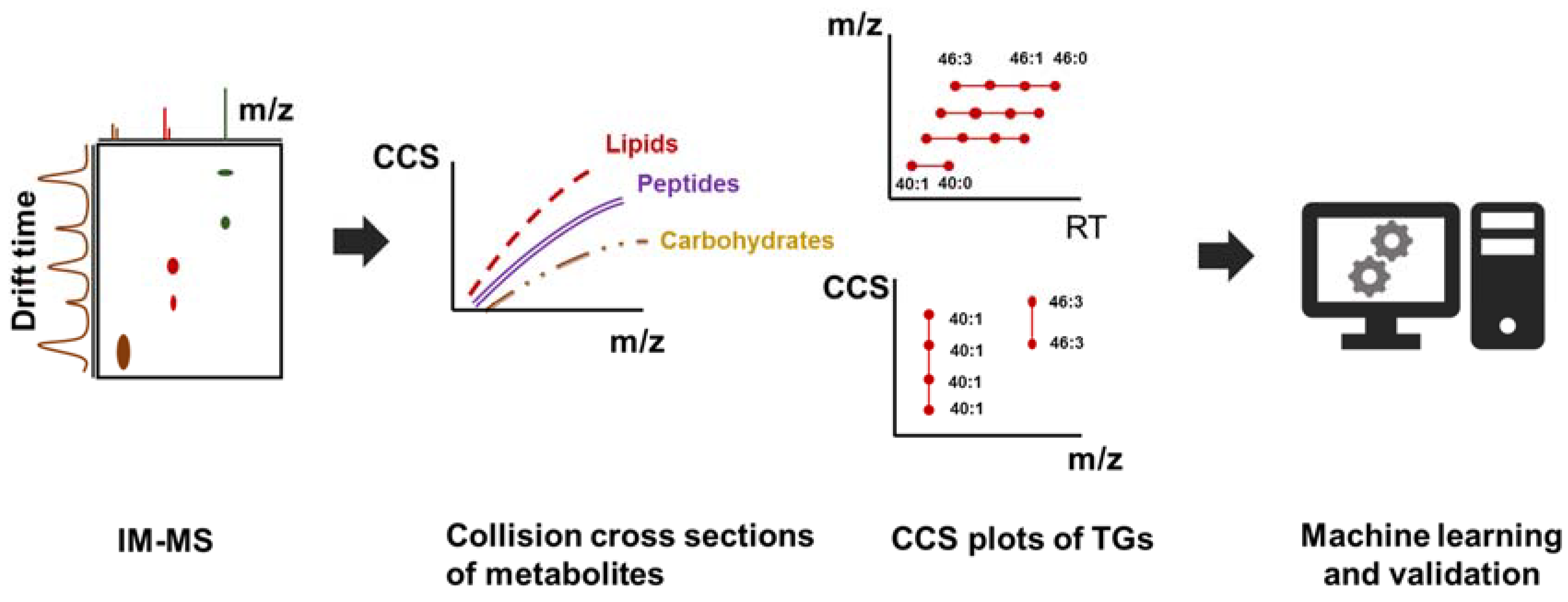
7. Ion Mobility and the Use of Collision Cross Section (CCS) Values
8. Compound Identification: Hybrid and Orthogonal Approaches
9. Critical Assessment of Small Molecule Analysis (CASMI)
10. Data Sharing and Data Retention
11. Conclusions and Outlook
Abbreviations and Glossary
| MSn | Multiple stage mass spectrometry |
| CASMI | Critical Assessment of Small Molecule Identification |
| CCS | Collisional cross-section |
| CFM-ID | Competitive Fragmentation Modeling for Metabolite Identification |
| FAHFAs | Fatty Acid ester of Hydroxyl Fatty Acids |
| Fragmentation tree | Mass spectral fragmentation pathway of a compound |
| GNPS | Global Natural Products Social molecular networking |
| HMDB | Human Metabolome Database |
| IM | Ion mobility |
| InChIKey | Hash key or short unique structure code |
| LipidBlast | In silico generated database for lipid identification |
| MassBank | Mass spectral database |
| MetaboBASE | Mass spectral library developed by Bruker |
| MoNA | MassBank of North America |
| NIST | National Institute of Standards and Technology |
| NMR | Nuclear Magnetic Resonance |
| ReSpect | RIKEN MSn spectral database for phytochemicals |
| SPLASH | Hashed code or unique identifier for mass spectra |
Author Contributions
Acknowledgments
Conflicts of Interest
References
- James, S.J.; Cutler, P.; Melnyk, S.; Jernigan, S.; Janak, L.; Gaylor, D.W.; Neubrander, J.A. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am. J. Clin. Nutr. 2004, 80, 1611–1617. [Google Scholar] [CrossRef] [PubMed]
- Vasan, R.S. Biomarkers of cardiovascular disease: Molecular basis and practical considerations. Circulation 2006, 113, 2335–2362. [Google Scholar] [CrossRef] [PubMed]
- Beger, R.D.; Dunn, W.; Schmidt, M.A.; Gross, S.S.; Kirwan, J.A.; Cascante, M.; Brennan, L.; Wishart, D.S.; Oresic, M.; Hankemeier, T.; et al. Metabolomics enables precision medicine: “A White Paper, Community Perspective”. Metabolomics 2016, 12, 149. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. Computational strategies for metabolite identification in metabolomics. Bioanalysis 2009, 1, 1579–1596. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, R.R.; Dorrestein, P.C.; Quinn, R.A. Illuminating the dark matter in metabolomics. Proc. Natl. Acad. Sci. USA 2015, 112, 12549–12550. [Google Scholar] [CrossRef] [PubMed]
- Peisl, L.; Schymanski, E.L.; Wilmes, P. Dark matter in host-microbiome metabolomics: Tackling the unknowns—A review. Anal. Chim. Acta 2017. [Google Scholar] [CrossRef]
- Uppal, K.; Walker, D.I.; Liu, K.; Li, S.; Go, Y.-M.; Jones, D.P. Computational metabolomics: A framework for the million metabolome. Chem. Res. Toxicol. 2016, 29, 1956–1975. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Choi, Y.H.; Verpoorte, R. NMR-based metabolomic analysis of plants. Nat. Protoc. 2010, 5, 536–549. [Google Scholar] [CrossRef] [PubMed]
- Eisenreich, W.; Bacher, A. Advances of high-resolution NMR techniques in the structural and metabolic analysis of plant biochemistry. Phytochemistry 2007, 68, 2799–2815. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Victoria, I.; Martín, J.; Reyes, F. Combined LC/UV/MS and NMR strategies for the dereplication of marine natural products. Planta Med. 2016, 82, 857–871. [Google Scholar] [CrossRef] [PubMed]
- Hubert, J.; Nuzillard, J.-M.; Renault, J.-H. Dereplication strategies in natural product research: How many tools and methodologies behind the same concept? Phytochem. Rev. 2017, 16, 55–95. [Google Scholar] [CrossRef]
- Schymanski, E.L.; Jeon, J.; Gulde, R.; Fenner, K.; Ruff, M.; Singer, H.P.; Hollender, J. Identifying small molecules via high resolution mass spectrometry: Communicating confidence. Environ. Sci. Technol. 2014, 48, 2097–2098. [Google Scholar] [CrossRef] [PubMed]
- Rochat, B. Proposed Confidence Scale and ID Score in the Identification of Known-Unknown Compounds Using High Resolution MS Data. J. Am. Soc. Mass Spectrom. 2017, 28, 709–723. [Google Scholar] [CrossRef] [PubMed]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.-M.; Fiehn, O.; Goodacre, R.; Griffin, J.L. Proposed minimum reporting standards for chemical analysis. Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Viant, M.R.; Kurland, I.J.; Jones, M.R.; Dunn, W.B. How close are we to complete annotation of metabolomes? Curr. Opin. Chem. Biol. 2017, 36, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Milman, B.L.; Zhurkovich, I.K. The chemical space for non-target analysis. TrAC Trends Anal. Chem. 2017, 97, 179–187. [Google Scholar] [CrossRef]
- Spicer, R.; Salek, R.M.; Moreno, P.; Canueto, D.; Steinbeck, C. Navigating freely-available software tools for metabolomics analysis. Metabolomics 2017, 13, 106. [Google Scholar] [CrossRef] [PubMed]
- Cajka, T.; Fiehn, O. Toward Merging Untargeted and Targeted Methods in Mass Spectrometry-Based Metabolomics and Lipidomics. Anal. Chem. 2016, 88, 524–545. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Tsugawa, H.; Cajka, T.; Ma, Y.; Lai, Z.; Mehta, S.S.; Wohlgemuth, G.; Barupal, D.K.; Showalter, M.R.; Arita, M.; et al. Identification of small molecules using accurate mass MS/MS search. Mass Spectrom. Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Almenara, X.; Montenegro-Burke, J.R.; Benton, H.P.; Siuzdak, G. Annotation: A Computational Solution for Streamlining Metabolomics Analysis. Anal. Chem. 2017, 90, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Fiehn, O. Advances in structure elucidation of small molecules using mass spectrometry. Bioanal. Rev. 2010, 2, 23–60. [Google Scholar] [CrossRef] [PubMed]
- Vaniya, A.; Fiehn, O. Using fragmentation trees and mass spectral trees for identifying unknown compounds in metabolomics. Trends Anal. Chem. 2015, 69, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Misra, B.B. New tools and resources in metabolomics: 2016–2017. Electrophoresis 2018. [Google Scholar] [CrossRef] [PubMed]
- Fenaille, F.; Saint-Hilaire, P.B.; Rousseau, K.; Junot, C. Data acquisition workflows in liquid chromatography coupled to high resolution mass spectrometry-based metabolomics: Where do we stand? J. Chromatogr. A 2017, 1526, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zaikin, V.; Halket, J.M. A Handbook of Derivatives for Mass Spectrometry; IM Publications: West Sussex, UK, 2009. [Google Scholar]
