Human Metabolic Network: Reconstruction, Simulation, and Applications in Systems Biology

{kind=link}
Abstract
:1. Introduction
2. Reconstruction of Global Human Metabolic Network
3. Modeling and Simulation Based on Human Metabolic Network
- Reactions: S (Stoichiometric Matrix), with m compounds (rows) and n reactions (columns). The stoichiometric coefficients are negative for the substrates of each reaction, and positive for the products.
- Flows: v (n by 1 vector) on all reactions
- Concentrations: X (m by 1 vector) of all compounds
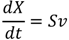
4. Systems Biology Applications of Human Metabolic Network
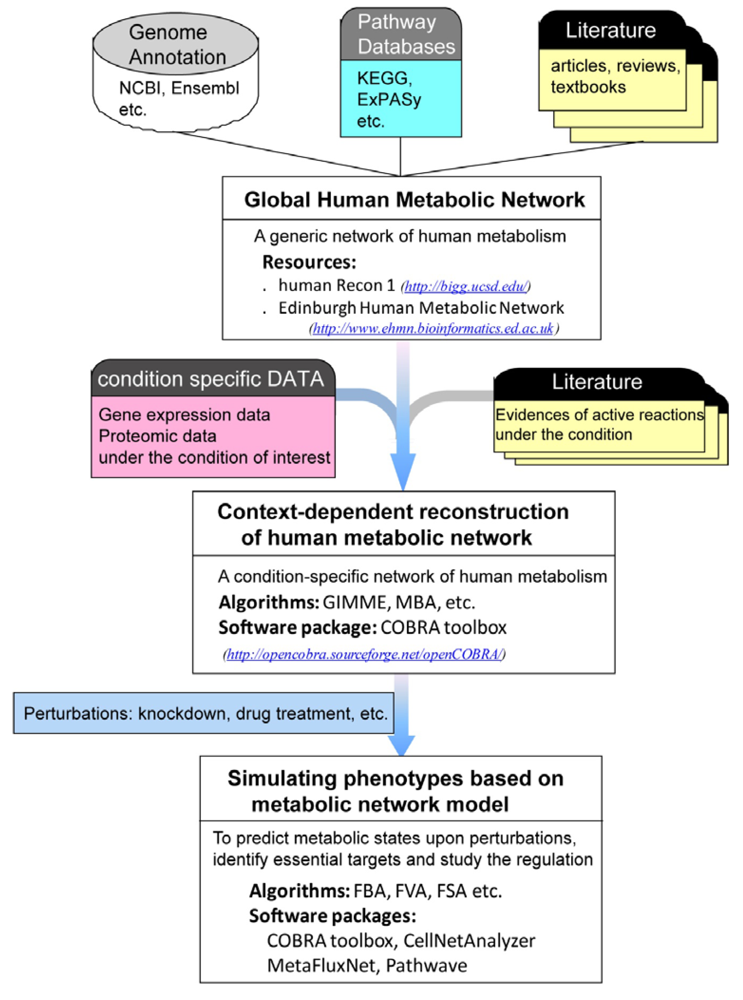
4.1. Reconstructing Context-Dependent Metabolic Network
4.2. Simulating Phenotypes Based on Metabolic Network Model
5. Challenges and Perspectives
5.1. The Consolidation of the Human Metabolic Network
5.2. Novel Approaches to Reconstructing Context-Dependent Networks
5.3. Reconstruction of Multi-Cellular Metabolic System
5.4. Incorporating Regulations on Multiple Layers
Acknowledgments
Conflict of Interest
References
- Zelezniak, A.; Pers, T.H.; Soares, S.; Patti, M.E.; Patil, K.R. Metabolic network topology Reveals transcriptional regulatory signatures of type 2 diabetes. PLoS Comput. Biol. 2010, 6, e1000729. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Lunt, S.Y.; Dayton, T.L.; Fiske, B.P.; Israelsen, W.J.; Mattaini, K.R.; Vokes, N.I.; Stephanopoulos, G.; Cantley, L.C.; Metallo, C.M.; et al. Metabolic pathway Alterations that support cell proliferation. Cold Spring Harbor Symp. Quant. Biol. 2012, 76. [Google Scholar]
- WARBURG, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar]
- Pollari, S.; Käkönen, S.-M.; Edgren, H.; Wolf, M.; Kohonen, P.; Sara, H.; Guise, T.; Nees, M.; Kallioniemi, O. Enhanced serine production by bone metastatic breast cancer cells stimulates osteoclastogenesis. Breast Cancer Res. Treat. 2011, 125, 421–430. [Google Scholar] [CrossRef]
- Serkova, N.J.; Spratlin, J.L.; Eckhardt, S.G. NMR-based metabolomics: Translational application and treatment of cancer. Curr. Opin. Mol. Ther. 2007, 9, 572–585. [Google Scholar]
- Galmarini, C.M.; Popowycz, F.; Joseph, B. Cytotoxic nucleoside analogues: Different strategies to improve their clinical efficacy. Curr. Med. Chem. 2008, 15, 1072–1082. [Google Scholar]
- Zhang, B.B.; Zhou, G.; Li, C. AMPK: An emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 2009, 9, 407–416. [Google Scholar]
- Berger, J.P.; Akiyama, T.E.; Meinke, P.T. PPARs: Therapeutic targets for metabolic disease. Trends Pharm. Sci. 2005, 26, 244–251. [Google Scholar] [CrossRef]
- Pfeiffer, T.; Soyer, O.S.; Bonhoeffer, S. The evolution of connectivity in metabolic networks. PLoS Biol. 2005, 3, e228. [Google Scholar]
- Duarte, N.C.; Becker, S.A.; Jamshidi, N.; Thiele, I.; Mo, M.L.; Vo, T.D.; Srivas, R.; Palsson, B.Ø. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc. Natl. Acad. Sci. USA 2007, 104, 1777–1782. [Google Scholar]
- Ma, H.; Sorokin, A.; Mazein, A.; Selkov, A.; Selkov, E.; Demin, O.; Goryanin, I. The Edinburgh human metabolic network reconstruction and its functional analysis. Mol. Syst. Biol. 2007, 3, 135. [Google Scholar]
