1. Introduction
Lipids are a structurally diverse group of naturally occurring amphipathic molecules that include fatty acids, glycerides, phospholipids, sphingolipids, sterols, prenols, and waxes. They are principal components of cellular membranes, store energy in the form of triacylglycerides, and function as signaling molecules. Ketoacyl and isoprene building blocks form lipid backbones, which can be coupled to sugars, amines, and other small molecules. For example, the glycerophospholipid phosphatidylinositol contains two esterified fatty acids at the glycerol
sn-1 and
sn-2 positions and an inositol sugar head group at
sn-3. 20-carbon (C20) polyunsaturated fatty acids (PUFAs) are liberated from
sn-2 by phospholipase A2 and oxidized into prostaglandins and other eicosanoids [
1,
2]. Prostaglandin-endoperoxide synthase (a.k.a. cyclooxygenase or Cox) catalyzes cyclopentane ring formation (
Figure 1A). Through enzymatic cascades, lipids are converted from one structural form to another, comprising pathways for modifying cell structure and function.
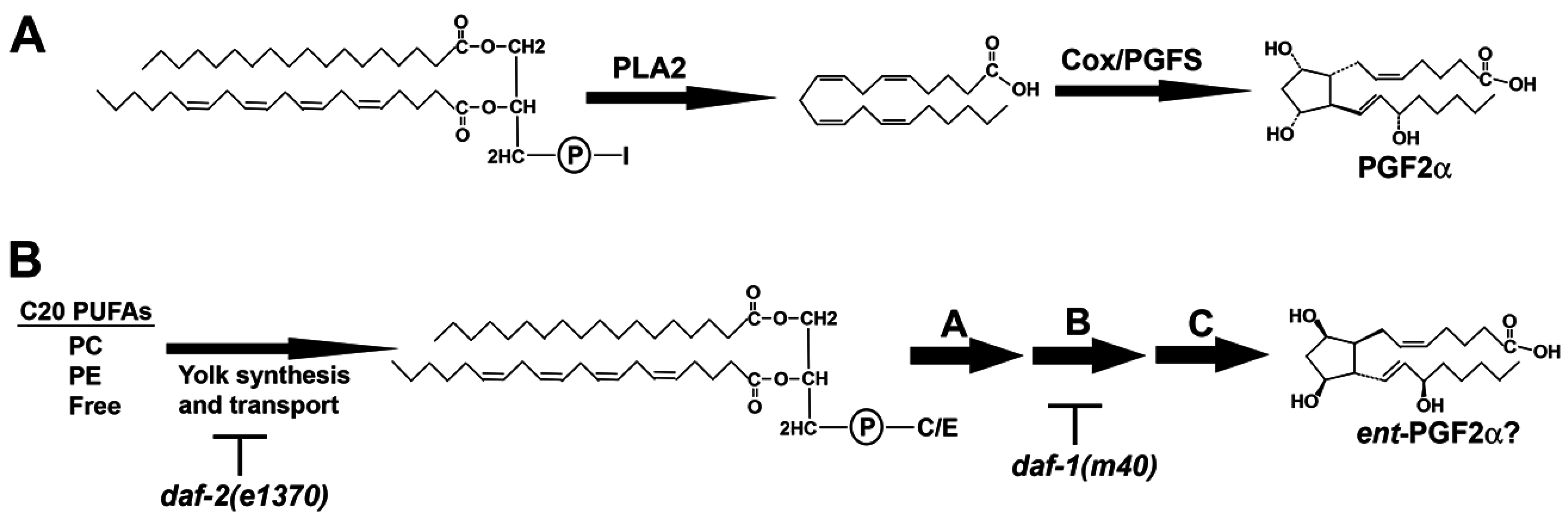
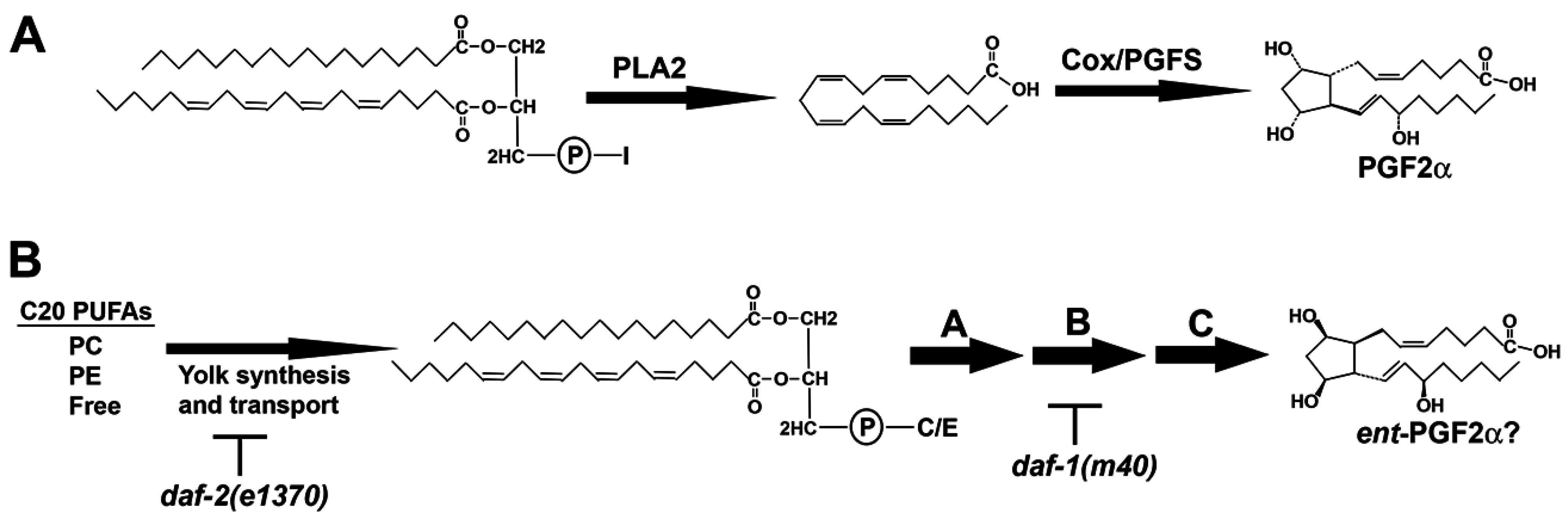
Figure 1.
Prostaglandin biosynthesis pathways. (A) In the Cox pathway, phospholipase A2 cleaves arachidonic acid (AA) from the sn-2 position of phospholipids like PI, generating lysoPI species lacking AA. PGF2α is synthesized from free AA by the sequential actions of Cox and prostaglandin F synthase (PGFS); (B) In C. elegans, C20 PUFAs are delivered to oocytes in yolk lipoprotein complexes. Yolk PUFAs are primarily found esterified to PC and PE, and in free forms. An incompletely understood metabolic pathway in oocytes, shown arbitrarily as steps A, B, and C, converts yolk lipids into F-series prostaglandins, such as ent-PGF2α (or a co-eluting stereoisomer). I, inositol; C, choline; E, ethanolamine; PC, phosphatidylcholine; PE, phosphatidylethanolamine; C20, 20-carbon.
The lipidome is the complete lipid composition of a tissue or organism, which can contain well over 10,000 different species. As a systems biology approach, lipidomics aims to determine and quantify this composition. Mass spectrometry is a widely used analytical platform for lipidomics because of its sensitivity, specificity, and accuracy [
3,
4,
5]. In contrast to targeted approaches that measure a set of predetermined lipids, untargeted lipidomics seeks to extend coverage to all detectable lipid species. This untargeted approach is greatly strengthened by high-resolution mass spectrometers that help discriminate among isobaric compounds, which have the same nominal mass, but different molecular formula. The “top-down” method is solely reliant on accurate mass detection of the parent lipid. It is unbiased toward lipid class, but biased toward the most abundant species. On the other hand, the “bottom-up” method relies on detection of structure-specific breakdown products using tandem mass spectrometry (MS/MS). It is biased toward lipid class, but capable of detecting species with low abundance. A major disadvantage of MS/MS is the targeted nature: one must anticipate which lipid classes are best candidates
a priori.
