Computational Methods for Metabolomic Data Analysis of Ion Mobility Spectrometry Data—Reviewing the State of the Art

Abstract
:1. Introduction
1.1. Overview: Ion Mobility Spectrometry

1.2. Outline

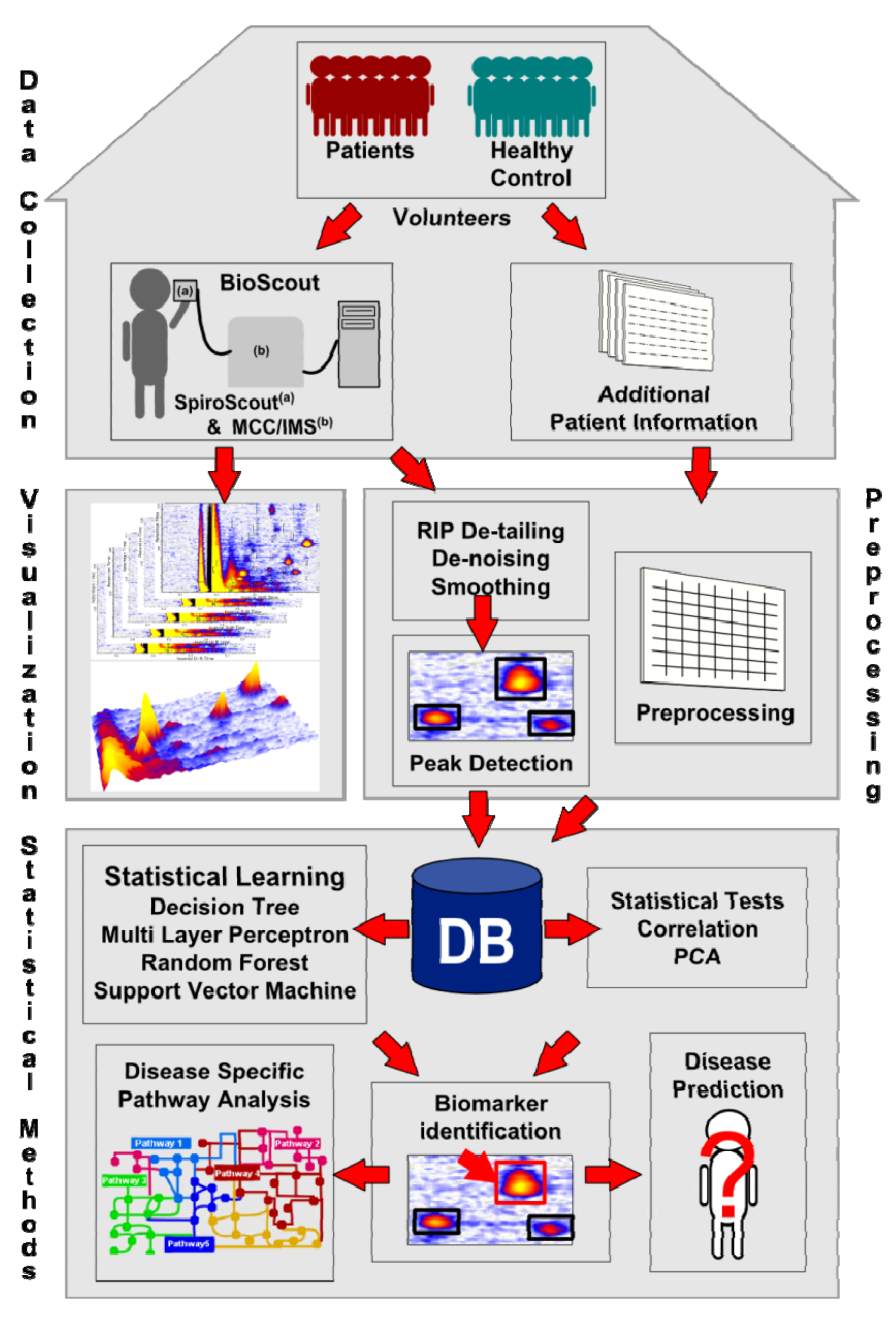
2. First Steps with IMS Data
2.1. Data Format
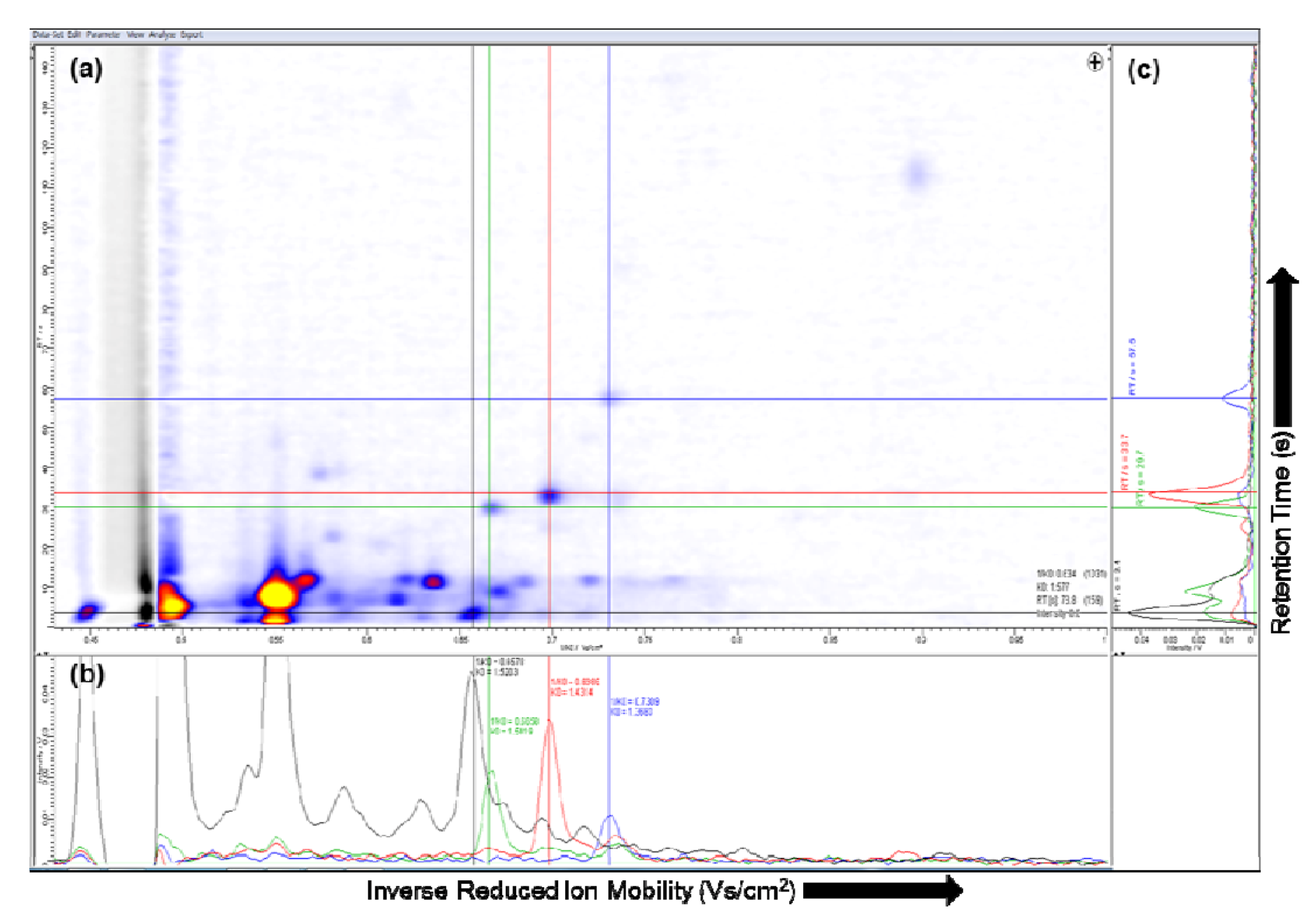
2.2. Visualization
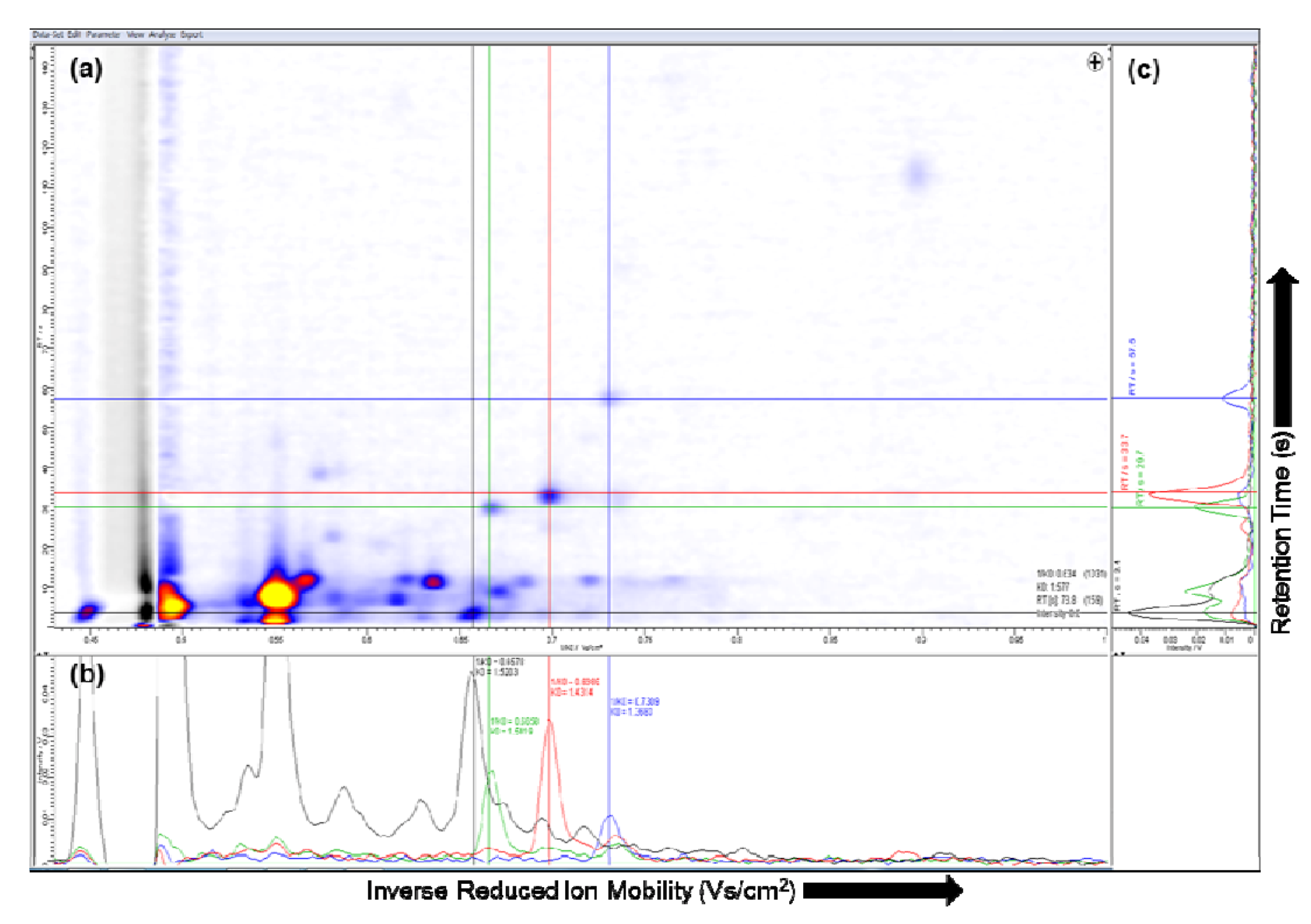
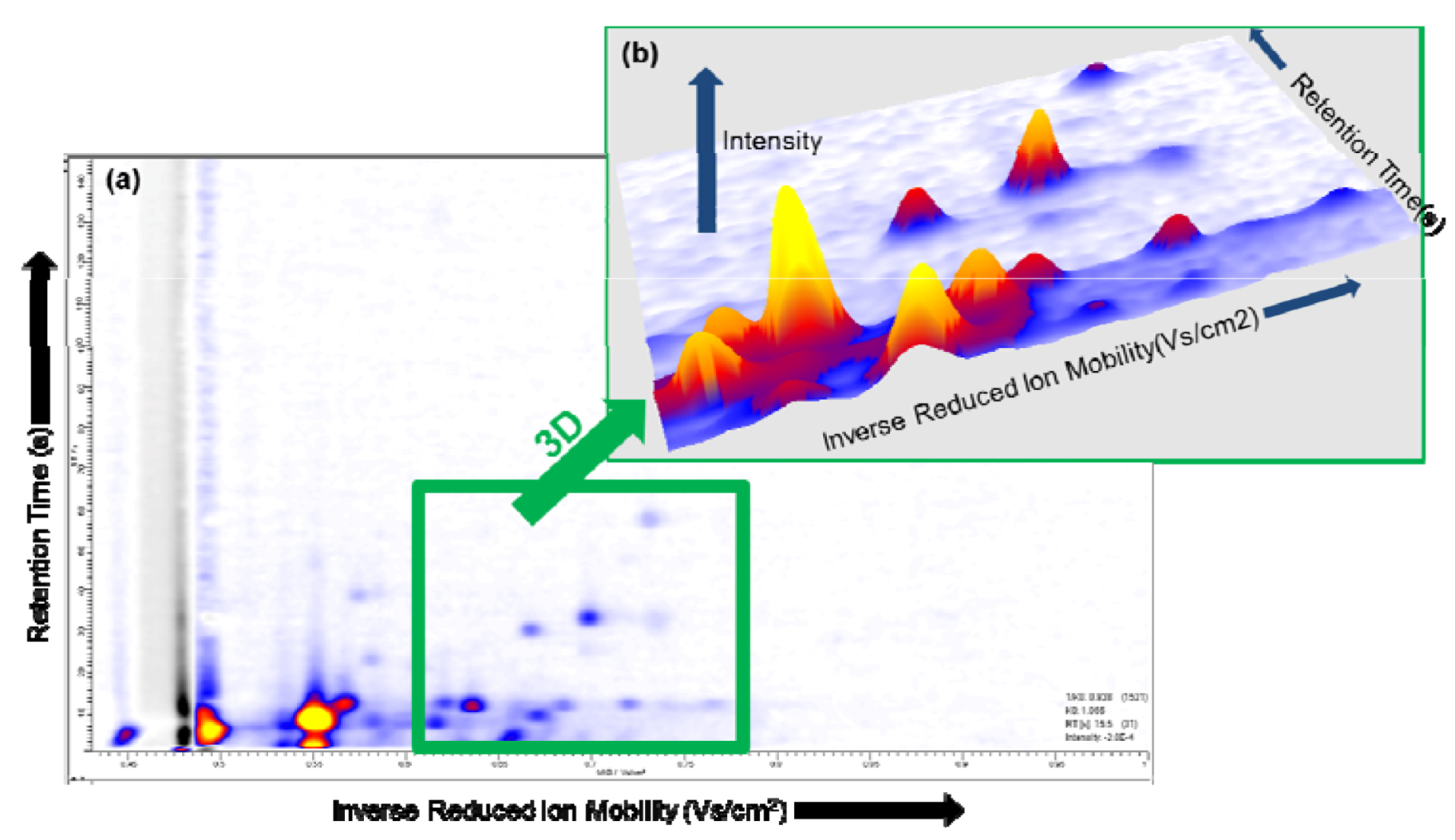
2.3. Pre-processing
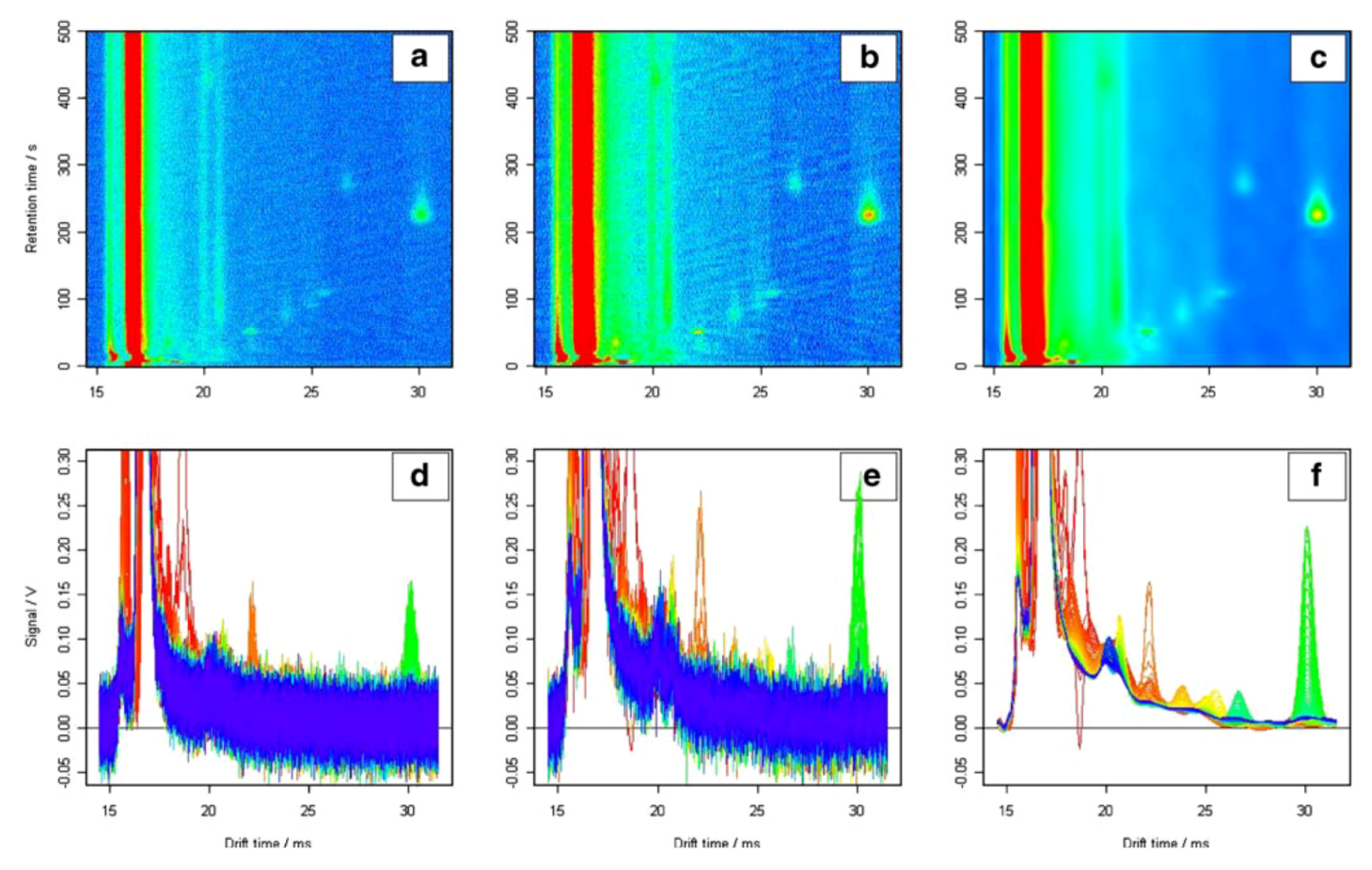
2.3.1. De-noising and Smoothing
2.3.2. Peak Detection
2.3.3. Merging Peak Sets
2.4. Database
3. Statistical Analysis
3.1. Statistical Tests
3.2. Correlation
3.3. Principal Component Analysis
4. Statistical Learning
4.1. Reduced Ion Mobility Prediction
4.2. Probabilistic Relational Learning

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Formula | Weight |
|---|---|---|
| 37 | pc7(M )⇒ bc ( M ) | 4.43 |
| 39 | pc11 (M )⇒ pc9(M ) | 4.82 |
| 44 | pc17 (M )∧ pc28 (M )⇒ pc21 (M ) | 5.05 |
| 46 | pc15 (M )∧ pc25 (M )⇒ pc5(M ) | −4.30 |
| 47 | pc17 (M )∧ pc19 (M )∧ pc20 (M )⇒ pc9(M ) | −8.98 |
| 53 | pc12 (M )∧ pc20 (M )∧ pc22 (M )⇒ pc11 (M ) | −8.14 |
| 57 | ¬pc1(M )∧¬pc18 (M )∧¬pc23 (M )∧ pc31 (M )⇒ bc ( M ) | 6.38 |
| 61 | ¬pc10 (M )∧ pc14 (M )∧¬pc18 (M )∧ pc21 (M )⇒ bc ( M ) | 7.15 |
| 62 | ¬pc12 (M )∧¬pc22 (M )∧¬pc30 (M )∧ pc31 (M )⇒ bc ( M ) | 7.49 |
| 66 | pc4(M )∧ pc26 (M )∧ pc28 (M )∧ pc29 (M )⇒ bc ( M ) | −5.62 |
| 68 | ¬pc9(M )∧¬pc13 (M )∧¬pc16 (M )∧ pc23 (M )∧¬pc29 (M )⇒ ¬bc ( M ) | 4.01 |
| 70 | pc1(M )∧ pc3(M )∧¬pc15 (M )∧¬pc23 (M )∧ pc26 (M )⇒ ¬bc ( M ) | −5.18 |
| 72 | pc0(M )∧¬pc11 (M )∧¬pc12 (M )∧¬pc21 (M )∧ pc22 (M )⇒ ¬bc ( M ) | 2.45 |
| 75 | pc5(M )∧ pc7(M )∧¬pc28 (M )∧¬pc29 (M )∧ pc31 (M )⇒ ¬bc ( M ) | −2.78 |
| 80 | pc0(M )∧¬pc12 (M )∧¬pc16 (M )∧ pc30 (M )∧¬pc32 (M )⇒ bc ( M ) | −5.55 |
| 81 | ¬pc6(M )∧¬pc13 (M )∧¬pc28 (M )∧ pc31 (M )∧ pc32 (M )⇒ ¬bc ( M ) | 5.61 |
| 82 | ¬pc3(M )∧¬pc4(M )∧ pc25 (M )∧¬pc28 (M )∧¬pc32 (M )⇒ ¬bc ( M ) | 8.77 |