- Gil de la Fuente, A.; Armitage, E.G.; Otero, A.; Barbas, C.; Godzien, J. Differentiating signals to make biological sense—A guide through databases for MS-based non-targeted metabolomics. Electrophoresis 2017. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, A.A.; da Silva, R.; Knight, R.; Lopes, N.P.; Dorrestein, P.C. Global chemical analysis of biology by mass spectrometry. Nat. Rev. Chem. 2017, 1, 0054. [Google Scholar] [CrossRef]
- Garg, N.; Luzzatto-Knaan, T.; Melnik, A.V.; Caraballo-Rodríguez, A.M.; Floros, D.J.; Petras, D.; Gregor, R.; Dorrestein, P.C.; Phelan, V.V. Natural products as mediators of disease. Nat. Prod. Rep. 2017, 34, 194–219. [Google Scholar] [CrossRef] [PubMed]
- Bloszies, C.S.; Fiehn, O. Using untargeted metabolomics for detecting exposome compounds. Curr. Opin. Toxicol. 2018, 8, 87–92. [Google Scholar] [CrossRef]
- Sud, M.; Fahy, E.; Cotter, D.; Azam, K.; Vadivelu, I.; Burant, C.; Edison, A.; Fiehn, O.; Higashi, R.; Nair, K.S. Metabolomics Workbench: An international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 2015, 44, D463–D470. [Google Scholar] [CrossRef] [PubMed]
- Haug, K.; Salek, R.M.; Conesa, P.; Hastings, J.; de Matos, P.; Rijnbeek, M.; Mahendraker, T.; Williams, M.; Neumann, S.; Rocca-Serra, P. MetaboLights—An open-access general-purpose repository for metabolomics studies and associated meta-data. Nucleic Acids Res. 2012, 41, D781–D786. [Google Scholar] [CrossRef] [PubMed]
- Bolton, E.E.; Wang, Y.; Thiessen, P.A.; Bryant, S.H. PubChem: Integrated platform of small molecules and biological activities. Annu. Rep. Comput. Chem. 2008, 4, 217–241. [Google Scholar]
- Pence, H.E.; Williams, A. ChemSpider: An online chemical information resource. J. Chem. Educ. 2010, 87, 1123–1124. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Foerster, H.; Fulcher, C.A.; Kaipa, P.; Krummenacker, M.; Latendresse, M.; Paley, S.; Rhee, S.Y.; Shearer, A.G.; Tissier, C. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2007, 36, D623–D631. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, I.; Jeske, L.; Ulbrich, M.; Placzek, S.; Chang, A.; Schomburg, D. The BRENDA enzyme information system–From a database to an expert system. J. Biotechnol. 2017, 261, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E. HMDB 3.0—The human metabolome database in 2013. Nucleic Acids Res. 2012, 41, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Degtyarenko, K.; De Matos, P.; Ennis, M.; Hastings, J.; Zbinden, M.; McNaught, A.; Alcántara, R.; Darsow, M.; Guedj, M.; Ashburner, M. ChEBI: A database and ontology for chemical entities of biological interest. Nucleic Acids Res. 2007, 36, D344–D350. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Gui, Y.; Chen, L.; Yuan, G.; Lu, H.-Z.; Xu, X. Use of natural products as chemical library for drug discovery and network pharmacology. PLoS ONE 2013, 8, e62839. [Google Scholar] [CrossRef] [PubMed]
- Jeffryes, J.G.; Colastani, R.L.; Elbadawi-Sidhu, M.; Kind, T.; Niehaus, T.D.; Broadbelt, L.J.; Hanson, A.D.; Fiehn, O.; Tyo, K.E.; Henry, C.S. MINEs: Open access databases of computationally predicted enzyme promiscuity products for untargeted metabolomics. J. Cheminform. 2015, 7, 44. [Google Scholar] [CrossRef] [PubMed]
- Pye, C.R.; Bertin, M.J.; Lokey, R.S.; Gerwick, W.H.; Linington, R.G. Retrospective analysis of natural products provides insights for future discovery trends. Proc. Natl. Acad. Sci. USA 2017, 114, 5601–5606. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, S.; Kell, D.B. Analysing and navigating natural products space for generating small, diverse, but representative chemical libraries. Biotechnol. J. 2018, 13, 1700503. [Google Scholar] [CrossRef] [PubMed]
- Warth, B.; Spangler, S.; Fang, M.; Johnson, C.H.; Forsberg, E.M.; Granados, A.; Martin, R.L.; Domingo-Almenara, X.; Huan, T.; Rinehart, D. Exposome-scale investigations guided by global metabolomics, pathway analysis, and cognitive computing. Anal. Chem. 2017, 89, 11505–11513. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.J.; Grulke, C.M.; Edwards, J.; McEachran, A.D.; Mansouri, K.; Baker, N.C.; Patlewicz, G.; Shah, I.; Wambaugh, J.F.; Judson, R.S. The CompTox Chemistry Dashboard: A community data resource for environmental chemistry. J. Cheminform. 2017, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.; Tsugawa, H.; Wohlgemuth, G.; Mehta, S.; Mueller, M.; Zheng, Y.; Ogiwara, A.; Meissen, J.; Showalter, M.; Takeuchi, K. Identifying metabolites by integrating metabolome databases with mass spectrometry cheminformatics. Nat. Methods 2018, 15, 53. [Google Scholar] [CrossRef] [PubMed]
- Stein, S. Mass spectral reference libraries: An ever-expanding resource for chemical identification. Anal. Chem. 2012, 84, 7274–7282. [Google Scholar] [CrossRef] [PubMed]
- Heller, S.; McNaught, A.; Stein, S.; Tchekhovskoi, D.; Pletnev, I. InChI-the worldwide chemical structure identifier standard. J. Cheminform. 2013, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Wohlgemuth, G.; Mehta, S.S.; Mejia, R.F.; Neumann, S.; Pedrosa, D.; Pluskal, T.; Schymanski, E.L.; Willighagen, E.L.; Wilson, M.; Wishart, D.S. SPLASH, a hashed identifier for mass spectra. Nat. Biotechnol. 2016, 34, 1099–1101. [Google Scholar] [CrossRef] [PubMed]
- Wallace, W.E.; Ji, W.; Tchekhovskoi, D.V.; Phinney, K.W.; Stein, S.E. Mass spectral library quality assurance by inter-library comparison. J. Am. Soc. Mass Spectrom. 2017, 28, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Vinaixa, M.; Schymanski, E.L.; Neumann, S.; Navarro, M.; Salek, R.M.; Yanes, O. Mass spectral databases for LC/MS-and GC/MS-based metabolomics: State of the field and future prospects. TrAC Trends Anal. Chem. 2016, 78, 23–35. [Google Scholar] [CrossRef]
- Guijas, C.; Montenegro-Burke, J.R.; Domingo-Almenara, X.; Palermo, A.; Warth, B.; Hermann, G.; Koellensperger, G.; Huan, T.; Uritboonthai, W.; Aisporna, A.E. METLIN: A Technology Platform for Identifying Knowns and Unknowns. Anal. Chem. 2018, 90, 3156–3164. [Google Scholar] [CrossRef] [PubMed]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Peake, D.A.; Mistrik, R.; Huang, Y. A platform to identify endogenous metabolites using a novel high performance Orbitrap MS and the mzCloud Library. Blood 2013, 4, 2–8. [Google Scholar]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Sawada, Y.; Nakabayashi, R.; Yamada, Y.; Suzuki, M.; Sato, M.; Sakata, A.; Akiyama, K.; Sakurai, T.; Matsuda, F.; Aoki, T. RIKEN tandem mass spectral database (ReSpect) for phytochemicals: A plant-specific MS/MS-based data resource and database. Phytochemistry 2012, 82, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Simon-Manso, Y.; Lowenthal, M.S.; Kilpatrick, L.E.; Sampson, M.L.; Telu, K.H.; Rudnick, P.A.; Mallard, W.G.; Bearden, D.W.; Schock, T.B.; Tchekhovskoi, D.V.; et al. Metabolite profiling of a NIST Standard Reference Material for human plasma (SRM 1950): GC-MS, LC-MS, NMR, and clinical laboratory analyses, libraries, and web-based resources. Anal. Chem. 2013, 85, 11725–11731. [Google Scholar] [CrossRef] [PubMed]