MS/MS with data-independent Sequential Window Acquisition of all Theoretical (SWATH) mass spectra provides a powerful combination of top-down and bottom-up methods [
6,
7]. Data-independent SWATH collects MS/MS spectra in 1
m/
z windows across the entire mass range simultaneously. SWATH using the AB SCIEX 5600 triple time-of-flight (TOF) mass spectrometer creates a comprehensive high mass resolution lipid library. This technology offers several strengths for comparative lipidomics between wild-type and mutant animals. Major strengths include: (1) it does not require
a priori assumptions; (2) library data are quantitative (relative); and (3) high mass resolution and MS/MS enable identification of many lipids. A particularly important feature is that the data can be searched retrospectively, eliminating the need to repeat sample preparation and analyses every time there is a new lipid of interest. For example, SWATH can be used to generate lipid libraries for rare or difficult to collect wild-type or mutant animal tissues. Researchers can then search the libraries using objective criteria, relying on software-based lipid annotations, or targeted approaches using neutral loss or precursor ion scans. The libraries provide a data-mining resource to generate testable models for biological discovery.
The nematode model
C. elegans is well suited for comprehensive lipidomics, although most studies have relied on data-dependent, targeted strategies [
8,
9]. Genetic screens and genome-editing technologies can be used to identify lipid-modifying enzymes or gene products impacting lipid metabolism [
10,
11,
12]. The DAF-1 type I TGF-β and DAF-2 insulin receptors transmit endocrine signals that promote reproduction and modulate lifespan [
13,
14]. Lipids appear to be central to multiple
daf-1 and
daf-2 functions, although the mechanisms are not well understood. Genetic loss of either signaling pathway causes altered transcription of lipid metabolism genes, triacylglyceride (TAG) accumulation, and ovarian prostaglandin deficiency [
15,
16,
17,
18,
19]. In the adult ovary, oocytes secrete multiple F-series prostaglandins that stimulate sperm motility (
Figure 1B) [
20,
21]. DAF-1 and DAF-2 promote prostaglandin synthesis through a metabolic pathway lacking Cox enzymes [
15,
16]. Liquid chromatography electrospray ionization tandem mass spectrometry (LC-MS/MS) data from mouse and zebrafish tissues, including
Cox-1;
Cox-2 double knockout mice provide evidence that this pathway is conserved [
16,
20]. However, the biochemical steps between arachidonic acid (AA) and PGF2 formation are not well understood.
Here we use MS/MSALL with SWATH analysis in C. elegans to evaluate the utility of comprehensive, searchable lipid libraries as a biological discovery tool. We provide selected examples for validating and searching the libraries. Disrupting daf-1 and daf-2 signaling causes specific alterations in numerous lipids, including TAG, phosphatidylcholine (PC), and phosphatidylinositol (PI) species. These lipids are potential downstream effectors and candidate markers to assess signaling activity. To investigate an incompletely understood metabolic pathway, we searched the libraries focusing on prostaglandin precursors. SWATH data raise the unexpected possibility that Cox-independent prostaglandins may be synthesized from lysophosphatidylcholine (LPC) intermediates. SWATH combined with classical genetics provides a powerful, untargeted approach to study lipids and generate novel hypotheses.
2. Results
A data-independent shotgun lipidomics workflow with SWATH acquisition was developed to perform comprehensive lipid analysis of wild-type and mutant
C. elegans. This mass spectrometry technique generated an archive of all detectable precursor ions and their MS/MS fragment ions in samples within a 6 min run time. The MS and MS/MS spectra generated by the AB SCIEX 5600 triple-TOFMS instrument are high resolution (>15,000–30,000) and high mass accuracy (<5 ppm). Neutral lipids were extracted from staged one-day adult wild-type,
daf-1(m40), and
daf-2(e1370) hermaphrodite worms shifted from 16 °C to 25 °C for 24 h. The