| 89 | ¬pc3(M )∧¬pc11 (M )∧ pc13 (M )∧¬pc17 (M )∧¬pc31 (M )⇒ ¬bc ( M ) | −5.15 |
4.3. Statistical Learning and Biomarkers
| Study | Disease | # | ACC | AUC | CV |
|---|---|---|---|---|---|
| Finthammer et al. 2010 [76] | BC | 158 | 90% | - | √ |
| Baumbach et al. 2007 [78] | BC | 107 | 99% | 99% | - |
| Westhoff et al. 2011 [66] | COPD | 130 | 94% | - | - |
| Hauschild et al. 2012 [79] | COPD and BC | 119 | 94% | 92% | √ |
5. Summary and Conclusion
| Computational requirements | Completed |
|---|---|
| Data format | *** |
| Visualization | *** |
| Pre-processing methods | ** |
| Peak detection methods | ** |
| Centralized data repository | * |
| Statistical approaches | *** |
| Statistical learning methods | * |
| Differentiation of diseases, infections, cancer, etc. | * |
| Disease pathway identification | - |
Acknowledgments
Conflict of Interest
References
- Ligor, T.; Ligor, M.; Amann, A.; Ager, C.; Bachler, M.; Dzien, A.; Buszewski, B. The analysis of healthy volunteers' exhaled breath by the use of solid-phase microextraction and GC-MS. J. Breath Res. 2008, 2, 046006:1–046006:8. [Google Scholar]
- Jünger, M.; Bödeker, B.; Baumbach, J.I. Peak assignment in multi-capillary column - ion mobility spectrometry using comparative studies with gas chromatography-mass spectrometry for exhalred breath analysis. Anal. Bioanal. Chem. 2010, 396, 471–482. [Google Scholar] [CrossRef]
- Mieth, M.; Schubert, J.K.; Groger, T.; Sabel, B.; Kischkel, S.; Fuchs, P.; Hein, D.; Zimmermann, R.; Miekisch, W. Automated Needle Trap Heart-Cut GC/MS and Needle Trap Comprehensive Two-Dimensional GC/TOF-MS for Breath Gas Analysis in the Clinical Environment. Anal. Chem. 2010, 82, 2541–2551. [Google Scholar]
- Kushch, I.; Schwarz, K.; Schwentner, L.; Baumann, B.; Dzien, A.; Schmid, A.; Unterkofler, K.; Gastl, G.; Spanel, P.; Smith, D.; et al. Compounds enhanced in a mass spectrometric profile of smokers' exhaled breath versus non-smokers as determined in a pilot study using PTR-MS. J. Breath Res. 2008, 2, 026002:1–026002:26. [Google Scholar]