- Psychogios, N.; Hau, D.D.; Peng, J.; Guo, A.C.; Mandal, R.; Bouatra, S.; Sinelnikov, I.; Krishnamurthy, R.; Eisner, R.; Gautam, B. The human serum metabolome. PLoS ONE 2011, 6, e16957. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523. [Google Scholar] [CrossRef] [PubMed]
- Moorthy, A.S.; Wallace, W.E.; Kearsley, A.J.; Tchekhovskoi, D.V.; Stein, S.E. Combining fragment-ion and neutral-loss matching during mass spectral library searching: A new general purpose algorithm applicable to illicit drug identification. Anal. Chem. 2017, 89, 13261–13268. [Google Scholar] [CrossRef] [PubMed]
- Schollée, J.E.; Schymanski, E.L.; Stravs, M.A.; Gulde, R.; Thomaidis, N.S.; Hollender, J. Similarity of High-Resolution Tandem Mass Spectrometry Spectra of Structurally Related Micropollutants and Transformation Products. J. Am. Soc. Mass Spectrom. 2017, 28, 2692–2704. [Google Scholar] [CrossRef] [PubMed]
- Depke, T.; Franke, R.; Brönstrup, M. Clustering of MS2 spectra using unsupervised methods to aid the identification of secondary metabolites from Pseudomonas aeruginosa. J. Chromatogr. B 2017, 1071, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Scheubert, K.; Hufsky, F.; Petras, D.; Wang, M.; Nothias, L.-F.; Dührkop, K.; Bandeira, N.; Dorrestein, P.C.; Böcker, S. Significance estimation for large scale metabolomics annotations by spectral matching. Nat. Commun. 2017, 8, 1494. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.; Phapale, P.; Chernyavsky, I.; Lavigne, R.; Fay, D.; Tarasov, A.; Kovalev, V.; Fuchser, J.; Nikolenko, S.; Pineau, C. FDR-controlled metabolite annotation for high-resolution imaging mass spectrometry. Nat. Methods 2017, 14, 57. [Google Scholar] [CrossRef] [PubMed]
- Stravs, M.A.; Schymanski, E.L.; Singer, H.P.; Hollender, J. Automatic recalibration and processing of tandem mass spectra using formula annotation. J. Mass Spectrom. 2013, 48, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Towards first principles calculation of electron impact mass spectra of molecules. Angew. Chem. Int. Ed. 2013, 52, 6306–6312. [Google Scholar] [CrossRef] [PubMed]
- Bauer, C.A.; Grimme, S. How to compute electron ionization mass spectra from first principles. J. Phys. Chem. A 2016, 120, 3755–3766. [Google Scholar] [CrossRef] [PubMed]
- Bauer, C.A.; Grimme, S. First principles calculation of electron ionization mass spectra for selected organic drug molecules. Org. Biomol. Chem. 2014, 12, 8737–8744. [Google Scholar] [CrossRef] [PubMed]
- Ásgeirsson, V.; Bauer, C.A.; Grimme, S. Unimolecular decomposition pathways of negatively charged nitriles by ab initio molecular dynamics. Phys. Chem. Chem. Phys. 2016, 18, 31017–31026. [Google Scholar] [CrossRef] [PubMed]
- Bauer, C.A.; Grimme, S. Elucidation of electron ionization induced fragmentations of adenine by semiempirical and density functional molecular dynamics. J. Phys. Chem. A 2014, 118, 11479–11484. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.; Greiner, R.; Wishart, D. Competitive fragmentation modeling of ESI-MS/MS spectra for putative metabolite identification. Metabolomics 2015, 11, 98–110. [Google Scholar] [CrossRef]
- Allen, F.; Pon, A.; Greiner, R.; Wishart, D. Computational prediction of electron ionization mass spectra to assist in GC/MS compound identification. Anal. Chem. 2016, 88, 7689–7697. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.; Pon, A.; Wilson, M.; Greiner, R.; Wishart, D. CFM-ID: A web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra. Nucleic Acids Res. 2014, 42, W94–W99. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Liu, K.-H.; Lee, D.Y.; DeFelice, B.; Meissen, J.K.; Fiehn, O. LipidBlast in silico tandem mass spectrometry database for lipid identification. Nat. Methods 2013, 10, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Okazaki, Y.; Saito, K.; Fiehn, O. LipidBlast templates as flexible tools for creating new in-silico tandem mass spectral libraries. Anal. Chem. 2014, 86, 11024–11027. [Google Scholar] [CrossRef] [PubMed]
- Spackman, P.R.; Bohman, B.; Karton, A.; Jayatilaka, D. Quantum chemical electron impact mass spectrum prediction for de novo structure elucidation: Assessment against experimental reference data and comparison to competitive fragmentation modeling. Int. J. Quantum Chem. 2017. [Google Scholar] [CrossRef]
- Dral, P.O.; Wu, X.; Spörkel, L.; Koslowski, A.; Thiel, W. Semiempirical quantum-chemical orthogonalization-corrected methods: Benchmarks for ground-state properties. J. Chem. Theory Comput. 2016, 12, 1097–1120. [Google Scholar] [CrossRef] [PubMed]
- Ásgeirsson, V.; Bauer, C.A.; Grimme, S. Quantum chemical calculation of electron ionization mass spectra for general organic and inorganic molecules. Chem. Sci. 2017, 8, 4879–4895. [Google Scholar] [CrossRef] [PubMed]
- Cautereels, J.; Claeys, M.; Geldof, D.; Blockhuys, F. Quantum chemical mass spectrometry: ab initio prediction of electron ionization mass spectra and identification of new fragmentation pathways. J. Mass Spectrom. 2016, 51, 602–614. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, N.F.; Díaz-Tendero, S.; Hervieux, P.-A.; Alcamí, M.; Martín, F. M3C: A Computational Approach to Describe Statistical Fragmentation of Excited Molecules and Clusters. J. Chem. Theory Comput. 2017, 13, 992–1009. [Google Scholar] [CrossRef] [PubMed]
- Pracht, P.; Bauer, C.A.; Grimme, S. Automated and efficient quantum chemical determination and energetic ranking of molecular protonation sites. J. Comput. Chem. 2017, 38, 2618–2631. [Google Scholar] [CrossRef] [PubMed]
- Janesko, B.G.; Li, L.; Mensing, R. Quantum Chemical Fragment Precursor Tests: Accelerating de novo annotation of tandem mass spectra. Anal. Chim. Acta 2017, 995, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Böcker, S. Searching molecular structure databases using tandem MS data: Are we there yet? Curr. Opin. Chem. Biol. 2017, 36, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Mogas, A.; Sales-Pardo, M.; Navarro, M.; Guimerà, R.; Yanes, O. imet: A network-based computational tool to assist in the annotation of metabolites from tandem mass spectra. Anal. Chem. 2017, 89, 3474–3482. [Google Scholar] [CrossRef] [PubMed]