daf-2(e1370) temperature-sensitive mutation causes reduced function at 25 °C [
22]. Lipids were extracted from three 500 mg frozen worm pellets per genotype. Directly infused samples were acquired in positive and negative ion modes. During SWATH acquisition, precursor ion isolation windows of 1 Da width selected in Q1 are fragmented in the Q2 collision cell and the generated product ions are monitored at high resolution by TOF. The SWATH data include a 250 ms survey scan of TOFMS from
m/
z 200–1200, followed by MS/MS of 1000 ion windows. Four independent injections were conducted for each genotype, comprising 12 data libraries.
Internal standards were not included in this study due to several complicating factors (see Discussion). The goal of this method is to compare lipid species across genotypes. Samples were analyzed both in positive and negative ion mode with appropriate blanks between samples. There was no significant carry-over from samples. To assess the influence of sample preparation and the analytical device on variability, each sample was analyzed in biological and technical replicates. The reproducibility of the method was evaluated by monitoring responses of technical replicates of extracted worm samples. The intensities of ions between replicates were very similar (
Supplemental Figure S1) with average coefficient of variation (CV) 5.67% in positive ion mode and 3.54% in negative ion mode. We conclude that the replicates are highly reproducible.
LipidView™ software 1.2 was used to search precursor- and fragment-ion masses against a lipid fragment database containing over 25,000 entries. To classify lipid species based on these database comparisons, the term annotated is used because absolute identification requires further analysis. Depending on structure, some lipids preferentially ionize in positive ion mode, whereas others preferentially ionize in negative ion mode. LipidView annotated 2817 lipid species covering a variety of classes, including 580 lipids in positive ion mode and 2237 in negative ion mode. The total independent species may be less because some lipids are detected in both ion modes.
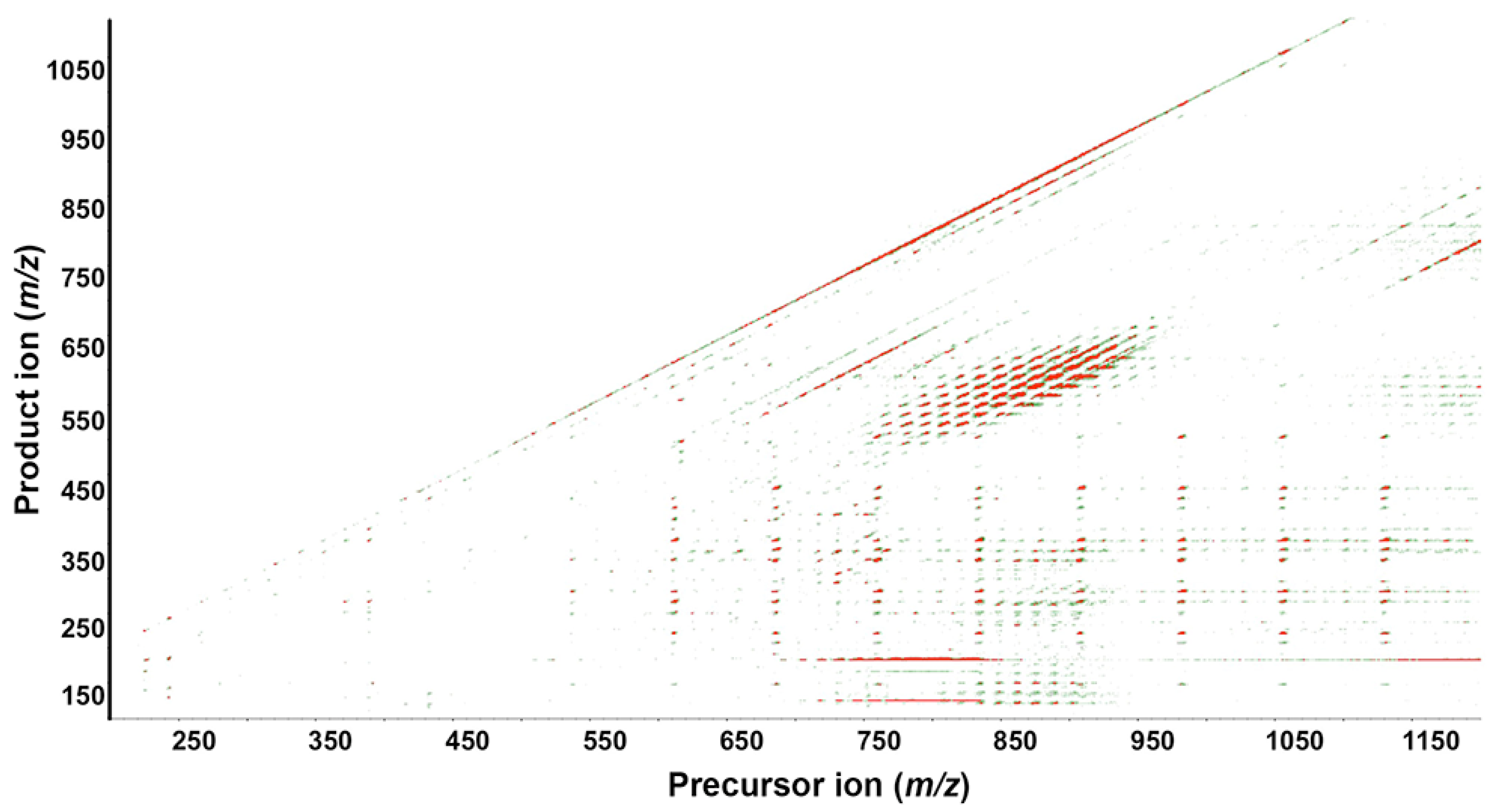
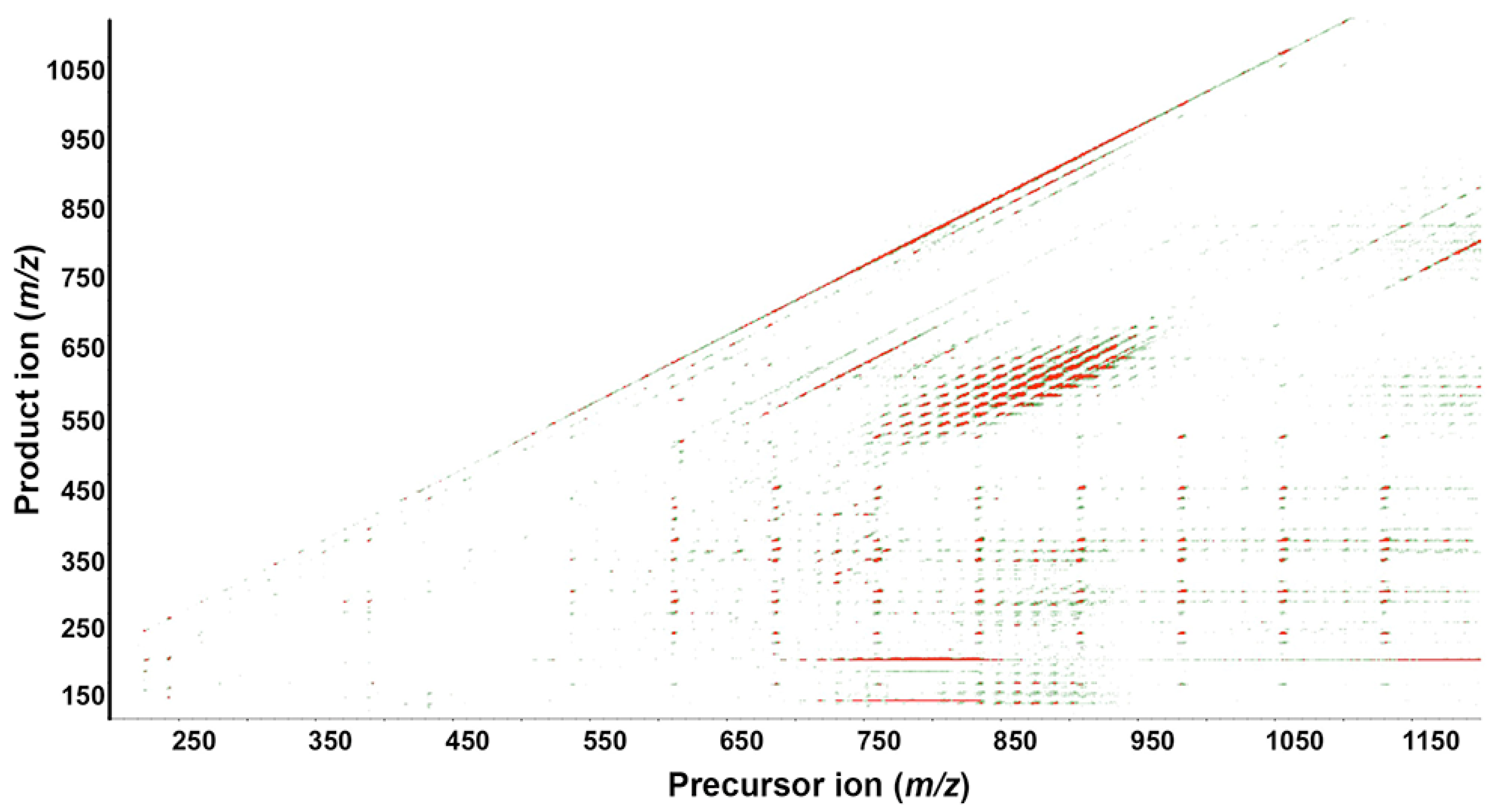
Figure 2 depicts a colored contour plot from a wild-type extract showing all precursor ions (
x-axis) and product ions (
y-axis) generated by SWATH acquisition in positive ion mode. It provides a snapshot of ion abundance. For example, the product ion
m/
z 184.0733 is a specific signature ion for phosphorylcholine moiety of glycerophospholipids and the precursor ion mass between
m/
z 700–850 shows strong ion intensity, as visualized by red color. Similarly, TAGs form a cluster of intense red peaks at precursor ion
m/
z 750–900 (
x-axis) and product ion
m/
z 450–600 (
y-axis).
C. elegans synthesizes odd chain fatty acids [
23,
24]. In positive ion mode, a cluster of TAG ions between
m/
z 800–900 containing an odd chain fatty acyl chain (e.g., C17:0 or C19:0) was observed. Phosphatidylserine (PS), PI, and phosphatidic acid (PA) molecular species containing odd chain fatty acids were also observed in negative ion mode (see below).
Figure 2.
Contour plot of a wild-type extract acquired in positive ion mode. The fragmentation map shows precursor ions with mass/charge (m/z) 200–1200 on the x-axis and product ions with m/z 100–1100 on the y-axis. Each dot represents an individual lipid species, with color from green to red indicating relative increasing abundance (3rd dimension).
Over 2500 ions (in positive and negative ion modes) were not annotated, perhaps reflecting software limitations, novel lipid species, or contaminating metabolites. The direct infusion method was unable to discriminate among isomers, including lipids with monomethyl branched chain fatty acids [
23]. The lipids annotated by LipidView were imported into MarkerView™ 1.21 software, where trends could be visualized across replicates and groups. PeakView™ 1.2 software was used for targeted neutral loss and precursor ion scans, as well as viewing MS/MS spectra.