- Ligor, M.; Ligor, T.; Bajtarevic, A.; Ager, C.; Pienz, M.; Klieber, M.; Denz, H.; Fiegl, M.; Hilbe, W.; Weiss, W.; et al. Determination of volatile organic compounds in exhaled breath of patients with lung cancer using solid phase microextraction and gas chromatography mass spectrometry. Clin. Chem. Lab. Med. 2009, 47, 550–560. [Google Scholar]
- Buszewski, B.; Ulanowska, A.; Ligor, T.; Denderz, N.; Amann, A. Analysis of exhaled breath from smokers, passive smokers and non-smokers by solid-phase microextraction gas chromatography/mass spectrometry. Biomed. Chromatogr. 2009, 23, 551–556. [Google Scholar] [CrossRef]
- Cheng, Z.J.; Warwick, G.; Yates, D.H.; Thomas, P.S. An electronic nose in the discrimination of breath from smokers and non-smokers: a model for toxin exposure. J. Breath Res. 2009, 3, 036003/036001–036003/036005. [Google Scholar]
- Dragonieri, S.; Annema, J.T.; Schot, R.; van der Schee, M.P.C.; Spanevello, A.; Carratu, P.; Resta, O.; Rabe, K.F.; Sterk, P.J. An electronic nose in the discrimination of patients with non-small cell lung cancer and COPD. Lung Cancer 2009, 64, 166–170. [Google Scholar] [CrossRef]
- Dragonieri, S.; Schot, R.; Mertens, B.J.A.; le Cessie, S.; Gauw, S.A.; Spanevello, A.; Resta, O.; Willard, N.P.; Vink, T.J.; Rabe, K.F.; et al. An electronic nose in the discrimination of patients with asthma and controls. J. Allergy Clin. Immun. 2007, 120, 856–862. [Google Scholar] [CrossRef]
- Horvath, I.; Lazar, Z.; Gyulai, N.; Kollai, M.; Losonczy, G. Exhaled biomarkers in lung cancer. Eur. Respir. J. 2009, 34, 261–275. [Google Scholar] [CrossRef]
- Beauchamp, J.; Kirsch, F.; Buettner, A. Real-time breath gas analysis for pharmacokinetics: monitoring exhaled breath by on-line proton-transfer-reaction mass spectrometry after ingestion of eucalyptol-containing capsules. J. Breath Res. 2010. [Google Scholar] [CrossRef]