- Ridder, L.; van der Hooft, J.J.; Verhoeven, S. Automatic compound annotation from mass spectrometry data using MAGMa. Mass Spectrom. 2014, 3, S0033. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kora, G.; Bowen, B.P.; Pan, C. MIDAS: A database-searching algorithm for metabolite identification in metabolomics. Anal. Chem. 2014, 86, 9496–9503. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.; Zeng, X. MIDAS-G: A computational platform for investigating fragmentation rules of tandem mass spectrometry in metabolomics. Metabolomics 2017, 13, 116. [Google Scholar] [CrossRef]
- Scheubert, K.; Hufsky, F.; Böcker, S. Computational mass spectrometry for small molecules. J. Cheminform. 2013, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Hufsky, F.; Scheubert, K.; Böcker, S. Computational mass spectrometry for small-molecule fragmentation. TrAC Trends Anal. Chem. 2014, 53, 41–48. [Google Scholar] [CrossRef]
- Hufsky, F.; Scheubert, K.; Böcker, S. New kids on the block: Novel informatics methods for natural product discovery. Nat. Prod. Rep. 2014, 31, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Hufsky, F.; Böcker, S. Mining molecular structure databases: Identification of small molecules based on fragmentation mass spectrometry data. Mass Spectrom. Rev. 2017, 36, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Ruttkies, C.; Schymanski, E.L.; Wolf, S.; Hollender, J.; Neumann, S. MetFrag relaunched: Incorporating strategies beyond in silico fragmentation. J. Cheminform. 2016, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Gerlich, M.; Neumann, S. MetFusion: Integration of compound identification strategies. J. Mass Spectrom. 2013, 48, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Witting, M.; Ruttkies, C.; Neumann, S.; Schmitt-Kopplin, P. LipidFrag: Improving reliability of in silico fragmentation of lipids and application to the Caenorhabditis elegans lipidome. PLoS ONE 2017, 12, e0172311. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Fiehn, O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinform. 2007, 8, 105. [Google Scholar] [CrossRef] [PubMed]
- Vaniya, A.; Samra, S.N.; Palazoglu, M.; Tsugawa, H.; Fiehn, O. Using MS-FINDER for identifying 19 natural products in the CASMI 2016 contest. Phytochem. Lett. 2017, 21, 306–312. [Google Scholar] [CrossRef]
- Tsugawa, H.; Kind, T.; Nakabayashi, R.; Yukihira, D.; Tanaka, W.; Cajka, T.; Saito, K.; Fiehn, O.; Arita, M. Hydrogen Rearrangement Rules: Computational MS/MS Fragmentation and Structure Elucidation Using MS-FINDER Software. Anal. Chem. 2016, 88, 7946–7958. [Google Scholar] [CrossRef] [PubMed]
- Duhrkop, K.; Shen, H.; Meusel, M.; Rousu, J.; Bocker, S. Searching molecular structure databases with tandem mass spectra using CSI:FingerID. Proc. Natl. Acad. Sci. USA 2015, 112, 12580–12585. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, M.; Shen, H.; Zamboni, N.; Rousu, J. Metabolite identification and molecular fingerprint prediction through machine learning. Bioinformatics 2012, 28, 2333–2341. [Google Scholar] [CrossRef] [PubMed]
- Brouard, C.; Shen, H.; Dührkop, K.; d’Alché-Buc, F.; Böcker, S.; Rousu, J. Fast metabolite identification with input output kernel regression. Bioinformatics 2016, 32, i28–i36. [Google Scholar] [CrossRef] [PubMed]
- Bocker, S.; Letzel, M.C.; Liptak, Z.; Pervukhin, A. SIRIUS: Decomposing isotope patterns for metabolite identification. Bioinformatics 2009, 25, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Rasche, F.; Svatoš, A.; Maddula, R.K.; Böttcher, C.; Böcker, S. Computing fragmentation trees from tandem mass spectrometry data. Anal. Chem. 2010, 83, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Laponogov, I.; Sadawi, N.; Galea, D.; Mirnezami, R.; Veselkov, K.A.; Wren, J. ChemDistiller: An engine for metabolite annotation in mass spectrometry. Bioinformatics 2018, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, A.; Butcher, P.; Maden, K.; Walker, S.; Widmer, M. Using In Silico Fragmentation to Improve Routine Residue Screening in Complex Matrices. J. Am. Soc. Mass Spectrom. 2017, 28, 2705–2715. [Google Scholar] [CrossRef] [PubMed]
- Blazenovic, I.; Kind, T.; Torbasinovic, H.; Obrenovic, S.; Mehta, S.S.; Tsugawa, H.; Wermuth, T.; Schauer, N.; Jahn, M.; Biedendieck, R.; et al. Comprehensive comparison of in silico MS/MS fragmentation tools of the CASMI contest: Database boosting is needed to achieve 93% accuracy. J. Cheminform. 2017, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Schymanski, E.L.; Ruttkies, C.; Krauss, M.; Brouard, C.; Kind, T.; Dührkop, K.; Allen, F.; Vaniya, A.; Verdegem, D.; Böcker, S.; et al. Critical Assessment of Small Molecule Identification 2016: Automated methods. J. Cheminform. 2017, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Kaliszan, R. QSRR: Quantitative structure-(chromatographic) retention relationships. Chem. Rev. 2007, 107, 3212–3246. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.E.; Babushok, V.I.; Brown, R.L.; Linstrom, P.J. Estimation of Kovats retention indices using group contributions. J. Chem. Inf. Model. 2007, 47, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Reig, M.; Ortiz-Villanueva, E.; Tauler, R.; Jaumot, J. Modelling of Hydrophilic Interaction Liquid Chromatography Stationary Phases Using Chemometric Approaches. Metabolites 2017, 7, 54. [Google Scholar] [CrossRef] [PubMed]
- Sahigara, F.; Mansouri, K.; Ballabio, D.; Mauri, A.; Consonni, V.; Todeschini, R. Comparison of different approaches to define the applicability domain of QSAR models. Molecules 2012, 17, 4791–4810. [Google Scholar] [CrossRef] [PubMed]
- Hall, L.M.; Hill, D.W.; Bugden, K.; Cawley, S.; Hall, L.H.; Chen, M.-H.; Grant, D.F. Development of a Reverse Phase HPLC Retention Index Model for Nontargeted Metabolomics Using Synthetic Compounds. J. Chem. Inf. Model. 2018, 58, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Barnes, B.B.; Wilson, M.B.; Carr, P.W.; Vitha, M.F.; Broeckling, C.D.; Heuberger, A.L.; Prenni, J.; Janis, G.C.; Corcoran, H.; Snow, N.H. “Retention projection” enables reliable use of shared gas chromatographic retention data across laboratories, instruments, and methods. Anal. Chem. 2013, 85, 11650–11657. [Google Scholar] [CrossRef] [PubMed]
