Shotgun Lipidomics by Sequential Precursor Ion Fragmentation on a Hybrid Quadrupole Time-of-Flight Mass Spectrometer

Abstract
:
1. Introduction
2. Results and Discussion
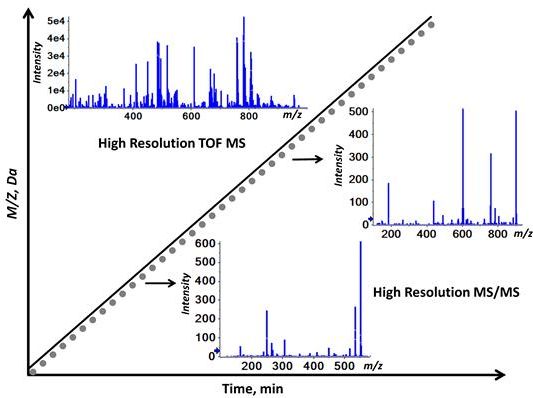
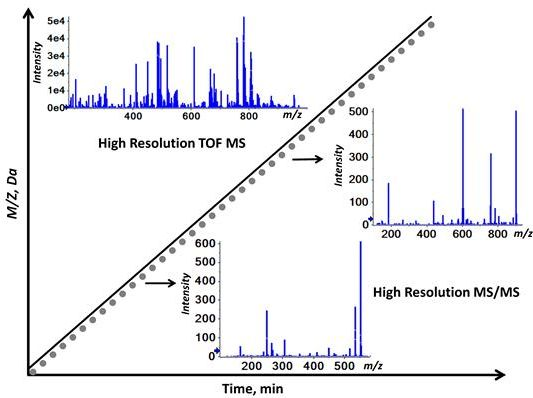
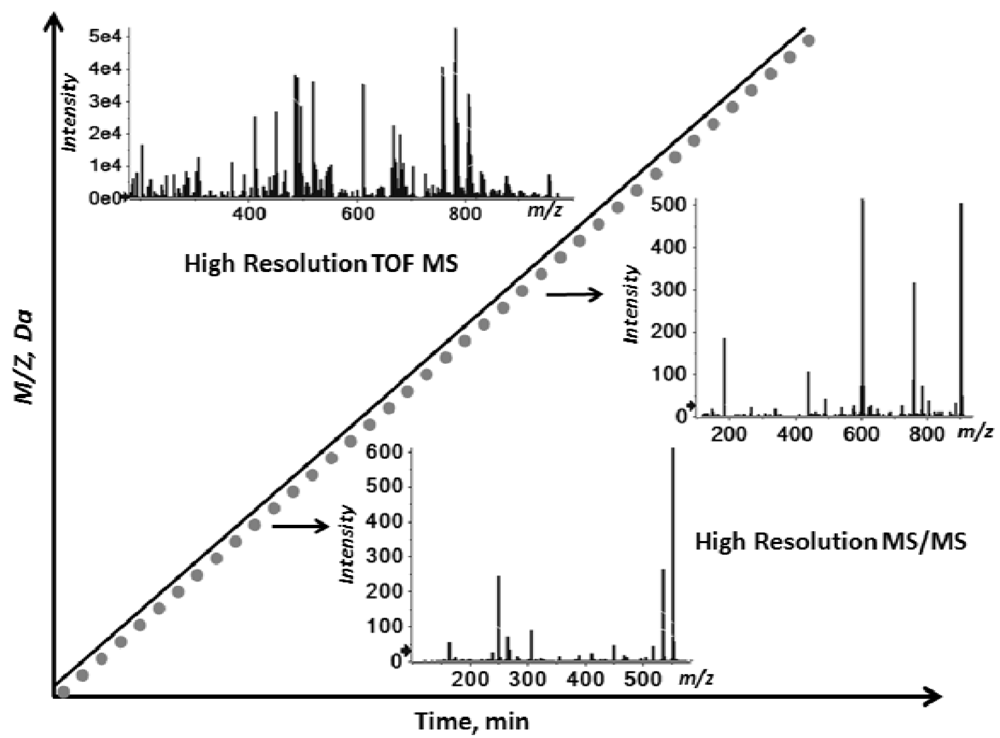
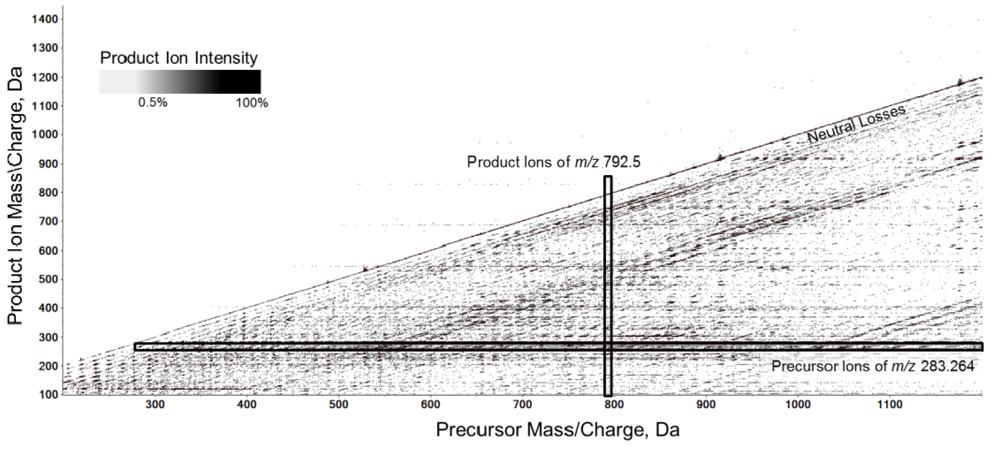
2.1. Workflow of the Sequential Precursor Ion Fragmentation
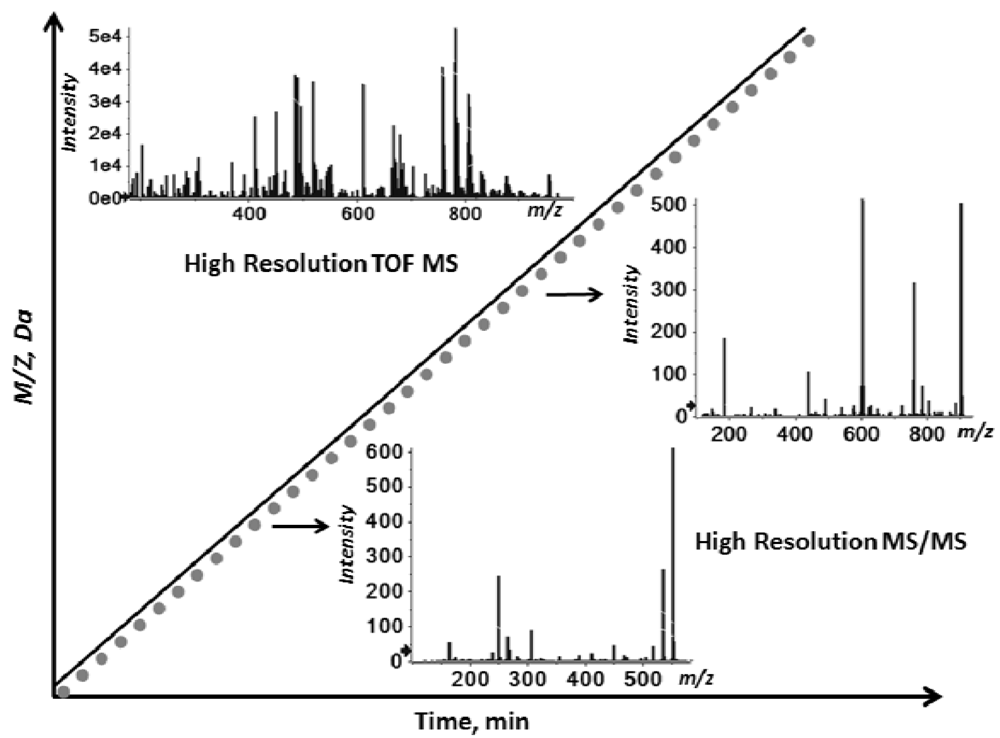
2.2. Overview of the Experimental Setup
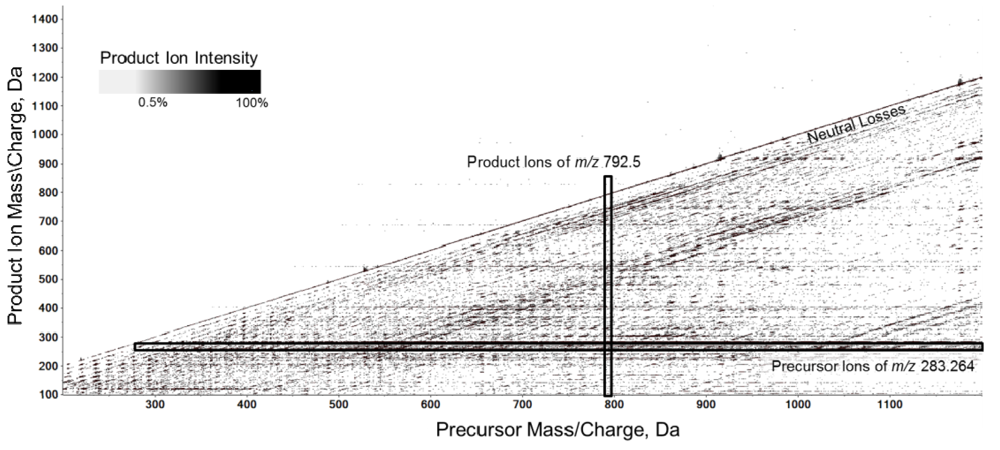
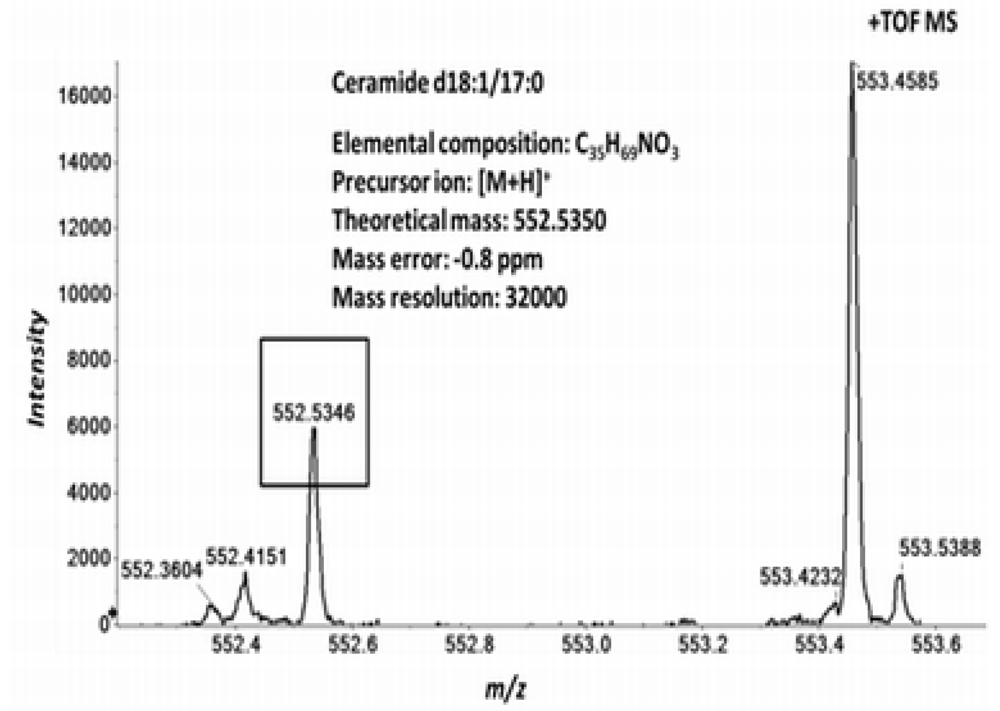
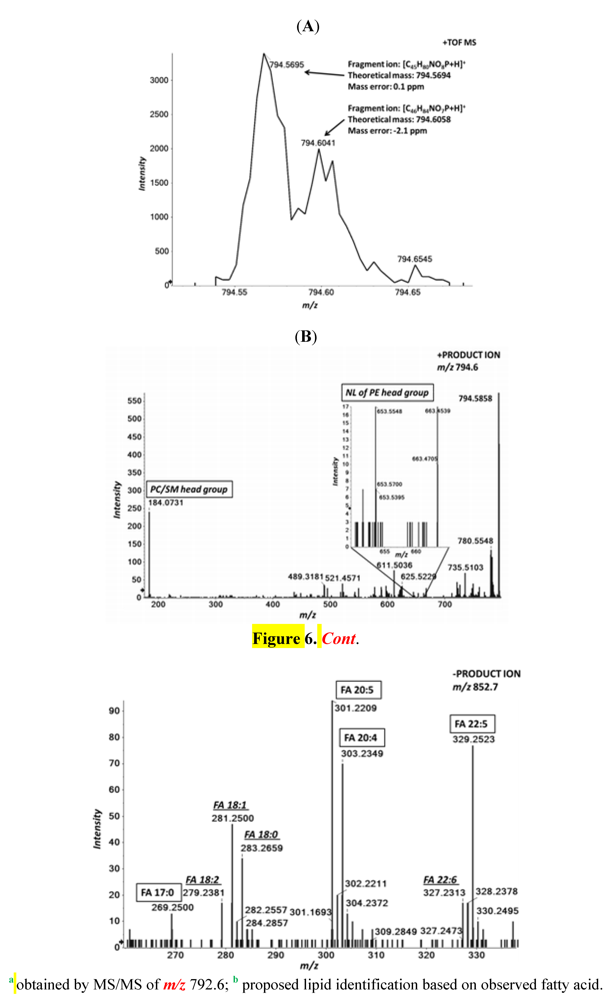
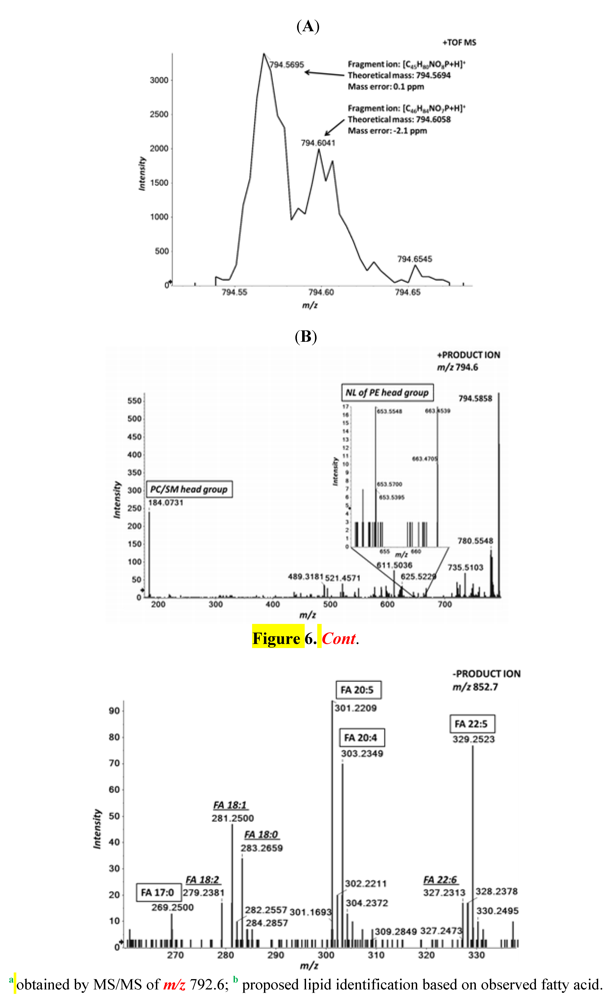
2.3. High Resolution Accurate MS Improves Lipid Identification but Does Not Reveal Underlying Molecular Species
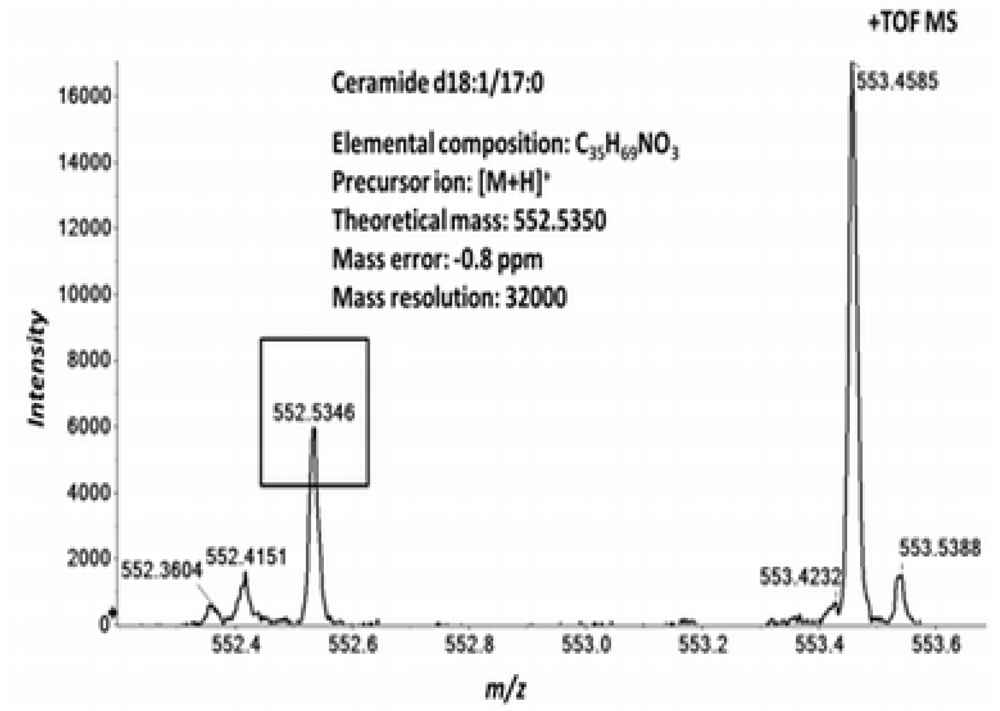
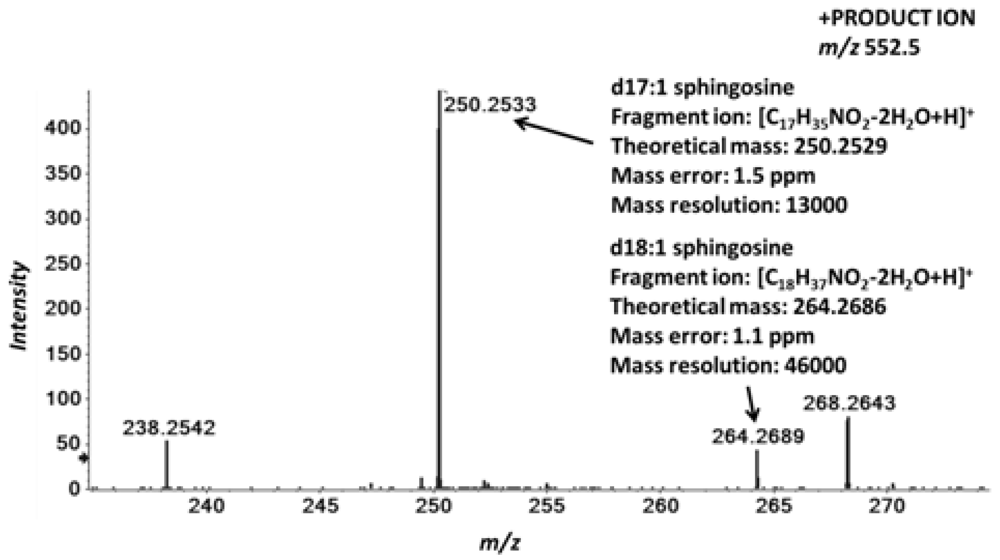
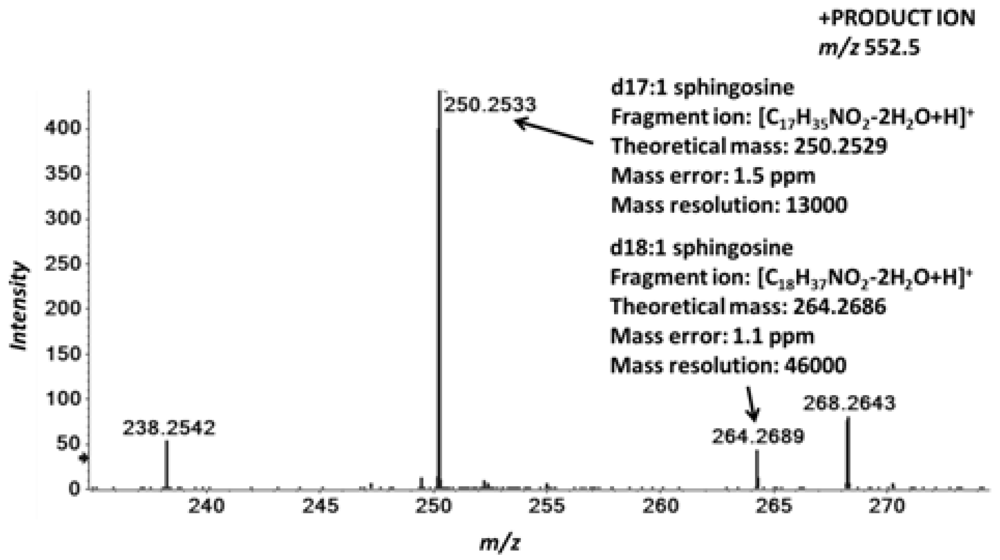
2.4. Accurate Mass Product Ion Data Are Required to Confirm Molecular Lipids
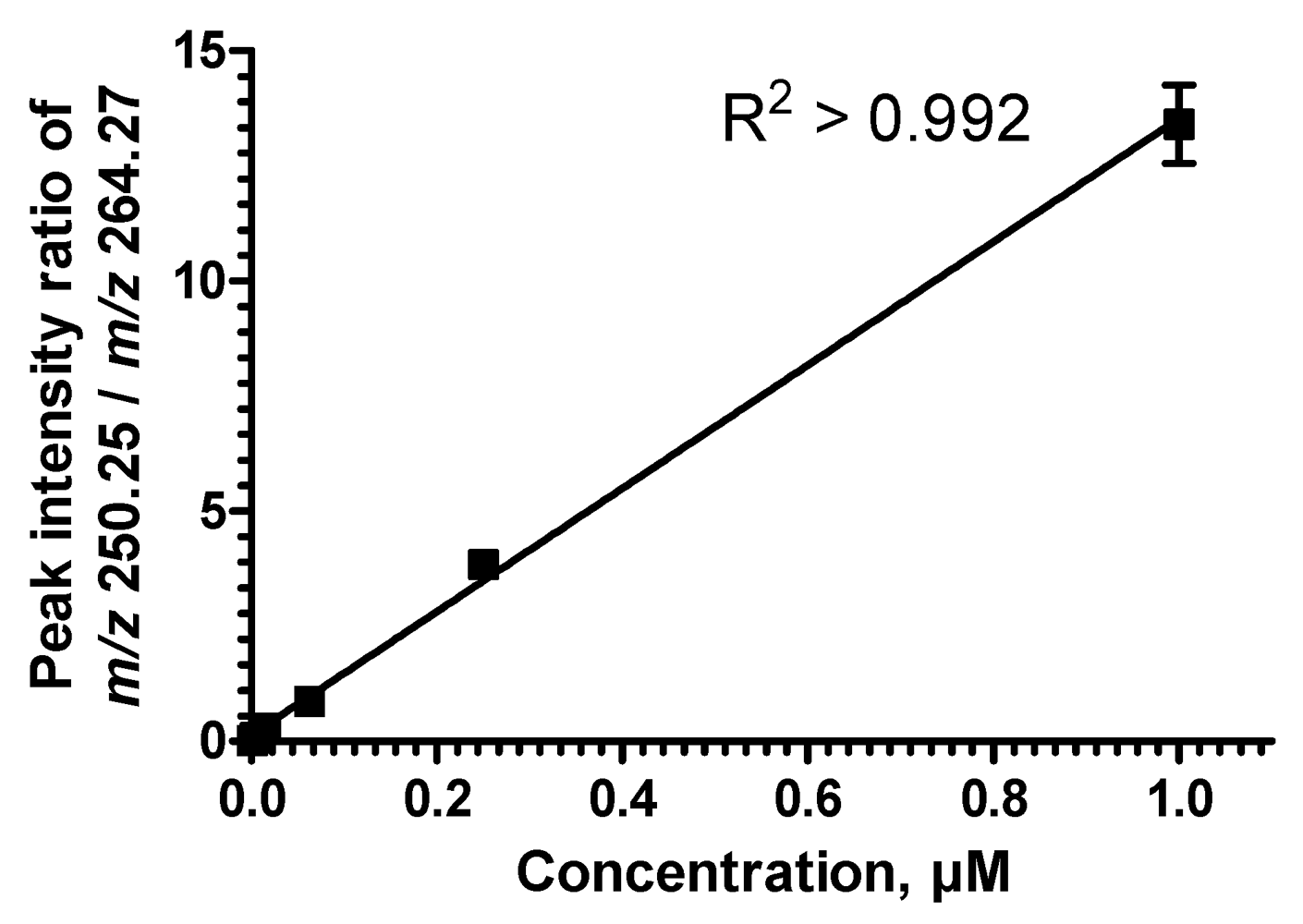
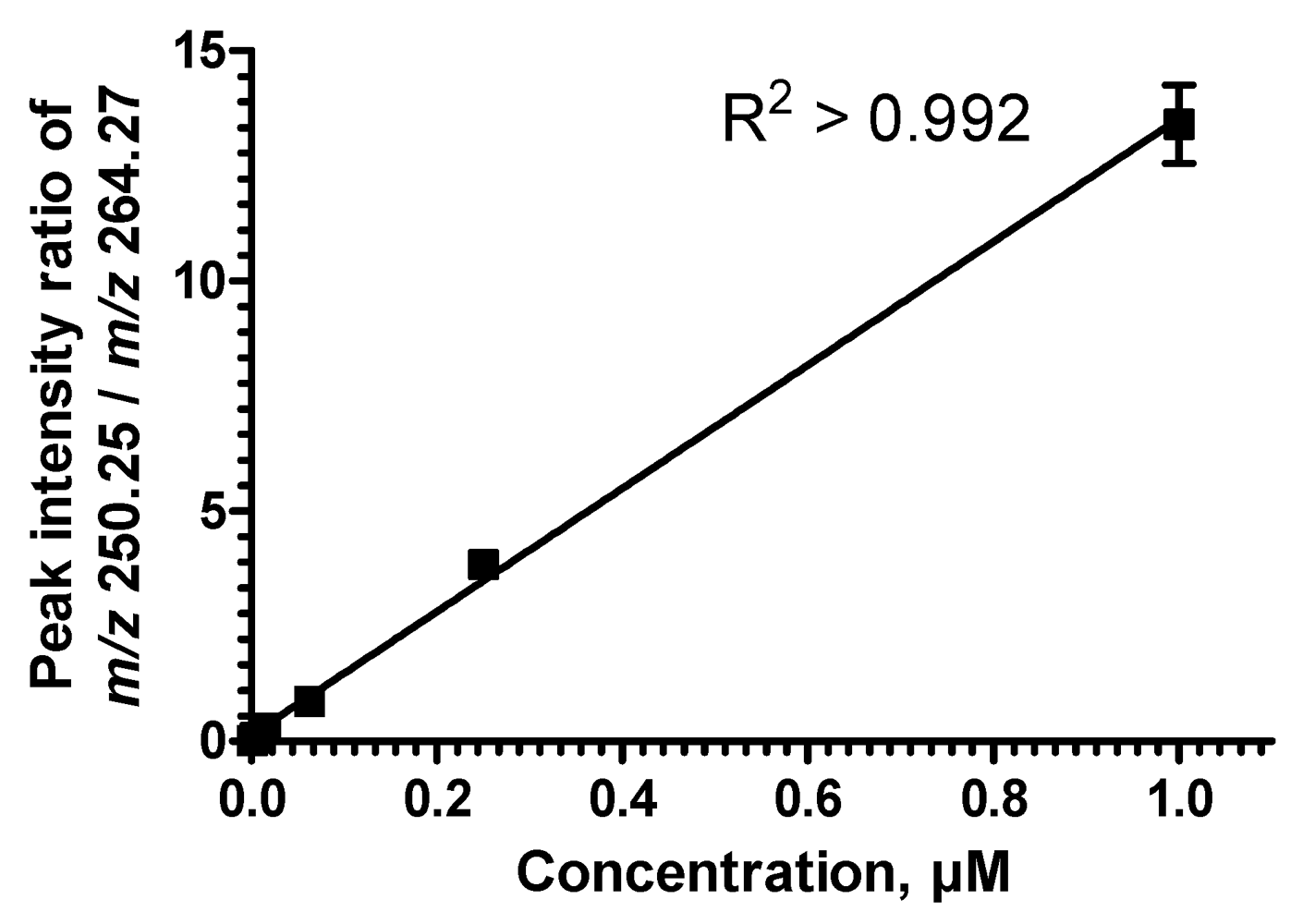
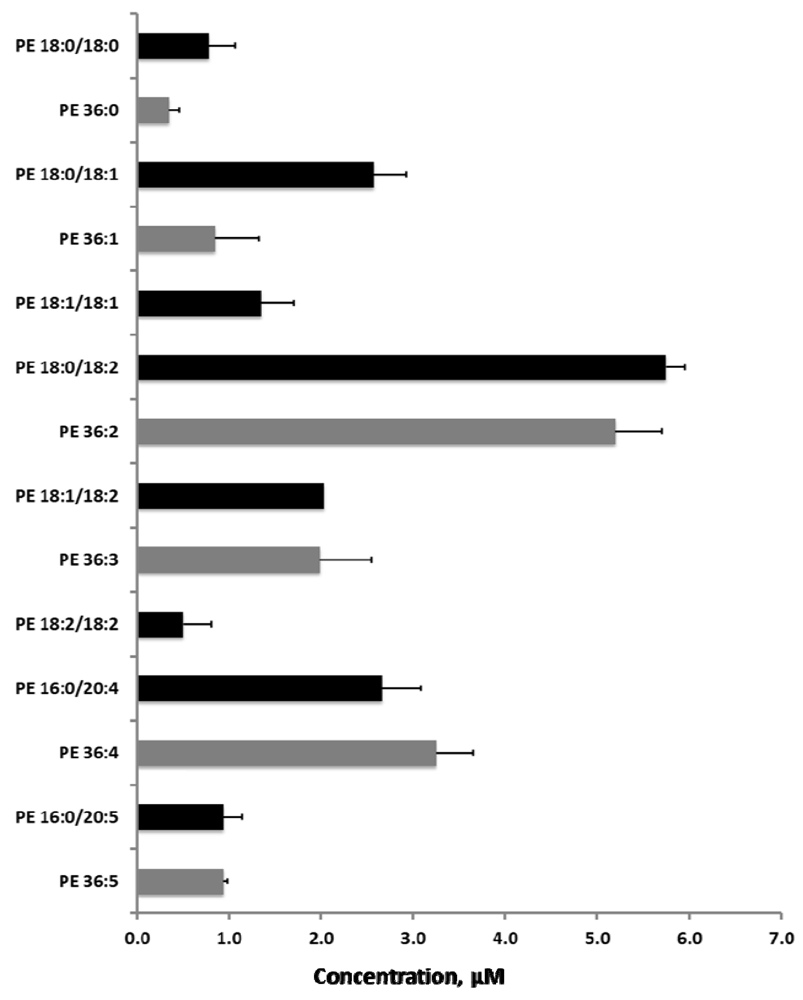
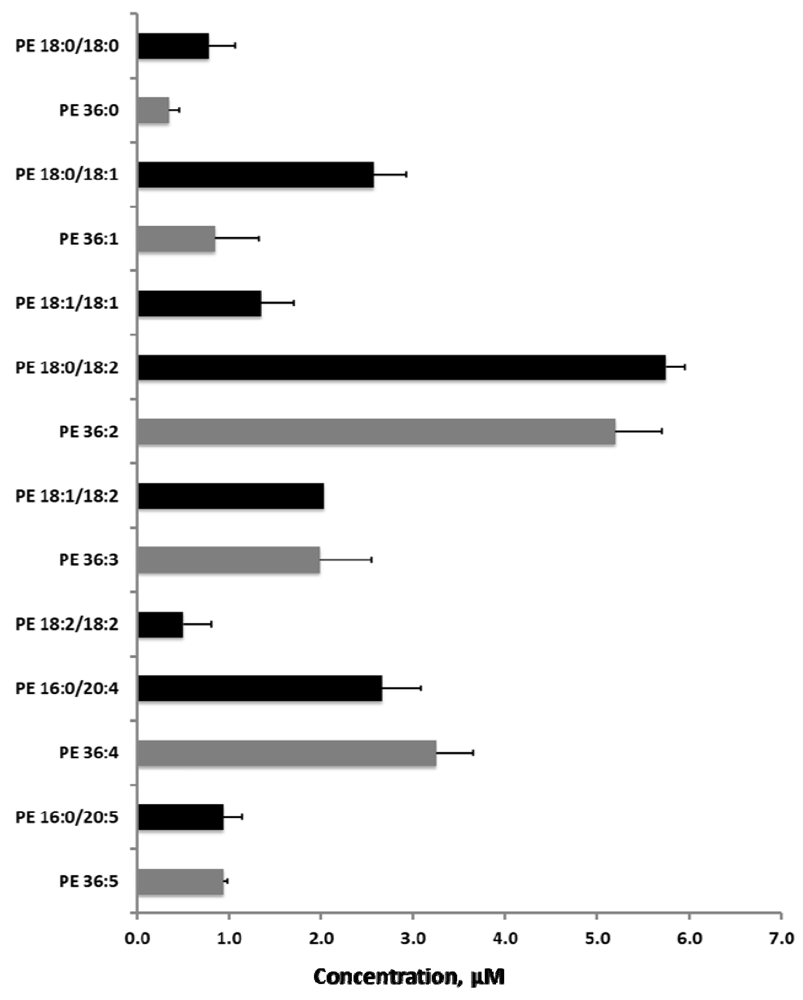