- Herbig, J.; Mueller, M.; Schallhart, S.; Titzmann, T.; Graus, M.; Hansel, A. On-line breath analysis with PTR-TOF. J. Breath Res. 2009, 3, 027004:1–027004:10. [Google Scholar]
- Perl, T.; Bödecker, B.; Jünger, M.; Nolte, J.; Vautz, W. Alignment of retention time obtained from multicapillary column gas chromatography used for VOC analysis with ion mobility spectrometry. Anal. Bioanal. Chem. 2010, 397, 2385–2394. [Google Scholar] [CrossRef]
- Baumbach, J.I. Process analysis using ion mobility spectrometry. Anal. Bioanal. Chem. 2006, 384, 1059–1070. [Google Scholar] [CrossRef]
- Baumbach, J.I.; Westhoff, M. Ion mobility spectrometry to detect lung cancer and airway infections. Spectrosc. Eur. 2006, 18, 22–27. [Google Scholar]
- Westhoff, M.; Litterst, P.; Freitag, L.; Baumbach, J.I. Ion mobility spectrometry in the diagnosis of Sarcoidosis: Results of a feasibility study. J. Physiol. Pharmacol. 2007, 58, 739–751. [Google Scholar]
- Vautz, W.; Nolte, J.; Fobbe, R.; Baumbach, J.I. Breath analysis—performance and potential of ion mobility spectrometry. J. Breath Res. 2009. [Google Scholar]
- Basanta, M.; Koimtzis, T.; Singh, D.; Wilson, I.; Thomas, C.L.P. An adaptive breath sampler for use with human subjects with an impaired respiratory function. Analyst 2007, 132, 153–163. [Google Scholar] [CrossRef]
- Basanta, M.; Koimtzis, T.; Thomas, C.L.P. Sampling and analysis of exhaled breath on human subjects with thermal desorption gas chromatography - differential mobility spectrometry. Int. J. Ion Mobility Spectrom. 2006, 9, 45–49. [Google Scholar]
- King, J.; Kupferthaler, A.; Frauscher, B.; Hackner, H.; Unterkofler, K.; Teschl, G.; Hinterhuber, H.; Amann, A.; Högl, B. Measurement of endogenous acetone and isoprene in exhaled breath during sleep. Physiol. Meas. 2012. [Google Scholar] [CrossRef]