- Bordbar, A.; Palsson, B.O. Using the reconstructed genome—Scale human metabolic network to study physiology and pathology. J. Int. Med. 2012, 271, 131–141. [Google Scholar] [CrossRef]
- Hao, T.; Ma, H.-W.; Zhao, X.-M.; Goryanin, I. Compartmentalization of the edinburgh human metabolic network. BMC Bioinformatics 2010, 11, 393. [Google Scholar]
- Thiele, I.; Palsson, B.Ø. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5. [Google Scholar]
- Stephanopoulos, G.N.; Aristidou, A.A.; Nielsen, J. Metabolic Engineering: PRINCIPLES and Methodologies, 1st ed.; Academic Press: San Diego, CA, USA, 1998. [Google Scholar]
- Orth, J.D.; Thiele, I.; Palsson, B.O. What is flux balance analysis? Nat. Biotech. 2010, 28, 245–248. [Google Scholar]
- Toya, Y.; Kono, N.; Arakawa, K.; Tomita, M. Metabolic flux analysis and visualization. J. Proteome Res. 2011, 10, 3313–3323. [Google Scholar] [CrossRef]
- Delgado, J.; Liao, J.C. Inverse flux analysis for reduction of acetate excretion in Escherichiacoli. Biotechnol. Progr. 1997, 13, 361–367. [Google Scholar] [CrossRef]
- Mahadevan, R.; Schilling, C.H. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab. Eng. 2003, 5, 264–276. [Google Scholar] [CrossRef]
- Park, J.M.; Kim, T.Y.; Lee, S.Y. Constraints-based genome-scale metabolic simulation for systems metabolic engineering. Biotechnol. Adv. 2009, 27, 979–988. [Google Scholar] [CrossRef]
- Schellenberger, J.; Que, R.; Fleming, R.M.T.; Thiele, I.; Orth, J.D.; Feist, A.M.; Zielinski, D.C.; Bordbar, A.; Lewis, N.E.; Rahmanian, S.; et al. Quantitative prediction of cellular metabolism with constraint-based models: The COBRA Toolbox v2.0. 0. Nat. Protoc. 2011, 6, 1290–1307. [Google Scholar] [CrossRef]
- Klamt, S.; Rodriguez, J.; Gilles, E. Structural and functional analysis of cellular networks with CellNetAnalyzer. BMC Syst. Biol. 2007, 1, 2. [Google Scholar]
- Lee, D.-Y.; Yun, H.; Park, S.; Lee, S.Y. MetaFluxNet: the management of metabolic reaction information and quantitative metabolic flux analysis. Bioinformatics 2003, 19, 2144–2146. [Google Scholar]
- Schramm, G.; Wiesberg, S.; Diessl, N.; Kranz, A.-L.; Sagulenko, V.; Oswald, M.; Reinelt, G.; Westermann, F.; Eils, R.; König, R. PathWave: discovering patterns of differentially regulated enzymes in metabolic pathways. Bioinformatics 2010, 26, 1225–1231. [Google Scholar]
- Gille, C.; Bölling, C.; Hoppe, A.; Bulik, S.; Hoffmann, S.; Hübner, K.; Karlstädt, A.; Ganeshan, R.; König, M.; Rother, K.; et al. HepatoNet1: A comprehensive metabolic reconstruction of the human hepatocyte for the analysis of liver physiology. Mol. Syst. Biol. 2010, 6, 411. [Google Scholar]
- Lewis, N.E.; Schramm, G.; Bordbar, A.; Schellenberger, J.; Andersen, M.P.; Cheng, J.K.; Patel, N.; Yee, A.; Lewis, R.A.; Eils, R.; et al. Large-scale in silico modeling of metabolic interactions between cell types in the human brain. Nat. Biotechnol. 2010, 28, 1279–1285. [Google Scholar]
- Becker, S.A.; Palsson, B.O. Context-specific metabolic networks are consistent with experiments. PLoS Comput. Biol. 2008, 4, e1000082. [Google Scholar] [CrossRef]
- Shlomi, T.; Cabili, M.N.; Herrgård, M.J.; Palsson, B. Network-based prediction of human tissue-specific metabolism. Nat. Biotechnol. 2008, 26, 1003–1010. [Google Scholar]