2.5. Sequential Precursor Ion Fragmentation Delivers Quantitative Data
2.6. Reproducibility of the Sequential Precursor Ion FragmentationTechnique
2.7. MS/MSALL Accompanied with High Resolution MS Increases the Confidence in Endogenous Lipid Identification

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Synthetic standard | Measured m/z | Peak intensity (cps) | Mean (n = 3) | CV (%) | ||
|---|---|---|---|---|---|---|
| Sample A | Sample B | Sample C | ||||
| LPC 17:0 | 184.07 | 1070 | 1165 | 966 | 1067 | 9.33 |
| D6-CE 18:0 | 369.35 | 810 | 916 | 896 | 874 | 6.44 |
| PC 17:0/17:0 | 184.07 | 12360 | 12056 | 12775 | 12397 | 2.91 |
| D3-GlcCER d18:1/16:0 | 264.27 | 282 | 302 | 282 | 288 | 4.00 |
| LPC 17:0 | 269.25 | 2875 | 2893 | 3496 | 3088 | 11.45 |
| PC 17:0/17:0 | 269.25 | 3559 | 4211 | 3821 | 3864 | 8.49 |
| PE 17:0/17:0 | 269.25 | 8682 | 8536 | 8629 | 8616 | 0.86 |
| PS 17:0717:0 | 269.25 | 3189 | 3332 | 3461 | 3327 | 4.09 |
| Molecular Ion | Calculated mass | PC 17:0/20:5 | PC P-18:0/20:4 | PC O-18:0/20:5 | PC O-16:0/22:5 | PE 18:0/22:5 |
|---|---|---|---|---|---|---|
| TOF MS | ||||||