- Lee, J.H.; Hwang, S.M.; Lee, D.W.; Heo, G.S. Determination of volatile organic compounds (VOCs) using Tedlar bag/solid-phase microextraction/gas chromatography/mass spectrometry (SPME/GC/MS) in ambient and workplace air. B Korean Chem. Soc. 2002, 23, 488–496. [Google Scholar]
- Schulz, K.; Jensen, M.L.; Balsley, B.B.; Davis, K.; Birks, J.W. Tedlar bag sampling technique for vertical profiling of carbon dioxide through the atmospheric boundary layer with high precision and accuracy. Environ. Sci. Technol. 2004, 38, 3683–3688. [Google Scholar] [CrossRef]
- Beauchamp, J.; Herbig, J.; Gutmann, R.; Hansel, A. On the use of Tedlar bags for breath-gas sampling and analysis. J. Breath Res. 2008, 046001: 1–046001:19. [Google Scholar]
- Filipiak, W.; Filipiak, A.; Ager, C.; Wiesenhofer, H.; Amann, A. Optimization of sampling parameters for collection and preconcentration of alveolar air by needle traps. J. Breath Res. 2012, 6, 027107. [Google Scholar] [CrossRef]
- Bajtarevic, A.; Ager, C.; Pienz, M.; Klieber, M.; Schwarz, K.; Ligor, M.; Ligor, T.; Filipiak, W.; Denz, H.; Fiegl, M.; et al. Noninvasive detection of lung cancer by analysis of exhaled breath. BMC Cancer 2009. [Google Scholar] [CrossRef]
- Miekisch, W.; Hengstenberg, A.; Kischkel, S.; Beckmann, U.; Mieth, M.; Schubert, J.K. Construction and Evaluation of a Versatile CO2 Controlled Breath Collection Device. Ieee Sen. J. 2010, 10, 211–215. [Google Scholar] [CrossRef]
- Baumbach, J.I.; Eiceman, G.A. Ion Mobility Spectrometry: Arriving On Site and Moving Beyond a Low Profile. Appl. Spectrosc. 1999, 53, 338A–355A. [Google Scholar] [CrossRef]
- Hill, H.H., Jr.; Siems, W.F.; St Louis, R.H.; McMinn, D.G. Ion mobility spectrometry. Anal. Chem. 1990, 62, 1201A–1209A. [Google Scholar]
- Ruzsanyi, V.; Baumbach, J.I.; Sielemann, S.; Litterst, P.; Westhoff, M.; Freitag, L. Detection of human metabolites using multi-capillary columns coupled to ion mobility spectrometers. J. Chromatogr. A 2005, 1084, 145–151. [Google Scholar] [CrossRef]