2.1. Characterization of Selected Lipids
We first sought to test whether SWATH could be used to discriminate among isobaric lipids. A prior targeted study using a data-dependent acquisition method characterized several TAG species in
C. elegans extracts [
9]. The most abundant TAGs contained multiple isobaric species with odd chain fatty acids. For example, TAG 51:3 was predominantly TAG 17:1/17:1/17:1, but also contained several other TAG species [
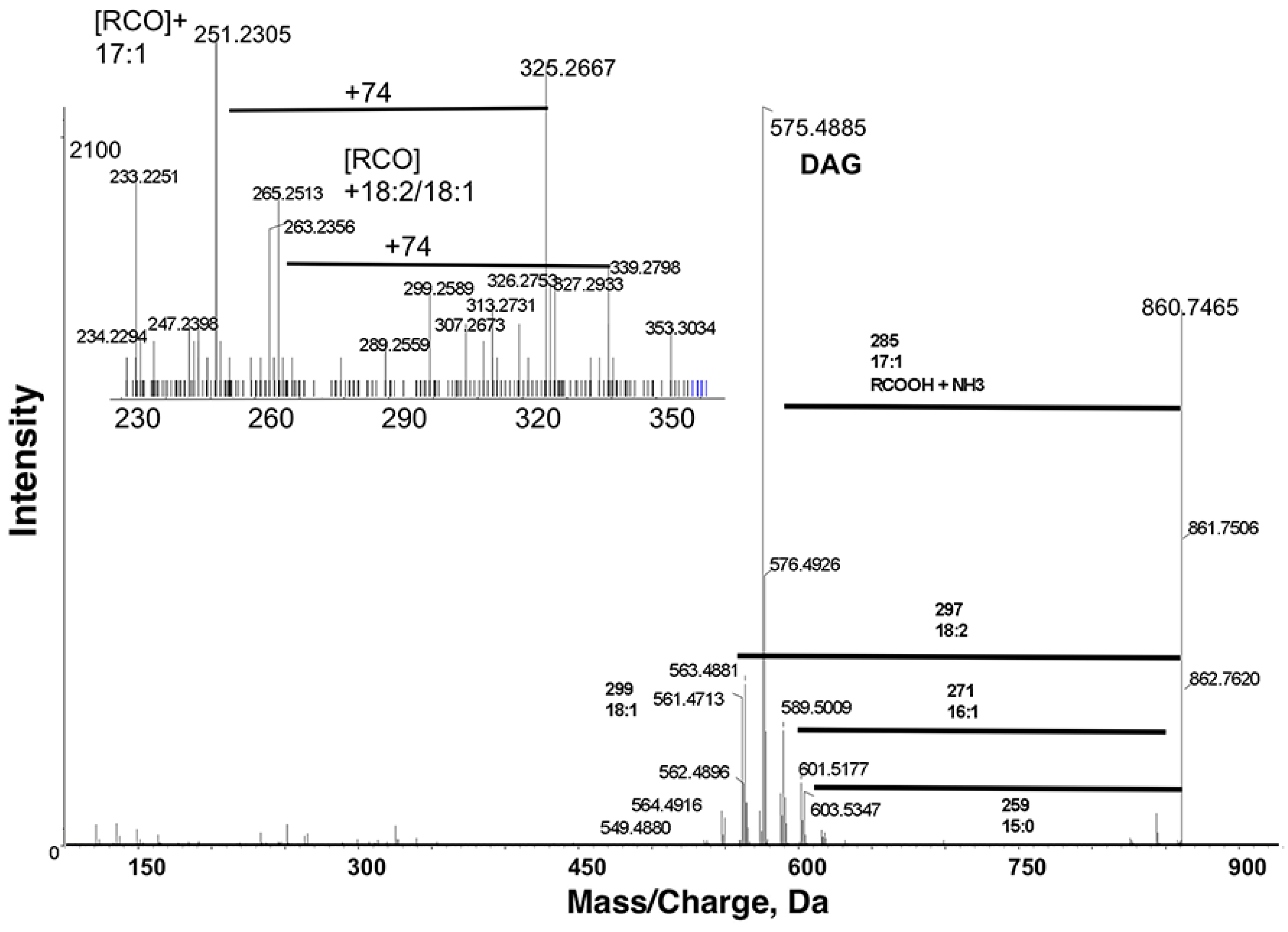
9]. According to LipidView annotations, the most abundant TAGs in our extracts contain odd chain fatty acids. The prominent ion
m/
z 860.7465 in positive ion mode was annotated as TAG 51:3 + NH
4. The ammonium adduct of TAG
m/
z 860.7465 [M+NH
4]
+ fragmented into a number of diacyl product ions, with the most intense product ion
m/
z 575.4885 after neutral losses of ammonia (17 Da) and the fatty acid 17:1 (
Figure 3). The other product ions
m/
z 547.4559, 549.4880, 561.4713, 563.4881, 589.5009 and 601.5177 corresponded to losses of 19:1, 19:0, 18:2, 18:1, 16:1 and 15:0, respectively, likely due to less abundant isobaric TAG species. TAG molecular species can be proposed based on fatty acyl group neutral loss in MS/MS of
m/
z 860.7465. Since the intensity of diacyl product ions due to loss of 17:1 is the highest, this lipid was tentatively identified as TAG 17:1/17:1/17:1. Therefore, SWATH can successfully discriminate among isobaric TAGs in
C. elegans extracts.
Figure 3.