- Stanstrup, J.; Neumann, S.; Vrhovsek, U. PredRet: Prediction of retention time by direct mapping between multiple chromatographic systems. Anal. Chem. 2015, 87, 9421–9428. [Google Scholar] [CrossRef] [PubMed]
- Boswell, P.G.; Abate-Pella, D.; Hewitt, J.T. Calculation of retention time tolerance windows with absolute confidence from shared liquid chromatographic retention data. J. Chromatogr. A 2015, 1412, 52–58. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mauri, A.; Consonni, V.; Pavan, M.; Todeschini, R. Dragon software: An easy approach to molecular descriptor calculations. Match 2006, 56, 237–248. [Google Scholar]
- Hong, H.; Xie, Q.; Ge, W.; Qian, F.; Fang, H.; Shi, L.; Su, Z.; Perkins, R.; Tong, W. Mold2, molecular descriptors from 2D structures for chemoinformatics and toxicoinformatics. J. Chem. Inf. Model. 2008, 48, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, N.-O.; de Bruyn Kops, C.; Flachsenberg, F.; Sommer, K.; Rarey, M.; Kirchmair, J. Benchmarking Commercial Conformer Ensemble Generators. J. Chem. Inf. Model. 2017, 57, 2719–2728. [Google Scholar] [CrossRef] [PubMed]
- Kanal, I.Y.; Keith, J.A.; Hutchison, G.R. A sobering assessment of small-molecule force field methods for low energy conformer predictions. Int. J. Quantum Chem. 2018, 118, e25512. [Google Scholar] [CrossRef]
- Ma, J.; Sheridan, R.P.; Liaw, A.; Dahl, G.E.; Svetnik, V. Deep neural nets as a method for quantitative structure–activity relationships. J. Chem. Inf. Model. 2015, 55, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Delgado, M.; Cernadas, E.; Barro, S.; Amorim, D. Do we need hundreds of classifiers to solve real world classification problems. J. Mach. Learn. Res. 2014, 15, 3133–3181. [Google Scholar]
- He, H.; Zhang, W.; Zhang, S. A novel ensemble method for credit scoring: Adaption of different imbalance ratios. Expert Syst. Appl. 2018, 98, 105–117. [Google Scholar] [CrossRef]
- Falchi, F.; Bertozzi, S.M.; Ottonello, G.; Ruda, G.F.; Colombano, G.; Fiorelli, C.; Martucci, C.; Bertorelli, R.; Scarpelli, R.; Cavalli, A. Kernel-based, partial least squares quantitative structure-retention relationship model for UPLC retention time prediction: A useful tool for metabolite identification. Anal. Chem. 2016, 88, 9510–9517. [Google Scholar] [CrossRef] [PubMed]
- Taraji, M.; Haddad, P.R.; Amos, R.I.; Talebi, M.; Szucs, R.; Dolan, J.W.; Pohl, C.A. Use of dual-filtering to create training sets leading to improved accuracy in quantitative structure-retention relationships modelling for hydrophilic interaction liquid chromatographic systems. J. Chromatogr. A 2017, 1507, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Taraji, M.; Haddad, P.R.; Amos, R.I.; Talebi, M.; Szucs, R.; Dolan, J.W.; Pohl, C.A. Prediction of retention in hydrophilic interaction liquid chromatography using solute molecular descriptors based on chemical structures. J. Chromatogr. A 2017, 1486, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Bruderer, T.; Varesio, E.; Hopfgartner, G. The use of LC predicted retention times to extend metabolites identification with SWATH data acquisition. J. Chromatogr. B 2017, 1071, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Creek, D.J.; Jankevics, A.; Breitling, R.; Watson, D.G.; Barrett, M.P.; Burgess, K.E. Toward global metabolomics analysis with hydrophilic interaction liquid chromatography–mass spectrometry: Improved metabolite identification by retention time prediction. Anal. Chem. 2011, 83, 8703–8710. [Google Scholar] [CrossRef] [PubMed]
- Aalizadeh, R.; Thomaidis, N.S.; Bletsou, A.A.; Gago-Ferrero, P. Quantitative Structure–Retention Relationship Models to Support Nontarget High-Resolution Mass Spectrometric Screening of Emerging Contaminants in Environmental Samples. J. Chem. Inf. Model. 2016, 56, 1384–1398. [Google Scholar] [CrossRef] [PubMed]
- Aicheler, F.; Li, J.; Hoene, M.; Lehmann, R.; Xu, G.; Kohlbacher, O. Retention time prediction improves identification in nontargeted lipidomics approaches. Anal. Chem. 2015, 87, 7698–7704. [Google Scholar] [CrossRef] [PubMed]
- Wolfer, A.M.; Lozano, S.; Umbdenstock, T.; Croixmarie, V.; Arrault, A.; Vayer, P. UPLC–MS retention time prediction: A machine learning approach to metabolite identification in untargeted profiling. Metabolomics 2016, 12, 8. [Google Scholar] [CrossRef]
- Cao, M.; Fraser, K.; Huege, J.; Featonby, T.; Rasmussen, S.; Jones, C. Predicting retention time in hydrophilic interaction liquid chromatography mass spectrometry and its use for peak annotation in metabolomics. Metabolomics 2015, 11, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Mollerup, C.B.; Mardal, M.; Dalsgaard, P.W.; Linnet, K.; Barron, L.P. Prediction of collision cross section and retention time for broad scope screening in gradient reversed-phase liquid chromatography-ion mobility-high resolution accurate mass spectrometry. J. Chromatogr. A 2018, 1542, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Hall, L.M.; Hall, L.H.; Kertesz, T.M.; Hill, D.W.; Sharp, T.R.; Oblak, E.Z.; Dong, Y.W.; Wishart, D.S.; Chen, M.H.; Grant, D.F. Development of Ecom50 and retention index models for nontargeted metabolomics: Identification of 1,3-dicyclohexylurea in human serum by HPLC/mass spectrometry. J. Chem. Inf. Model. 2012, 52, 1222–1237. [Google Scholar] [CrossRef] [PubMed]
- Eugster, P.J.; Boccard, J.; Debrus, B.; Bréant, L.; Wolfender, J.-L.; Martel, S.; Carrupt, P.-A. Retention time prediction for dereplication of natural products (CxHyOz) in LC–MS metabolite profiling. Phytochemistry 2014, 108, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Chouinard, C.D.; Cruzeiro, V.W.D.; Beekman, C.R.; Roitberg, A.E.; Yost, R.A. Investigating Differences in Gas-Phase Conformations of 25-Hydroxyvitamin D3 Sodiated Epimers using Ion Mobility-Mass Spectrometry and Theoretical Modeling. J. Am. Soc. Mass Spectrom. 2017, 28, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- May, J.C.; Goodwin, C.R.; Lareau, N.M.; Leaptrot, K.L.; Morris, C.B.; Kurulugama, R.T.; Mordehai, A.; Klein, C.; Barry, W.; Darland, E. Conformational ordering of biomolecules in the gas phase: Nitrogen collision cross sections measured on a prototype high resolution drift tube ion mobility-mass spectrometer. Anal. Chem. 2014, 86, 2107–2116. [Google Scholar] [CrossRef] [PubMed]
- May, J.C.; McLean, J.A. Ion mobility-mass spectrometry: Time-dispersive instrumentation. Anal. Chem. 2015, 87, 1422–1436. [Google Scholar] [CrossRef] [PubMed]