- Baumbach, J.I. Ion Mobility Spectrometry coupled with Multi-Capillary Columns for Metabolic Profiling of Human Breath. J. Breath Res. 2009, 3, 1–16. [Google Scholar]
- Maddula, S.; Blank, L.; Schmid, A.; Baumbach, J.I. Detection of volatile metabolites of Escherichia coli by multi capillary column coupled ion mobility spectrometry. Anal. Bioanal. Chem. 2009, 394, 791–800. [Google Scholar] [CrossRef]
- Eiceman, G.A.; Karpas, Z. Ion Mobility Spectrometry, 2nd ed; CRC Press, Taylor & Francis: Boca Raton, FL, USA, 2005; Volume 1, p. 337. [Google Scholar]
- Vautz, W.; Bödecker, B.; Bader, S.; Baumbach, J.I. Recommendation of a standard format for data sets from GC/IMS with sensor-controlled sampling. Int. J. Ion Mobility Spectrom. 2008, 11, 71–76. [Google Scholar] [CrossRef]
- Maddula, S.; Rupp, K.; Baumbach, J.I. Recommendation for an upgrade to the standard format in order to cross-link the GC/MSD and the MCC/IMS data. Int. J. Ion Mobility Spectrom. 2012, 15, 79–81. [Google Scholar] [CrossRef]
- Bunkowski, A. Software tool for coupling chromatographic total ion current dependencies of GC/MSD and MCC/IMS. Int. J. Ion Mobility Spectrom. 2010, 13, 169–175. [Google Scholar] [CrossRef]
- Bödeker, B.; Vautz, W.; Baumbach, J.I. Visualisation of MCC/IMS—Data. Int. J. Ion Mobility Spectrom. 2008, 11, 77–82. [Google Scholar] [CrossRef]
- Borsdorf, H.; Eiceman, G.A. Ion Mobility Spectrometry: Principles and Applications. Appl. Spectrosc. Rev. 2006, 41, 323–375. [Google Scholar] [CrossRef]
- Cumeras, R.; Schneider, T.; Favrod, P.; Figueras, E.; Gracia, I.; Maddula, S.; Baumbach, J.I. Stability and alignment of MCC/IMS devices. Int. J. Ion Mobility Spectrom. 2012, 15, 41–46. [Google Scholar] [CrossRef]
- Bader, S. Identification and Quantification of Peaks in Spectrometric Data. Technical University of Dortmund: Dortmund, Germany, 2008. [Google Scholar]
- Bunkowski, A. MCC-IMS data analysis using automated spectra processing and explorative visualization methods.