| [M+H]+ | 794.5694/794.6058 | 794.5674 | 794.6037 | 794.6037 | 794.6037 | 794.5674 |
| [M+CH3COO]− | 852.5760/852.6124 | 852.5745 | 852.5745 b | 852.5745 b | 852.5745 b | 792.5408 |
| [C5H15NO4P]+ | 184.0733 | 184.0731 | 184.0731 | 184.0731 | 184.0731 | |
| [M+H−C2H8NO4P]+ | 653.5503 | 653.5414 | ||||
| MS/MS | ||||||
| [C17H33O2]− | 269.2486 | 269.2534 | ||||
| [C18H35O2]− | 283.2642 | 283.2689 a | ||||
| [C20H29O2]− | 301.2173 | 301.2209 | 301.2209 | |||
| [C20H31O2]− | 303.233 | 303.2373 | ||||
| [C22H33O2]− | 329.2486 | 329.2506 | 329.2546 a |
2.8. MS/MSALL is a Valid Methodology for the Assessment of Molecular Lipidomes

3. Experimental
3.1. Materials
3.2. Sample Preparation and Extraction
3.3. MS and MS/MS Acquisitions on Hybrid Quadrupole Time-of-Flight
3.4. Accurate MassData Processing
3.5. QqQ Shotgun Lipidomics Analyses
4. Conclusions
Supplementary Materials
Acknowledgments
Conflict of Interest
References and Notes
- Han, X.; Gross, R.W. Global analyses of cellular lipidomes directly from crude extracts of biological samples by ESI mass spectrometry: A bridge to lipidomics. J. Lipid Res. 2003, 44, 1071–1079. [Google Scholar] [CrossRef]