- D’Atri, V.; Causon, T.; Hernandez-Alba, O.; Mutabazi, A.; Veuthey, J.L.; Cianferani, S.; Guillarme, D. Adding a new separation dimension to MS and LC–MS: What is the utility of ion mobility spectrometry? J. Sep. Sci. 2018, 41, 20–67. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Aly, N.A.; Zhou, Y.; Dupuis, K.T.; Bilbao, A.; Paurus, V.L.; Orton, D.J.; Wilson, R.; Payne, S.H.; Smith, R.D.; et al. A structural examination and collision cross section database for over 500 metabolites and xenobiotics using drift tube ion mobility spectrometry. Chem. Sci. 2017, 8, 7724–7736. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Casey, C.P.; Zheng, X.; Ibrahim, Y.M.; Wilkins, C.S.; Renslow, R.S.; Thomas, D.G.; Payne, S.H.; Monroe, M.E.; Smith, R.D.; et al. PIXiE: An algorithm for automated ion mobility arrival time extraction and collision cross section calculation using global data association. Bioinformatics 2017, 33, 2715–2722. [Google Scholar] [CrossRef] [PubMed]
- Ewing, M.A.; Glover, M.S.; Clemmer, D.E. Hybrid ion mobility and mass spectrometry as a separation tool. J. Chromatogr. A 2016, 1439, 3–25. [Google Scholar] [CrossRef] [PubMed]
- May, J.C.; McLean, J.A. Advanced multidimensional separations in mass spectrometry: Navigating the big data deluge. Annu. Rev. Anal. Chem. 2016, 9, 387–409. [Google Scholar] [CrossRef] [PubMed]
- Lapthorn, C.; Pullen, F.; Chowdhry, B.Z. Ion mobility spectrometry-mass spectrometry (IMS-MS) of small molecules: Separating and assigning structures to ions. Mass Spectrom. Rev. 2013, 32, 43–71. [Google Scholar] [CrossRef] [PubMed]
- Canterbury, J.D.; Yi, X.; Hoopmann, M.R.; MacCoss, M.J. Assessing the dynamic range and peak capacity of nanoflow LC−FAIMS−MS on an ion trap mass spectrometer for proteomics. Anal. Chem. 2008, 80, 6888–6897. [Google Scholar] [CrossRef] [PubMed]
- Hines, K.M.; Ross, D.H.; Davidson, K.L.; Bush, M.F.; Xu, L. Large-scale structural characterization of drug and drug-like compounds by high-throughput ion mobility-mass spectrometry. Anal. Chem. 2017, 89, 9023–9030. [Google Scholar] [CrossRef] [PubMed]
- Nichols, C.M.; May, J.C.; Sherrod, S.D.; McLean, J.A. Automated flow injection method for the high precision determination of drift tube ion mobility collision cross sections. Analyst 2018, 143, 1556–1559. [Google Scholar] [CrossRef] [PubMed]
- Mairinger, T.; Causon, T.J.; Hann, S. The potential of ion mobility–mass spectrometry for non-targeted metabolomics. Curr. Opin. Chem. Biol. 2018, 42, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Metz, T.O.; Baker, E.S.; Schymanski, E.L.; Renslow, R.S.; Thomas, D.G.; Causon, T.J.; Webb, I.K.; Hann, S.; Smith, R.D.; Teeguarden, J.G. Integrating ion mobility spectrometry into mass spectrometry-based exposome measurements: What can it add and how far can it go? Bioanalysis 2017, 9, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Lapthorn, C.; Pullen, F.S.; Chowdhry, B.Z.; Wright, P.; Perkins, G.L.; Heredia, Y. How useful is molecular modelling in combination with ion mobility mass spectrometry for ‘small molecule’ ion mobility collision cross-sections? Analyst 2015, 140, 6814–6823. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Tu, J.; Zhu, Z.-J. Advancing the large-scale CCS database for metabolomics and lipidomics at the machine-learning era. Curr. Opin. Chem. Biol. 2018, 42, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Bijlsma, L.; Bade, R.; Celma, A.; Mullin, L.; Cleland, G.; Stead, S.; Hernandez, F.; Sancho, J.V. Prediction of collision cross-section values for small molecules: Application to pesticide residue analysis. Anal. Chem. 2017, 89, 6583–6589. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Shen, X.; Tu, J.; Zhu, Z.-J. Large-scale prediction of collision cross-section values for metabolites in ion mobility-mass spectrometry. Anal. Chem. 2016, 88, 11084–11091. [Google Scholar] [CrossRef] [PubMed]
- Kyle, J.E.; Zhang, X.; Weitz, K.K.; Monroe, M.E.; Ibrahim, Y.M.; Moore, R.J.; Cha, J.; Sun, X.; Lovelace, E.S.; Wagoner, J. Uncovering biologically significant lipid isomers with liquid chromatography, ion mobility spectrometry and mass spectrometry. Analyst 2016, 141, 1649–1659. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Tu, J.; Xiong, X.; Shen, X.; Zhu, Z.-J. LipidCCS: Prediction of Collision Cross-Section Values for Lipids with High Precision to Support Ion Mobility–Mass Spectrometry-Based Lipidomics. Anal. Chem. 2017, 89, 9559–9566. [Google Scholar] [CrossRef] [PubMed]
- Paglia, G.; Williams, J.P.; Menikarachchi, L.; Thompson, J.W.; Tyldesley-Worster, R.; Halldórsson, S.; Rolfsson, O.; Moseley, A.; Grant, D.; Langridge, J.; et al. Ion mobility derived collision cross sections to support metabolomics applications. Anal. Chem. 2014, 86, 3985–3993. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Mesa, M.; Le Bizec, B.; Monteau, F.; García-Campaña, A.M.; Dervilly-Pinel, G. Collision Cross Section (CCS) database: An additional measure to characterize steroids. Anal. Chem. 2018, 90, 4616–4625. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Dupuis, K.T.; Aly, N.A.; Zhou, Y.; Smith, F.B.; Tang, K.; Smith, R.D.; Baker, E.S. Utilizing ion mobility spectrometry and mass spectrometry for the analysis of polycyclic aromatic hydrocarbons, polychlorinated biphenyls, polybrominated diphenyl ethers and their metabolites. Anal. Chim. Acta 2018. [Google Scholar] [CrossRef]
- Gabelica, V.; Marklund, E. Fundamentals of ion mobility spectrometry. Curr. Opin. Chem. Biol. 2018, 42, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Wyttenbach, T.; Pierson, N.A.; Clemmer, D.E.; Bowers, M.T. Ion mobility analysis of molecular dynamics. Annu. Rev. Phys. Chem. 2014, 65, 175–196. [Google Scholar] [CrossRef] [PubMed]
- Warnke, S.; Seo, J.; Boschmans, J.; Sobott, F.; Scrivens, J.H.; Bleiholder, C.; Bowers, M.T.; Gewinner, S.; Schöllkopf, W.; Pagel, K. Protomers of benzocaine: Solvent and permittivity dependence. J. Am. Chem. Soc. 2015, 137, 4236–4242. [Google Scholar] [CrossRef] [PubMed]
- Lapthorn, C.; Dines, T.J.; Chowdhry, B.Z.; Perkins, G.L.; Pullen, F.S. Can ion mobility mass spectrometry and density functional theory help elucidate protonation sites in'small' molecules? Rapid Commun. Mass Spectrom. 2013, 27, 2399–2410. [Google Scholar] [CrossRef] [PubMed]