- Bader, S.; Urfer, W.; Baumbach, J.I. Preprocessing of Ion Mobility Spectra by Lognormal Detailing and Wavelet Transform. Int. J. Ion Mobility Spectrom. 2008, 11, 43–50. [Google Scholar] [CrossRef]
- Urbas, A.A.; Harrington, P.B. Two-dimensional wavelet compression of ion mobility spectra. Anal. Chim. Acta 2001, 446, 393–412. [Google Scholar]
- Cai, C.; de B. Harrington, P. Different Discrete Wavelet Transforms Applied to Denoising Analytical Data. J. Chem. Inf. Comp. Sci. 1998, 38, 1161–1170. [Google Scholar] [CrossRef]
- Savitzky, A.; Golay, M.J.E. Smoothing and Differentiation of Data by Simplified Least Squares Procedures. Anal. Chem. 1964, 36, 1627–1639. [Google Scholar] [CrossRef]
- Haddad, R.A.; Akansu, A.N. A class of fast Gaussian binomial filters for speech and image-processing. IEEE T. Signal Proces. 1991, 39, 723–727. [Google Scholar] [CrossRef]
- Cleveland, W.S. Robust locally weighted regression and smoothing scatterplots. J. Am. Stat. Assoc. 1979, 74, 829–836. [Google Scholar] [CrossRef]
- Bruce, J.; Balch, T.; Veloso, M. Intelligent Robots and Systems, 2000. (IROS 2000). In proceedings of Ieee Fast and inexpensive color image segmentation for interactive robots, Takamatsu, Japan, 31 Oct–05 Nov; 2000. [Google Scholar]
- Randolph, T.W.; Yasui, Y. Multiscale processing of mass spectrometry data. Biometrics 2006, 62, 589–597. [Google Scholar] [CrossRef]
- Wegner, S.; Sahlström, A.; Pleißner, K.P.; Oswald, H.; Fleck, E. Bildverarbeitung für die Medizin. Eine hierarchische Wasserscheidentransformation für die Spotdetektion in 2D-Gel-Elektrophorese-Bildern. In Proceedings: Bildverarbeitung für die Medizin Aachen, Germany, 26–27 March, 1998.
- Meyer, F.; Beucher, S. Morphological segmentation. J. Vis. Commun. Image R 1990, 1, 21–46. [Google Scholar] [CrossRef]
- Vincent, L. Watersheds in Digital Spaces: An Efficient Algorithm Based on Immersion Simulations. IEEE T. Pattern. Anal. 1991, 13, 583–598. [Google Scholar] [CrossRef]
- Bödeker, B.; Vautz, W.; Baumbach, J.I. Peak Finding and Referencing in MCC/IMS - Data. Int. J. Ion Mobility Spectrom. 2008, 11, 83–88. [Google Scholar] [CrossRef]
- Ward, J.H. Hierarchical grouping to optimize an objective function. J. Am. Stat. Assoc. 1963. [Google Scholar] [CrossRef]
- Sturm, M.; Bertsch, A.; Gröpl, C.; Hildebrandt, A.; Hussong, R.; Lange, E.; Pfeifer, N.; Schulz-Trieglaff, O.; Zerck, A.; Reinert, K.; et al. OpenMS-An open-source software framework for mass spectrometry. BMC Bioinformatics 2008. [Google Scholar] [CrossRef]
- Lesniak, T. Entwurf, Erprobung und Bewertung eines Informationsschemas für Untersuchungen von Metaboliten.
- Chen, R.S.; Nadkarni, P.; Marenco, L.; Levin, F.; Erdos, J.; Miller, P.L. Exploring Performance Issues for a Clinical Database Organized Using an Entity-Attribute-Value Representation. J. Am. Med. Inform. Assn. 2000, 7, 475–487. [Google Scholar]
- Vogtland, D.; Baumbach, J.I. Breit-Wigner-Function and IMS-Signals. Int. J. Ion Mobility Spectrom. 2009, 12, 109–114. [Google Scholar] [CrossRef]
- Bessa, V.; Darwiche, K.; Teschler, H.; Sommerwerck, U.; Rabis, T.; Baumbach, J.I.; Freitag, L. Detection of volatile organic compounds (VOCs) in exhaled breath of patients with chronic obstructive pulmonary disease (COPD) by ion mobility spectrometry. Int. J. Ion Mobility Spectrom. 2011, 14, 7–13. [Google Scholar] [CrossRef]
- Koczulla, R.; Hattesohl, A.; Schmid, S.; Bödeker, B.; Maddula, S.; Baumbach, J.I. MCC/IMS as potential noninvasive technique in the diagnosis of patients with COPD with and without alpha 1-antitrypsin deficiency. Int. J. Ion Mobility Spectrom. 2011, 14, 177–185. [Google Scholar] [CrossRef]
- Rabis, T.; Sommerwerck, U.; Anhenn, O.; Darwiche, K.; Freitag, L.; Teschler, H.; Bödeker, B.; Maddula, S.; Baumbach, J.I. Detection of infectious agents in the airways by ion mobility spectrometry of exhaled breath. Int. J. Ion Mobility Spectrom. 2011, 11, 187–195. [Google Scholar]
- Maddula, S.; Rabis, T.; Sommerwerck, U.; Anhenn, O.; Darwiche, K.; Freitag, L.; Teschler, H.; Baumbach, J.I. Correlation analysis on data sets to detect infectious agents in the airways by ion mobility spectrometry of exhaled breath. Int. J. Ion Mobility Spectrom. 2011, 14, 197–206. [Google Scholar] [CrossRef]
- Kreuder, A.-E.; Buchinger, H.; Kreuer, S.; Volk, T.; Maddula, S.; Baumbach, J.I. Characterization of propofol in human breath of patients undergoing anesthesia. Int. J. Ion Mobility Spectrom. 2011, 14, 167–175. [Google Scholar] [CrossRef]
- Carstens, E.; Hirn, A.; Quintel, M.; Nolte, J.; Juenger, M.; Perl, T.; Vautz, W. On-line determination of serum propofol concentrations by expired air analysis. Int. J. Ion Mobility Spectrom. 2010, 13, 37–40. [Google Scholar] [CrossRef]
- Pearson, K. On lines and planes of closest fit to systems of points in space. Philosophical Magazine 1901, 2, 559–572. [Google Scholar]
- Westhoff, M.; Litterst, P.; Maddula, S.; Bödecker, B.; Rahmann, S.; Davies, A.N.; Baumbach, J.I. Differentiation of chronic obstructive pulmonary disease (COPD) including lung cancer from healthy control group by breath analysis using ion mobility spectrometry. Int. J. Ion Mobility Spectrom. 2010, 13, 131–139. [Google Scholar] [CrossRef]
- Westhoff, M.; Litterst, P.; Maddula, S.; Bödeker, B.; Baumbach, J.I. Statistical and bioinformatical methods to differentiate chronic obstructive pulmonary disease (COPD) including lung cancer from healthy control by breath analysis using ion mobility spectrometr. Int. J. Ion Mobility Spectrom. 2011, 11, 139–149. [Google Scholar]
- Cheung, W.; Xu, Y.; Thomas, C.L.P.; Goodacre, R. Discrimination of bacteria using pyrolysis-gas chromatography-differential mobility spectrometry (Py-GC-DMS) and chemometrics. Analyst 2009, 134, 557–563. [Google Scholar] [CrossRef]
- Team, R.D.C. R: A language and environment for statistical computing. Available online: http://www.google.com.hk/url?sa=t&rct=j&q=A+language+and+environment+for+statistical+computing&source=web&cd=1&ved=0CCoQFjAA&url=http%3A%2F%2Fwww.lsw.uni-heidelberg. de%2Fusers%2Fchristlieb%2Fteaching%2FUKStaSS10%2FR-refman.pdf&ei=_753UMmgJ--eiAeu7IGoAQ&usg=AFQjCNHz-GCuz9CII0JC8onCa91DwwVoHQ&cad=rjt/ (accessed on 12 October 2012).