- Shevchenko, A.; Simons, K. Lipidomics: Coming to grips with lipid diversity. Nat. Rev. Mol. Cell. Biol. 2010, 11, 593–598. [Google Scholar] [CrossRef]
- Lingwood, D.; Binnington, B.; Rog, T.; Vattulainen, I.; Grzybek, M.; Coskun, U.; Lingwood, C.A.; Simons, K. Cholesterol modulates glycolipid conformation and receptor activity. Nat. Chem. Biol. 2011, 7, 260–262. [Google Scholar]
- Menuz, V.; Howell, K.S.; Gentina, S.; Epstein, S.; Riezman, I.; Fornallaz-Mulhauser, M.; Hengartner, M.O.; Gomez, M.; Riezman, H.; Martinou, J.C. Protection of C. elegans from anoxia by HYL-2 ceramide synthase. Science 2009, 324, 381–384. [Google Scholar]
- Perman, J.C.; Bostrom, P.; Lindbom, M.; Lidberg, U.; StAhlman, M.; Hagg, D.; Lindskog, H.; Scharin Tang, M.; Omerovic, E.; Mattsson Hulten, L.; et al. The VLDL receptor promotes lipotoxicity and increases mortality in mice following an acute myocardial infarction. J. Clin. Invest. 2011, 121, 2625–2640. [Google Scholar]
- Quehenberger, O.; Armando, A.M.; Brown, A.H.; Milne, S.B.; Myers, D.S.; Merrill, A.H.; Bandyopadhyay, S.; Jones, K.N.; Kelly, S.; Shaner, R.L.; et al. Lipidomics reveals a remarkable diversity of lipids in human plasma. J. Lipid Res. 2010, 51, 3299–3305. [Google Scholar] [CrossRef]