Product ion spectra obtained from MS/MS of m/z 860.7465 in positive ion mode. m/z 860.7465 [M+NH4]+ fragmented into a number of diacyl product ions, with the most intense product ion at m/z 575.4885 after neutral losses of ammonia (17 Da) and the fatty acid 17:1. The other less abundant product ions correspond to losses of 19:1, 19:0, 18:2 (shown), 18:1, 16:1 (shown), and 15:0 (shown). These spectra indicate that m/z 860.7465 corresponds to triacylglyceride (TAG) 51:3 + NH4.
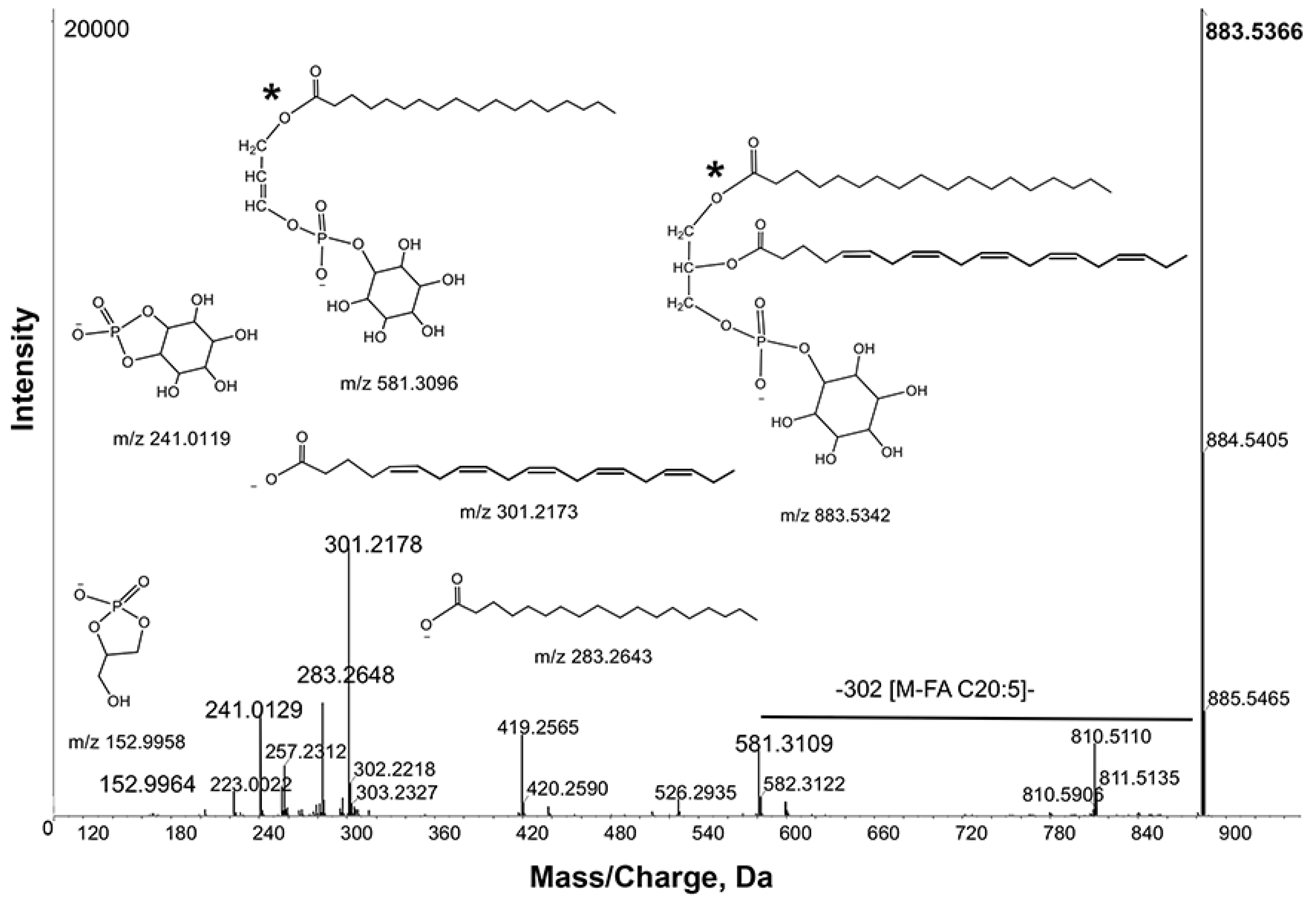
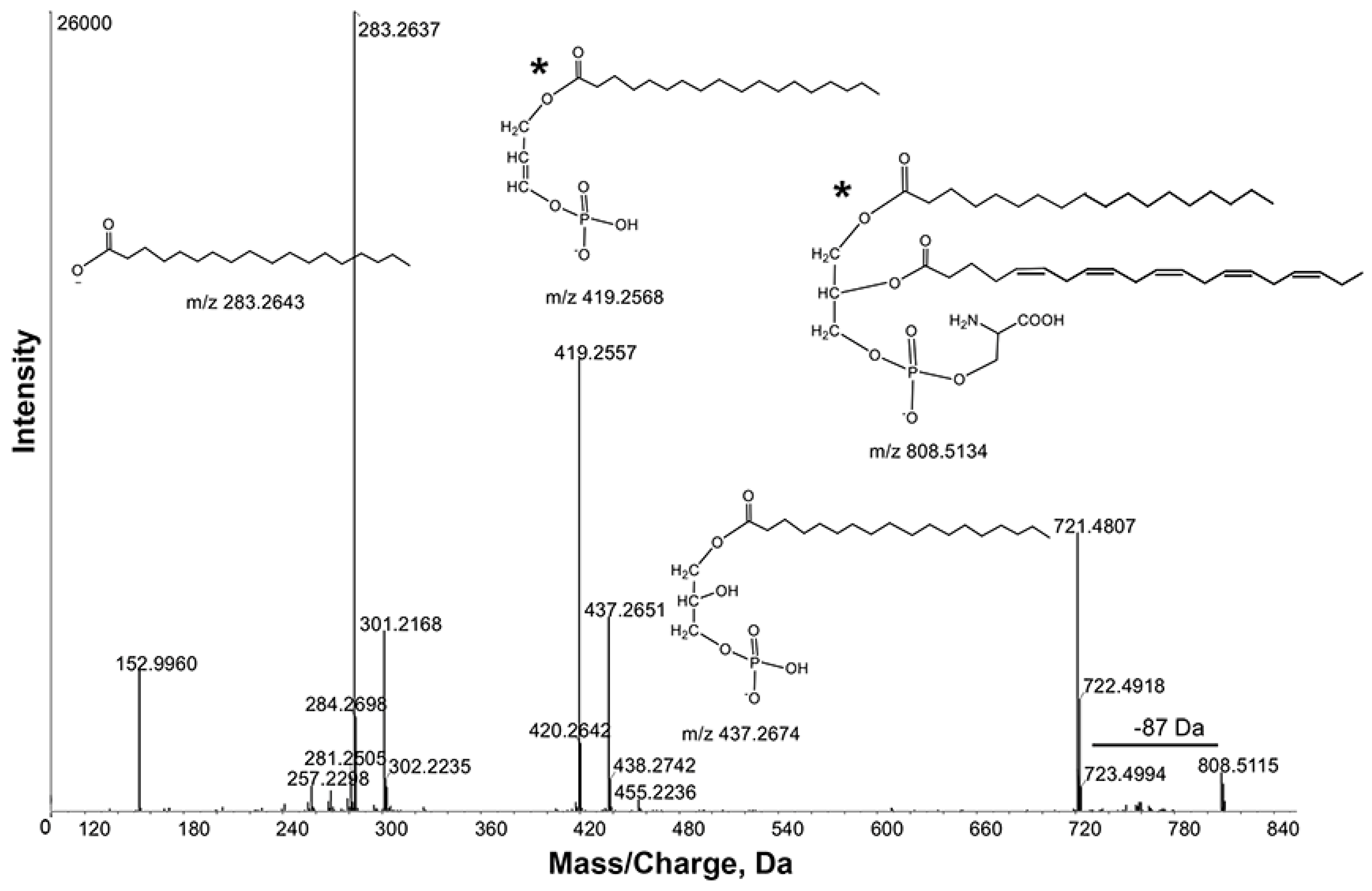
Schwudke
et al., also characterized PS and PC species using respective neutral loss and precursor ion scans in positive ion mode [
9]. We were able to characterize these lipids in both positive and negative ion modes. In positive ion mode, MS/MS of PS precursor ion [M+H]
+ produces a product ion [M+H-185]
+, resulting from loss of the phosphoserine moiety. In negative ion mode, the presence of the [M−H-87]
− product ion is characteristic of PS. The negative ion spectra are easier to interpret because they lack sodium adducts. Neutral loss scan Δ
m/z 87.08 showed abundant PS species in the mass range
m/
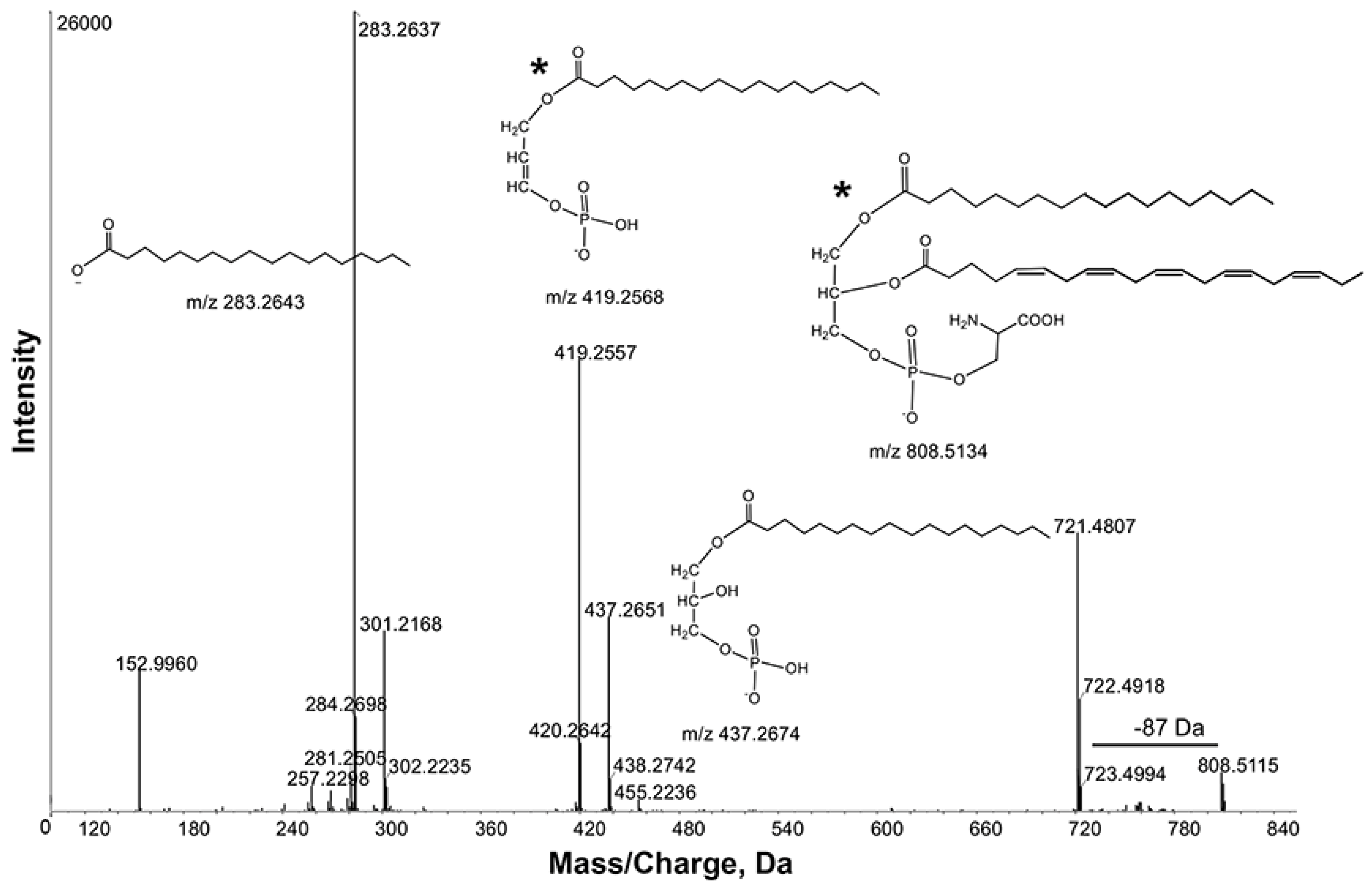
z 700–900. The intense ion
m/
z 808.5115 contained product ions
m/
z 721.4807, 437.2651 and 419.2557 due to losses of [M-88], [M-88-301] and [M-88-283], respectively (
Figure 4). These observations were further supported by product ions at
m/
z 301.2168 and 283.2637, corresponding to C20:5 and 18:0 fatty acyl chains. Thus, the ion
m/
z 808.5115 was tentatively identified as PS 38:5. Consistent with this interpretation, Schwudke
et al., identified PS 38:5 as the most abundant PS in wild-type extracts [
9].