- Revercomb, H.E.; Mason, E.A. Theory of Plasma Chromatography Gaseous Electrophoresis - Review. Anal. Chem. 1975, 47, 970–983. [Google Scholar] [CrossRef]
- Benezra, S.A. Separation of Mixtures of Aromatic Ketones in Sub-Nanogram Range by Plasma Chromatography. J. Chromatogr. Sci. 1976, 14, 122–125. [Google Scholar]
- Karasek, F.W.; Kim, S.H.; Rokushika, S. Plasma chromatography of alkyl amines. Anal. Chem. 1978, 50, 2013–2016. [Google Scholar] [CrossRef]
- Wessel, M.D.; Sutter, J.M.; Jurs, P.C. Prediction of Reduced Ion Mobility Constants of Organic Compounds from Molecular Structure. Anal. Chem. 1996, 68, 4237–4243. [Google Scholar] [CrossRef]
- Liu, H.; Yao, X.; Liu, M.; Hu, Z.; Fan, B. Prediction of gas-phase reduced ion mobility constants (K0) based on the multiple linear regression and projection pursuit regression. Talanta 2007, 71, 258–263. [Google Scholar] [CrossRef]
- Hariharan, C.B.; Baumbach, J.I.; Vautz, W. Linearized Equations for the Reduced Ion Mobilities of Polar Aliphatic Organic Compounds. Anal. Chem. 2010, 82, 427–431. [Google Scholar] [CrossRef]
- Friedman, N.; Getoor, L.; Koller, D.; Pfeffer, A. Learning Probabilistic Relational Models. In Proceedings of the Sixteenth International Joint Conferences on Artificial Intelligence (IJCAI-99), Stockholm, Sweden, July 31–August 6 1999; pp. 1300–1309.
- Finthammer, M.; Beierle, C.; Fisseler, J.; Kern-Isberner, G.; Möller, B.; Baumbach, J.I. Probabilistic Relational Learning for Medical Diagnosis Based on Ion Mobility Spectrometry. Int. J. Ion Mobility Spectrom. 2010, 13, 83–92. [Google Scholar] [CrossRef]
- Muggleton, S.; de Raedt, L. Inductive logic programming: Theory and methods. J. Logic Program. 1994, 19/20, 629–679. [Google Scholar] [CrossRef]
- Baumbach, J.; Bunkowski, A.; Lange, S.; Oberwahrenbrock, T.; Kleinboelting, N.; Rahmann, S.; Baumbach, J.I. IMS2—An integrated medical software system for early lung cancer detection using ion mobility spectrometry data of human breath. J. Integr. Bioinformatics 2007, 4, 75. [Google Scholar]
- Hauschild, A.-C.; Baumbach, J.I.; Baumbach, J. Integrated Statistical Learning of Metabolic Ion Mobility - Spectrometry Profiles for Pulmonary Disease Identification. Genet. Mol. Res. 2012, 11, 2733–2744. [Google Scholar] [CrossRef]
- Khatri, P.; Sirota, M.; Butte, A.J. Ten Years of Pathway Analysis: Current Approaches and Outstanding Challenges. PLoS Comput. Biol. 2012, 8, e1002375. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hauschild, A.-C.; Schneider, T.; Pauling, J.; Rupp, K.; Jang, M.; Baumbach, J.I.; Baumbach, J. Computational Methods for Metabolomic Data Analysis of Ion Mobility Spectrometry Data—Reviewing the State of the Art. Metabolites 2012, 2, 733-755. https://doi.org/10.3390/metabo2040733
Hauschild A-C, Schneider T, Pauling J, Rupp K, Jang M, Baumbach JI, Baumbach J. Computational Methods for Metabolomic Data Analysis of Ion Mobility Spectrometry Data—Reviewing the State of the Art. Metabolites. 2012; 2(4):733-755. https://doi.org/10.3390/metabo2040733
Chicago/Turabian StyleHauschild, Anne-Christin, Till Schneider, Josch Pauling, Kathrin Rupp, Mi Jang, Jörg Ingo Baumbach, and Jan Baumbach. 2012. "Computational Methods for Metabolomic Data Analysis of Ion Mobility Spectrometry Data—Reviewing the State of the Art" Metabolites 2, no. 4: 733-755. https://doi.org/10.3390/metabo2040733
APA StyleHauschild, A.-C., Schneider, T., Pauling, J., Rupp, K., Jang, M., Baumbach, J. I., & Baumbach, J. (2012). Computational Methods for Metabolomic Data Analysis of Ion Mobility Spectrometry Data—Reviewing the State of the Art. Metabolites, 2(4), 733-755. https://doi.org/10.3390/metabo2040733