- Jung, H.R.; Sylvanne, T.; Koistinen, K.M.; Tarasov, K.; Kauhanen, D.; Ekroos, K. High throughput quantitative molecular lipidomics. Biochim. Biophys. Acta 1811, 925–934. [Google Scholar]
- Taguchi, R.; Houjou, T.; Nakanishi, H.; Yamazaki, T.; Ishida, M.; Imagawa, M.; Shimizu, T. Focused lipidomics by tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2005, 823, 26–36. [Google Scholar] [CrossRef]
- Sullards, M.C. Analysis of sphingomyelin, glucosylceramide, ceramide, sphingosine, and sphingosine 1-phosphate by tandem mass spectrometry. Methods Enzymol. 2000, 312, 32–45. [Google Scholar] [CrossRef]
- Han, X.; Gross, R.W. Electrospray ionization mass spectroscopic analysis of human erythrocyte plasma membrane phospholipids. Proc. Natl. Acad. Sci. USA 1994, 91, 10635–10639. [Google Scholar] [CrossRef]
- Brugger, B.; Erben, G.; Sandhoff, R.; Wieland, F.T.; Lehmann, W.D. Quantitative analysis of biological membrane lipids at the low picomole level by nano-electrospray ionization tandem mass spectrometry. Proc. Natl. Acad. Sci. USA 1997, 94, 2339–2344. [Google Scholar] [CrossRef]