- Boschmans, J.; Jacobs, S.; Williams, J.P.; Palmer, M.; Richardson, K.; Giles, K.; Lapthorn, C.; Herrebout, W.A.; Lemière, F.; Sobott, F. Combining density functional theory (DFT) and collision cross-section (CCS) calculations to analyze the gas-phase behaviour of small molecules and their protonation site isomers. Analyst 2016, 141, 4044–4054. [Google Scholar] [CrossRef] [PubMed]
- Stow, S.M.; Causon, T.J.; Zheng, X.; Kurulugama, R.T.; Mairinger, T.; May, J.C.; Rennie, E.E.; Baker, E.S.; Smith, R.D.; McLean, J.A. An interlaboratory evaluation of drift tube ion mobility–mass spectrometry collision cross section measurements. Anal. Chem. 2017, 89, 9048–9055. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Xiong, X.; Zhu, Z.-J. MetCCS predictor: A web server for predicting collision cross-section values of metabolites in ion mobility-mass spectrometry based metabolomics. Bioinformatics 2017, 33, 2235–2237. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, Y.M.; Hamid, A.M.; Cox, J.T.; Garimella, S.V.; Smith, R.D. Ion Elevators and Escalators in Multilevel Structures for Lossless Ion Manipulations. Anal. Chem. 2017, 89, 1972–1977. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, Y.M.; Hamid, A.M.; Deng, L.; Garimella, S.V.; Webb, I.K.; Baker, E.S.; Smith, R.D. New frontiers for mass spectrometry based upon structures for lossless ion manipulations. Analyst 2017, 142, 1010–1021. [Google Scholar] [CrossRef] [PubMed]
- Weber, R.J.M.; Lawson, T.N.; Salek, R.M.; Ebbels, T.M.D.; Glen, R.C.; Goodacre, R.; Griffin, J.L.; Haug, K.; Koulman, A.; Moreno, P.; et al. Computational tools and workflows in metabolomics: An international survey highlights the opportunity for harmonisation through Galaxy. Metabolomics 2017, 13, 12. [Google Scholar] [CrossRef] [PubMed]
- Allard, P.-M.; Péresse, T.; Bisson, J.; Gindro, K.; Marcourt, L.; Pham, V.C.; Roussi, F.; Litaudon, M.; Wolfender, J.-L. Integration of molecular networking and in-silico MS/MS fragmentation for natural products dereplication. Anal. Chem. 2016, 88, 3317–3323. [Google Scholar] [CrossRef] [PubMed]
- Allard, P.-M.; Genta-Jouve, G.; Wolfender, J.-L. Deep metabolome annotation in natural products research: Towards a virtuous cycle in metabolite identification. Curr. Opin. Chem. Biol. 2017, 36, 40–49. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, A.G.; Godzien, J.; López, M.F.; Rupérez, F.J.; Barbas, C.; Otero, A. Knowledge-based metabolite annotation tool: CEU Mass Mediator. J. Pharm. Biomed. Anal. 2018, 154, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Broeckling, C.D.; Ganna, A.; Layer, M.; Brown, K.; Sutton, B.; Ingelsson, E.; Peers, G.; Prenni, J.E. Enabling Efficient and Confident Annotation of LC−MS Metabolomics Data through MS1 Spectrum and Time Prediction. Anal. Chem. 2016, 88, 9226–9234. [Google Scholar] [CrossRef] [PubMed]
- Uppal, K.; Walker, D.I.; Jones, D.P. xMSannotator: An R package for network-based annotation of high-resolution metabolomics data. Anal. Chem. 2017, 89, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Van Der Hooft, J.J.J.; Wandy, J.; Barrett, M.P.; Burgess, K.E.; Rogers, S. Topic modeling for untargeted substructure exploration in metabolomics. Proc. Natl. Acad. Sci. USA 2016, 113, 13738–13743. [Google Scholar] [CrossRef] [PubMed]
- Naz, S.; Gallart-Ayala, H.; Reinke, S.N.; Mathon, C.; Blankley, R.; Chaleckis, R.; Wheelock, C.E. Development of a Liquid Chromatography–High Resolution Mass Spectrometry Metabolomics Method with High Specificity for Metabolite Identification Using All Ion Fragmentation Acquisition. Anal. Chem. 2017, 89, 7933–7942. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Müller, E.; Schymanski, E.L.; Ruttkies, C.; Schulze, T.; Brack, W.; Krauss, M. Performance of combined fragmentation and retention prediction for the identification of organic micropollutants by LC-HRMS. Anal. Bioanal. Chem. 2018, 410, 1931–1941. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, C.; Méret, M.; Schmitt, C.A.; Lisec, J. Compound annotation in liquid chromatography/high-resolution mass spectrometry based metabolomics: Robust adduct ion determination as a prerequisite to structure prediction in electrospray ionization mass spectra. Rapid Commun. Mass Spectrom. 2017, 31, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Zani, C.L.; Carroll, A.R. Database for rapid dereplication of known natural products using data from MS and fast NMR experiments. J. Nat. Prod. 2017, 80, 1758–1766. [Google Scholar] [CrossRef] [PubMed]
- De Vijlder, T.; Valkenborg, D.; Lemière, F.; Romijn, E.P.; Laukens, K.; Cuyckens, F. A tutorial in small molecule identification via electrospray ionization-mass spectrometry: The practical art of structural elucidation. Mass Spectrom. Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Werner, E.; Heilier, J.-F.; Ducruix, C.; Ezan, E.; Junot, C.; Tabet, J.-C. Mass spectrometry for the identification of the discriminating signals from metabolomics: Current status and future trends. J. Chromatogr. B 2008, 871, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, B.L.; Mahapatra, S.; Farmer, D.K.; McNeil, M.R.; Casero, R.A., Jr.; Belisle, J.T. Elucidating the Structure of N1-Acetylisoputreanine: A Novel Polyamine Catabolite in Human Urine. ACS Omega 2017, 2, 3921–3930. [Google Scholar] [CrossRef] [PubMed]
- Cajka, T.; Garay, L.A.; Sitepu, I.R.; Boundy-Mills, K.L.; Fiehn, O. Multiplatform mass spectrometry-based approach identifies extracellular glycolipids of the yeast Rhodotorula babjevae UCDFST 04-877. J. Nat. Prod. 2016, 79, 2580–2589. [Google Scholar] [CrossRef] [PubMed]
- Nikolić, D. CASMI 2016: A manual approach for dereplication of natural products using tandem mass spectrometry. Phytochem. Lett. 2017, 21, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Ruttkies, C.; Gerlich, M.; Neumann, S. Tackling CASMI 2012: Solutions from MetFrag and MetFusion. Metabolites 2013, 3, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, T.; Kasama, T.; Kinumi, T.; Makabe, H.; Matsuda, F.; Miura, D.; Miyashita, M.; Nakamura, T.; Tanaka, K.; Yamamoto, A. Winners of CASMI2013: Automated tools and challenge data. Mass Spectrom. 2014, 3, S0039. [Google Scholar] [CrossRef] [PubMed]
- Newsome, A.G.; Nikolic, D. CASMI 2013: Identification of small molecules by tandem mass spectrometry combined with database and literature mining. Mass Spectrom. 2014, 3, S0034. [Google Scholar] [CrossRef] [PubMed]