Figure 4.
Product ion spectra obtained from MS/MS of m/z 808.5115 in negative ion mode. Key product ion structures with corresponding m/z are shown. These spectra identified m/z 808.5115 as Phosphatidylserine (PS) 38:5 (20:5_18:0). *, the sn-1 and sn-2 fatty acid positions indicated in the structures are arbitrary and cannot be inferred from the data.
PCs generate an abundant product ion
m/
z 184.0733 in positive ion mode, corresponding to protonated phosphorylcholine. Precursor ion scan
m/
z 184.0733 detected a large cluster of PC species in the mass range
m/
z 700–850 (
Supplemental Figure S2), consistent with the targeted approach [
9]. In negative ion mode, PCs produce abundant [M-15]
− and [M+OAc]
− ions in the presence of ammonium acetate buffer [
9,
25]. We conclude that SWATH provides results comparable to targeted methods, yet with increased flexibility.
2.3. Delineating Novel Lipid Metabolism Pathways
Above we described untargeted and targeted analyses of SWATH libraries, focusing on selected lipid classes. The comparative method also provides an objective opportunity to investigate lipid metabolism pathways. SWATH might be used to identify candidate lipid intermediates based on empirical data from other sources. The Cox-independent prostaglandin pathway was used as an example.
daf-1 and
daf-2 are both required for Cox-independent prostaglandin synthesis, but act at different steps (
Figure 1B).
daf-2(e1370) disrupts prostaglandin precursor transport to oocytes [
15], whereas
daf-1(m40) inhibits a downstream biochemical step, likely through reduced expression of an enzyme [
16]. When enzyme activity is blocked, a strong increase in substrate abundance results. For example, knockout of the omega-3 desaturase enzyme
fat-1 causes a 12.8 fold increase in the substrate AA or 20:4 [
28]. Relative to the wild type,
daf-1 mutants are predicted to contain strongly increased levels of an unknown prostaglandin intermediate, which is reduced in
daf-2 mutants.
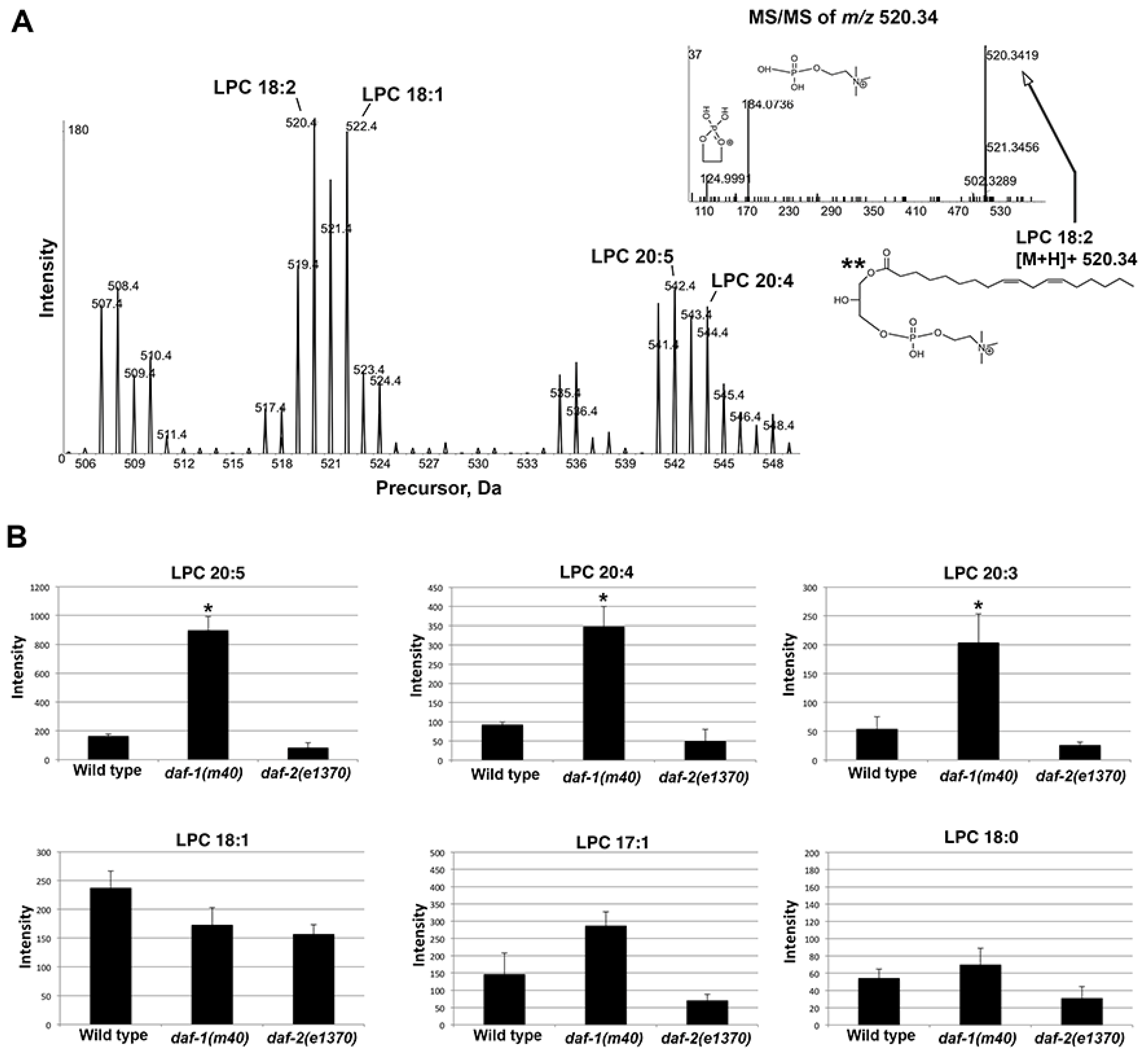
First, using the same staged nematode growth conditions as those used for SWATH, we confirmed that
daf-1(m40) hermaphrodites have reduced prostaglandin synthesis. Consistent with a prior study [
16], LC-MS/MS operated in multiple-reaction monitoring (MRM) mode showed strong reductions in F-series prostaglandins derived from DGLA (20:3), AA (20:4), and EPA (20:5) (
Supplemental Figure S7). Next, we used MarkerView to search the SWATH libraries for all lipids specifically altered in
daf-1 mutants
versus the other two strains. MarkerView identified LPC 20:5 and LPC 20:4 as two lipids with the most robust changes. PCA showed clear LPC separation among the genotypes (
Supplemental Figure S8). To validate these data, we used precursor ion scan
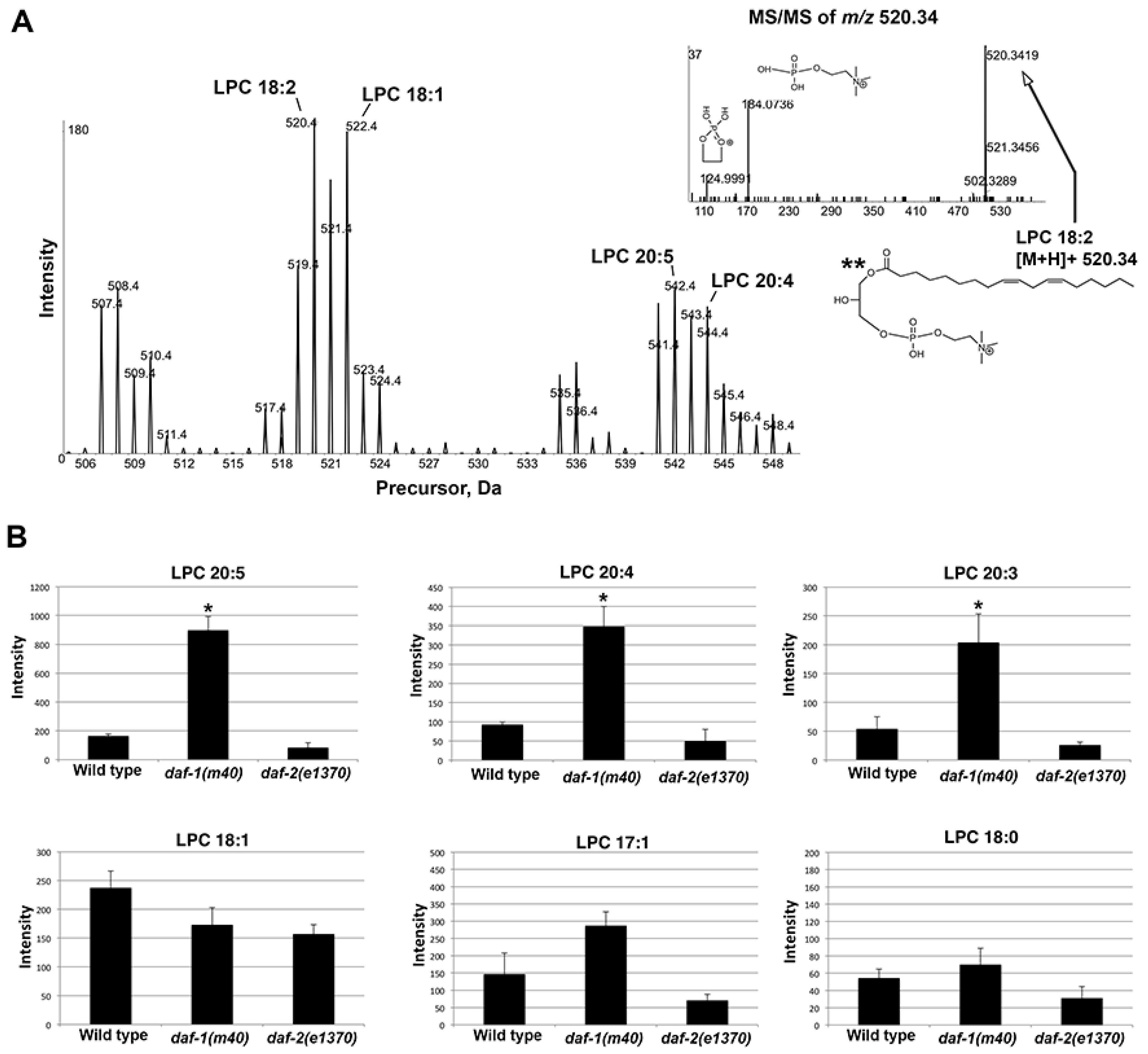
m/
z 184.0733 in positive ion mode. A series of ions was detected between
m/
z 505–550. MS/MS confirmed LPC identity (
Figure 7A). LPC 20:3, LPC 20:4, and LPC 20:5 increased by 3–6 fold in
daf-1 mutant
versus wild-type extracts (