- Jerby, L.; Shlomi, T.; Ruppin, E. Computational reconstruction of tissue-specific metabolic models: application to human liver metabolism. Mol. Syst. Biol. 2010, 6, 401. [Google Scholar]
- Oberhardt, M.A.; Palsson, B.Ø.; Papin, J.A. Applications of genome-scale metabolic reconstructions. Mol. Syst. Biol. 2009, 5, 320. [Google Scholar]
- Chang, R.L.; Xie, L.; Xie, L.; Bourne, P.E.; Palsson, B. Drug off-target effects predicted using structural analysis in the context of a metabolic network model. PLoS Comput. Biol. 2010, 6, e1000938. [Google Scholar]
- Folger, O.; Jerby, L.; Frezza, C.; Gottlieb, E.; Ruppin, E.; Shlomi, T. Predicting selective drug targets in cancer through metabolic networks. Mol. Syst. Biol. 2011, 7, 501. [Google Scholar]
- Shlomi, T.; Benyamini, T.; Gottlieb, E.; Sharan, R.; Ruppin, E. Genome-scale metabolic modeling elucidates the role of proliferative adaptation in causing the warburg effect. PLoS Comput. Biol. 2011, 7, e1002018. [Google Scholar] [CrossRef]
- Herrgård, M.J.; Swainston, N.; Dobson, P.; Dunn, W.B.; Arga, K.Y.; Arvas, M.; Blüthgen, N.; Borger, S.; Costenoble, R.; Heinemann, M.; et al. A consensus yeast metabolic network reconstruction obtained from a community approach to systems biology. Nat. Biotechnol. 2008, 26, 1155–1160. [Google Scholar]
- Thiele, I.; Palsson, B.Ø. Reconstruction annotation jamborees: A community approach to systems biology. Mol. Syst. Biol. 2010, 6, 361. [Google Scholar]
- Colijn, C.; Brandes, A.; Zucker, J.; Lun, D.S.; Weiner, B.; Farhat, M.R.; Cheng, T.-Y.; Moody, D.B.; Murray, M.; Galagan, J.E. Interpreting expression data with metabolic flux models: Predicting mycobacterium tuberculosis mycolic acid production. PLoS Comput. Biol. 2009, 5, e1000489. [Google Scholar]
- Walther, J.L.; Metallo, C.M.; Zhang, J.; Stephanopoulos, G. Optimization of (13)C isotopic tracers for metabolic flux analysis in mammalian cells. Metab. Eng. 2011. [Google Scholar]
- Gaglio, D.; Metallo, C.M.; Gameiro, P.A.; Hiller, K.; Danna, L.S.; Balestrieri, C.; Alberghina, L.; Stephanopoulos, G.; Chiaradonna, F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol. Syst. Biol. 2011, 7, 523. [Google Scholar]
- Bordbar, A.; Lewis, N.E.; Schellenberger, J.; Palsson, B.Ø.; Jamshidi, N. Insight into human alveolar macrophage and M. tuberculosis interactions via metabolic reconstructions. Mol. Syst. Biol. 2010, 6, 422. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wu, M.; Chan, C. Human Metabolic Network: Reconstruction, Simulation, and Applications in Systems Biology. Metabolites 2012, 2, 242-253. https://doi.org/10.3390/metabo2010242
Wu M, Chan C. Human Metabolic Network: Reconstruction, Simulation, and Applications in Systems Biology. Metabolites. 2012; 2(1):242-253. https://doi.org/10.3390/metabo2010242
Chicago/Turabian StyleWu, Ming, and Christina Chan. 2012. "Human Metabolic Network: Reconstruction, Simulation, and Applications in Systems Biology" Metabolites 2, no. 1: 242-253. https://doi.org/10.3390/metabo2010242
APA StyleWu, M., & Chan, C. (2012). Human Metabolic Network: Reconstruction, Simulation, and Applications in Systems Biology. Metabolites, 2(1), 242-253. https://doi.org/10.3390/metabo2010242