- Dührkop, K.; Scheubert, K.; Böcker, S. Molecular formula identification with SIRIUS. Metabolites 2013, 3, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Zamboni, N.; Heinonen, M.; Rousu, J. Metabolite identification through machine learning—Tackling casmi challenge using FingerID. Metabolites 2013, 3, 484–505. [Google Scholar] [CrossRef] [PubMed]
- Gewin, V. Data sharing: An open mind on open data. Nature 2016, 529, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Spicer, R.A.; Steinbeck, C. A lost opportunity for science: Journals promote data sharing in metabolomics but do not enforce it. Metabolomics 2018, 14, 16. [Google Scholar] [CrossRef] [PubMed]
- Rübel, O.; Greiner, A.; Cholia, S.; Louie, K.; Bethel, E.W.; Northen, T.R.; Bowen, B.P. OpenMSI: A high-performance web-based platform for mass spectrometry imaging. Anal. Chem. 2013, 85, 10354–10361. [Google Scholar] [CrossRef] [PubMed]
- Djoumbou Feunang, Y.; Eisner, R.; Knox, C.; Chepelev, L.; Hastings, J.; Owen, G.; Fahy, E.; Steinbeck, C.; Subramanian, S.; Bolton, E.; et al. ClassyFire: Automated chemical classification with a comprehensive, computable taxonomy. J. Cheminform. 2016, 8, 61. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Confidence Level | Description | Minimum Data Requirements |
|---|---|---|
| Level 0 | Unambigous 3D structure: Isolated, pure compound, including full stereochemistry | Following natural product guidelines, determination of 3D structure |
| Level 1 | Confident 2D structure: uses reference standard match or full 2D structure elucidation | At least two orthogonal techniques defining 2D structure confidently, such as MS/MS and RT or CCS |
| Level 2 | Probable structure: matched to literature data or databases by diagnostic evidence | At least two orthogonal pieces of information, including evidence that excludes all other candidates |
| Level 3 | Possible structure or class: Most likely structure, isomers possible, substance class or substructure match | One or several candidates possible, requires at least one piece of information supporting the proposed candidate |
| Level 4 | Unkown feature of insterest: | Presence in sample |
| Database | Targets | Description |
|---|---|---|
| PubChem [32] | All small molecules | Small molecules, metadata |
| ChemSpider [33] | All small molecules | Small molecules, curated data |
| KEGG [34] | Metabolites | Pathway database, multiple species |
| MetaCyc [35] | Metabolites | Pathway database, multiple species |
| BRENDA [36] | Enzymes | Enzyme and metabolism data |
| HMDB [37] | Metabolites | Human metabolites |
| CHEBI [38] | Small molecules | Molecules of biological interest |
| UNPD [39] | Metabolites | Secondary plant metabolites |
| MINE [40] | Metabolites | In silico predicted metabolites |
| Database | Targets | Description |
|---|---|---|
| NIST | EI-MS, CID-MS/MS | Curated DB, graphical interface |
| WILEY | EI-MS, CID-MS/MS | Largest collection of EI-MS data |
| METLIN [51] | CID-MS/MS | Developed for QTOF instruments |
| MoNA | EI, MS/MS, MSn | Autocurated collection of spectra |
| MassBank [52] | EI, MS/MS, MSn | Longest standing community database |
| mzCloud [53] | MSn | Multiple stage MSn |
| GNPS [54] | MS/MS | Community database |
| ReSpect [55] | MS/MS, RT | Plant metabolomics database |
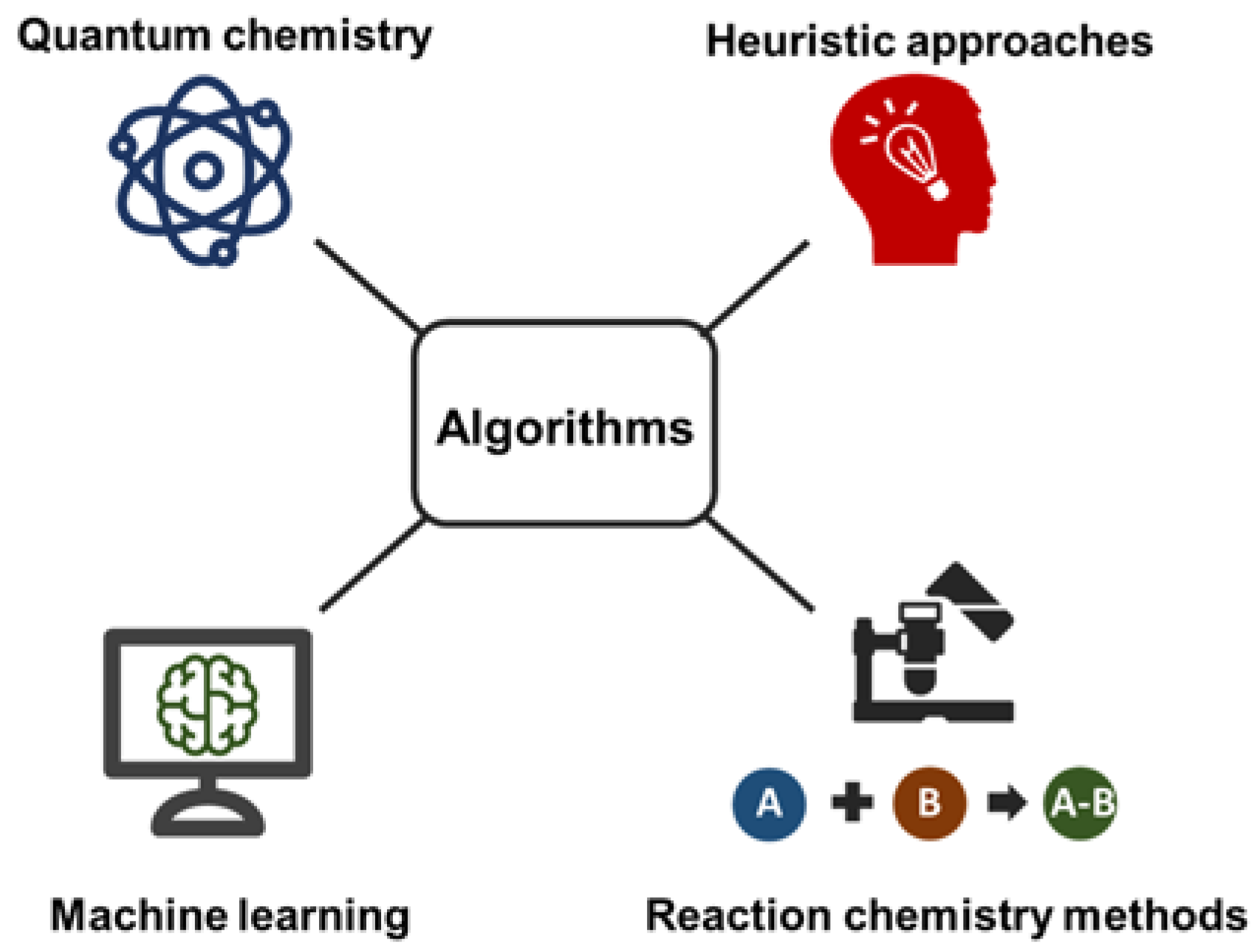
| In Silico Method | Software | Platform | Description |
|---|---|---|---|
| Quantum chemistry | QCEIMS | EI-MS | Uses chemistry first principles; requires cluster computations |
| Machine learning | CFM-ID/CSI:FingerID | EI-MS CID-MS/MS | Requires diverse training sets; Fast method |
| Heuristic approaches | LipidBlast | CID-MS/MS | for specific compound classes (lipids); Fast method |
| Reaction chemistry methods | MassFrontier | EI-MSCID-MS/MS | generates only bar code spectra; Covers experimental gas phase reactions |
| Tools | Fragmentation Method | Compound DB | Type of Interfacce |
|---|---|---|---|
| MS-FINDER | Rule-based (hydrogen rearrangement rules) | 15 integrated target DBs plus MINE and PubChem | Windows GUI |
| CFM-ID | Hybrid rule-based machine learning | KEGG, HMDB | Web application and command line tool |
| MetFrag | Hybrid rule-based combinatorial | HMDB, KEGG, PubChem | Web application, command line tool, |
| Mass Frontier | Rule-based (literature reaction mechanisms) | Internal MS database | Windows GUI |
| ChemDistiller | Fingerprint and spectral machine learning | 17 different target databases, 130 Mio compounds total | Command line, web-based output |
| MAGMa, MAGMa+ | Rule-based | PubChem, KEGG, HMDB | Web application, command line tool |
| CSI:FingerID | Combination of fragmentation trees and machine learning | PubChem and multiple bio databases | Platform independent GUI, command line tool |
| Data Sharing | Link | Description |
|---|---|---|
| GitHub | github.com | Software development platform |
| BitBucket | bitbucket.org | Collaborative software sharing |
| SourceForge | sourceforge.net | Collaborative software sharing |
| Zenodo | zenodo.org | Open research data repository |
| Figshare | figshare.com | Online research data repository |
| Metabolomics Workbench | metabolomicsworkbench.org | Experimental metabolomics data |
| MetaboLights | ebi.ac.uk/metabolights | European metabolomics repository |
| OpenMSI | openmsi.nersc.gov | Mass spectral imaging data |
| MetaSpace | metaspace2020.eu | Mass spectral imaging data |
| GNPS | gnps.ucsd.edu | Mass spectral data sharing |
| MassBank | massbank.jp | Mass spectral data sharing |
| MoNA | massbank.us | Mass spectral sharing community |
| Norman MassBank | massbank.eu | Mass spectral data sharing |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blaženović, I.; Kind, T.; Ji, J.; Fiehn, O. Software Tools and Approaches for Compound Identification of LC-MS/MS Data in Metabolomics. Metabolites 2018, 8, 31. https://doi.org/10.3390/metabo8020031
Blaženović I, Kind T, Ji J, Fiehn O. Software Tools and Approaches for Compound Identification of LC-MS/MS Data in Metabolomics. Metabolites. 2018; 8(2):31. https://doi.org/10.3390/metabo8020031
Chicago/Turabian StyleBlaženović, Ivana, Tobias Kind, Jian Ji, and Oliver Fiehn. 2018. "Software Tools and Approaches for Compound Identification of LC-MS/MS Data in Metabolomics" Metabolites 8, no. 2: 31. https://doi.org/10.3390/metabo8020031
APA StyleBlaženović, I., Kind, T., Ji, J., & Fiehn, O. (2018). Software Tools and Approaches for Compound Identification of LC-MS/MS Data in Metabolomics. Metabolites, 8(2), 31. https://doi.org/10.3390/metabo8020031