Figure 7B). LPCs containing monounsaturated and saturated fatty acyl chains were much less affected, although saturated LPCs were close to the detection level (
Figure 7B). LPCs in
daf-2 mutants were reduced by approximately 50% compared to the wild type (
Figure 7B). We conclude that untargeted and targeted SWATH approaches identified LPCs containing prostaglandin precursors as specifically increased in
daf-1 mutants.
Prostaglandin precursors are transported in yolk lipoprotein complexes, which primarily contain C20 PUFAs in PC, PE, and free forms [
21]. Although MarkerView did not detect statistically significant changes in LPE and free PUFAs, we considered the possibility that low levels might confound LipidView assignments. First, we used PeakView to evaluate LPEs manually. Neutral loss scan
m/
z 141.00 conducted in positive ion mode identified LPE species between
m/
z 460–510 in
daf-1 mutants. Among the three groups, LPEs containing prostaglandin precursors showed the same trend as LPCs, except LPE levels were close to the detection limit. For quantification of free PUFAs, we developed an LC-MS/MS method operated in MRM mode. MRM analysis showed that free 20:4 and 20:5 levels were increased by 20.2% and 60.3%, respectively in
daf-1(m40) extracts compared to wild-type extracts (
Supplemental Figure S9). These data indicate that LipidView may miss less abundant or less ionizable lipids detectable using targeted approaches. In summary, comparative SWATH lipidomics identified LPCs and possibly LPEs containing PUFAs as strongly up-regulated in
daf-1 mutants. Other lipids containing prostaglandin precursors were much less affected (
i.e., free 20:4 and 20:5) or unaffected.
Figure 7.
LPC species in wild-type and mutant extracts. (A) Precursor ion scan m/z 184.0733 in positive ion mode detects a series of LPCs between m/z 505–550. MS/MS in the inset (upper right) shows LPC 18:2 product ion spectra with corresponding structures. (B) Selected LPCs quantified from precursor ion scan m/z 184.0733 in positive ion mode. C. elegans prostaglandins are synthesized from dihomo-gamma-linolenic acid (20:3), AA (20:4), and EPA (20:5).
*, p < 0.005. **, the sn-1 fatty acid position indicated in the structure is arbitrary (sn-1 vs. sn-2) and cannot be inferred from the data.
3. Discussion
A key feature of this study is the combined use of data-independent SWATH and genetic manipulation to investigate lipid biology. In
C. elegans, mutational or RNA-mediated interference screens can be designed to identify enzymes and other lipid regulators, based on phenotypes resulting from pathway dysfunction. For example, TGF-β pathway components were identified in screens for sperm motility defects, which occur when Cox-independent prostaglandin synthesis is disrupted [
15,
16]. CRISPR/Cas9 and other genome-editing technologies are available to modify or delete gene products of interest [
29,
30,
31]. The comparative SWATH approaches outlined here are well suited for discovery of lipid biomarkers, enzyme substrates, and metabolic pathways.
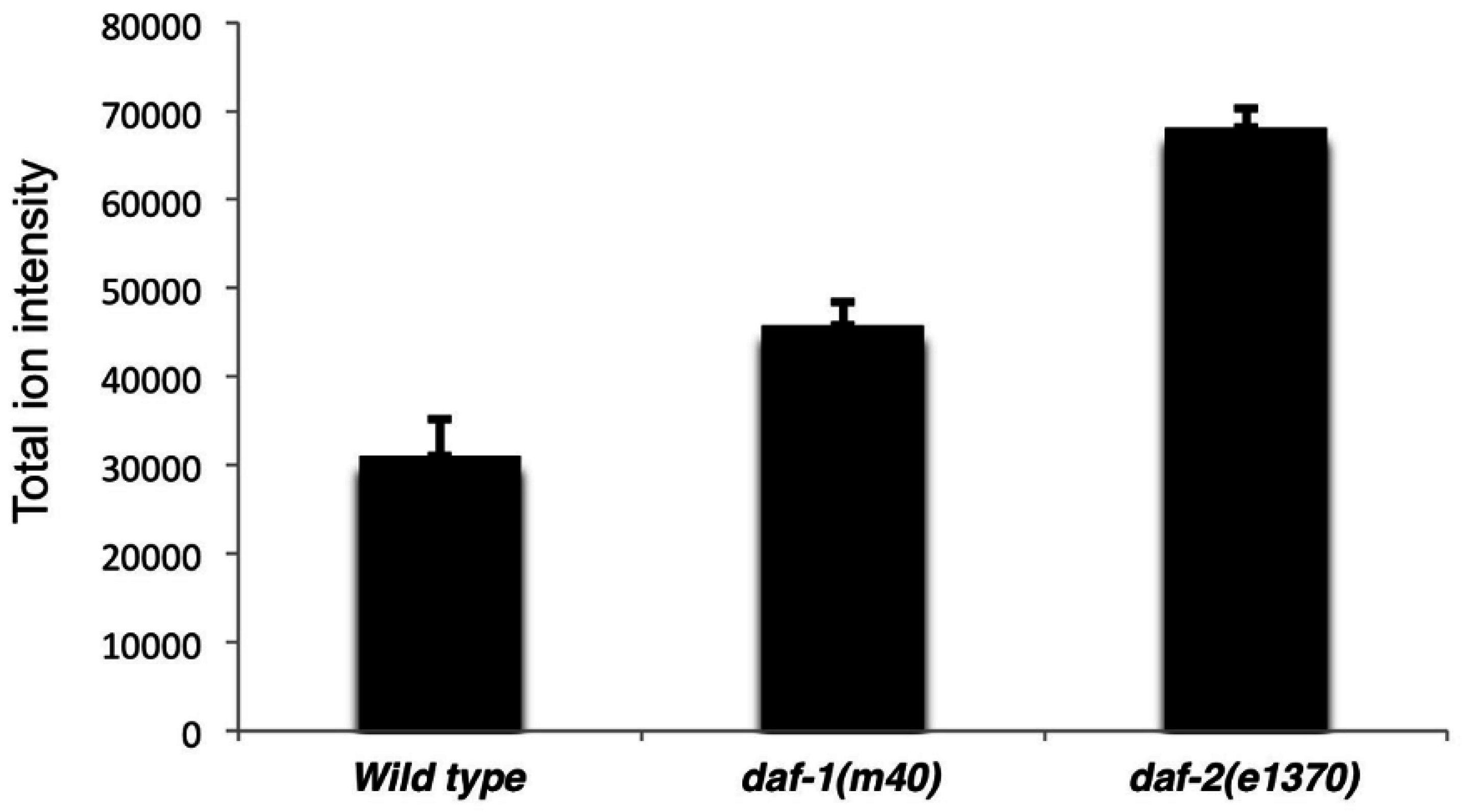
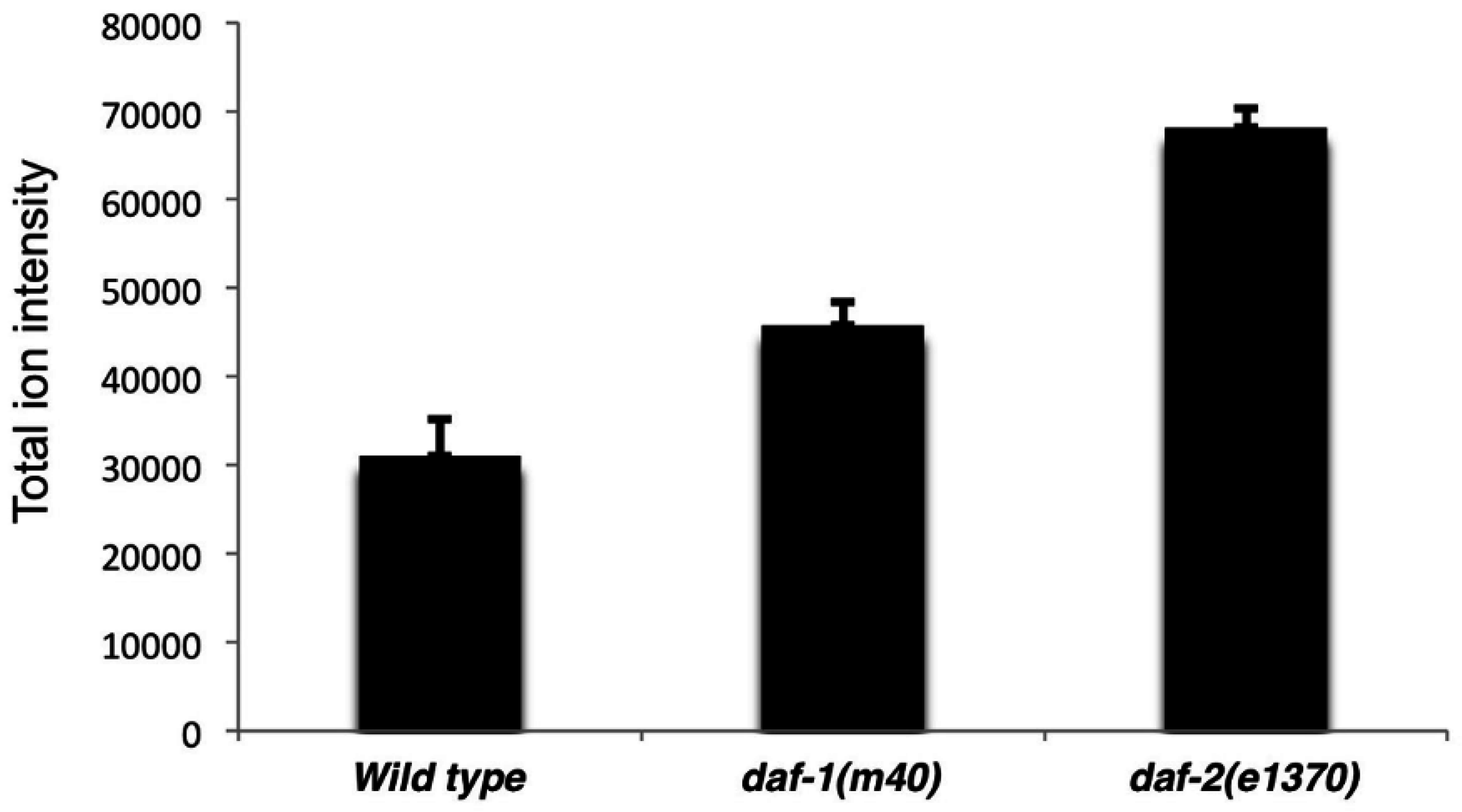
We focused on two well-studied mutant strains to evaluate method effectiveness. Disrupting insulin and TGF-β signaling causes overlapping phenotypes, but the molecular mechanisms are distinct [
13,
14]. Similarly, the mutants have different lipid profiles. These profiles could be used to identify downstream lipid mediators or develop “lipid signatures” for assessing signaling activity. Previous studies have demonstrated elevated TAG levels in
daf-1 and
daf-2 mutants. Our SWATH method identified increases in total TAG ion intensity in the mutants, as well as identified the individual molecular TAG species. SWATH also provided an unexpected model for Cox-independent prostaglandin metabolism: LPCs and other lyso glycerophospholipids may be key intermediates. As shown in the
Figure 1B example, we can predict that biochemical step A occurs via phospholipase activity, producing LPC 20:4. During step B, TGF-β signaling promotes expression of an enzyme that acts on LPC 20:4. The number of steps between LPC 20:4 and PGF2 isomers are unknown and could be large. This testable model is consistent with failure of dietary 20:4 supplementation to rescue
daf-1(m40) sperm motility defects [
32]. Future genetic screening and SWATH could help identify enzymes and substrates, providing a framework for the pathway. Stable isotope labeling studies could also be used to trace pathway flux.
In targeted approaches, internal standards are often included to account for variation in lipid extraction efficiency, ionization efficiency, etc. However, the untargeted nature of SWATH makes the choice of internal standards difficult. Lipids containing odd-chain fatty acids or stable isotopes are used as internal standards in mammalian studies. C. elegans synthesizes odd-chain fatty acids, so these inexpensive options are not good choices. More problematic, lipids containing stable isotopes are expensive and unavailable for most lipid classes. With the present limited selection, we suggest excluding internal standards for C. elegans SWATH. To avoid confounding issues, multiple independent replicates should be conducted to assess variability. Lipids exhibiting high variation among replicates or replicates with high coefficient of variation should be interpreted with extreme caution. When a set of altered lipids is chosen for further studies, a targeted approach using the best available internal standards can be developed. It is important to remember that SWATH provides an objective method for discovery. SWATH is not a substitute for MRM or other methods capable of absolute quantification.
There are shortcomings to the SWATH approach. First, the coverage of lipids is not complete. Although SWATH detected several thousand species, extraction conditions and mass spectrometer settings, such as collision energy will influence coverage. For example, our neutral lipid extraction method is not efficient at extracting oxidized lipids and multiple conditions should be explored. Second, LipidView does not always accurately annotate lipid species, which should be confirmed through MS/MS interpretation. Third, as mentioned above, SWATH does not provide absolute quantification. Despite these shortcomings, the advantages are powerful. In particular, the ability to retrospectively search SWATH libraries is important for lipid identification and biological discovery. Changes in a specific lipid species between two samples might suggest changes in precursors or related lipids. As new hypotheses form, they can be tested across all generated libraries. SWATH libraries can be searched by any researcher with appropriate software, obviating the need to repeat sample preparation and analysis. In genetic models where tissue is limiting, a well-constructed library provides a valuable resource for the entire community.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}