- Rhee, E.P.; Cheng, S.; Larson, M.G.; Walford, G.A.; Lewis, G.D.; McCabe, E.; Yang, E.; Farrell, L.; Fox, C.S.; O'Donnell, C.J.; et al. Lipid profiling identifies a triacylglycerol signature of insulin resistance and improves diabetes prediction in humans. J. Clin. Invest. 2011, 121, 1402–1411. [Google Scholar] [CrossRef]
- Meikle, P.J.; Wong, G.; Tsorotes, D.; Barlow, C.K.; Weir, J.M.; Christopher, M.J.; MacIntosh, G.L.; Goudey, B.; Stern, L.; Kowalczyk, A.; et al. Plasma lipidomic analysis of stable and unstable coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2723–2732. [Google Scholar] [CrossRef]
- Hiukka, A.; Stahlman, M.; Pettersson, C.; Levin, M.; Adiels, M.; Teneberg, S.; Leinonen, E.S.; Hulten, L.M.; Wiklund, O.; Oresic, M.; et al. ApoCIII-enriched LDL in type 2 diabetes displays altered lipid composition, increased susceptibility for sphingomyelinase, and increased binding to biglycan. Diabetes 2009, 58, 2018–2026. [Google Scholar] [CrossRef]
- Ejsing, C.S.; Sampaio, J.L.; Surendranath, V.; Duchoslav, E.; Ekroos, K.; Klemm, R.W.; Simons, K.; Shevchenko, A. Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc. Natl. Acad. Sci. USA 2009, 106, 2136–2141. [Google Scholar]
- Linden, D.; William-Olsson, L.; Ahnmark, A.; Ekroos, K.; Hallberg, C.; Sjogren, H.P.; Becker, B.; Svensson, L.; Clapham, J.C.; Oscarsson, J.; Schreyer, S. Liver-directed overexpression of mitochondrial glycerol-3-phosphate acyltransferase results in hepatic steatosis, increased triacylglycerol secretion and reduced fatty acid oxidation. FASEB J. 2006, 20, 434–443. [Google Scholar] [CrossRef]
- Geiger, T.; Cox, J.; Mann, M. Proteomics on an Orbitrap benchtop mass spectrometer using all-ion fragmentation. Mol. Cell. Proteomics 2010, 9, 2252–2261. [Google Scholar] [CrossRef]
- Chen, Y.Q.; Kuo, M.S.; Li, S.; Bui, H.H.; Peake, D.A.; Sanders, P.E.; Thibodeaux, S.J.; Chu, S.; Qian, Y.W.; Zhao, Y.; et al. AGPAT6 is a novel microsomal glycerol-3-phosphate acyltransferase. J. Biol. Chem. 2008, 283, 10048–10057. [Google Scholar]
- Liebisch, G.; Drobnik, W.; Reil, M.; Trumbach, B.; Arnecke, R.; Olgemoller, B.; Roscher, A.; Schmitz, G. Quantitative measurement of different ceramide species from crude cellular extracts byelectrospray ionization tandem mass spectrometry (ESI-MS/MS). J. Lipid Res. 1999, 40, 1539–1546. [Google Scholar]
- Poad, B.L.; Pham, H.T.; Thomas, M.C.; Nealon, J.R.; Campbell, J.L.; Mitchell, T.W.; Blanksby, S.J. Ozone-induced dissociation on a modified tandem linear ion-trap: Observations of different reactivity for isomeric lipids. J. Am. Soc. Mass Spectrom. 2010, 21, 1989–1999. [Google Scholar] [CrossRef]
- Ekroos, K.; Ejsing, C.S.; Bahr, U.; Karas, M.; Simons, K.; Shevchenko, A. Charting molecular composition of phosphatidylcholines by fatty acid scanning and ion trap MS3 fragmentation. J. Lipid Res. 2003, 44, 2181–2192. [Google Scholar] [CrossRef]
- Merrill, A.H., Jr.; Sullards, M.C.; Allegood, J.C.; Kelly, S.; Wang, E. Sphingolipidomics: High-throughput, structure-specific, and quantitative analysis of sphingolipids by liquid chromatography tandem mass spectromet. Methods 2005, 36, 207–224. [Google Scholar] [CrossRef]
- Ekroos, K.; Chernushevich, I.V.; Simons, K.; Shevchenko, A. Quantitative profiling of phospholipids by multiple precursor ion scanning on a hybrid quadrupole time-of-flight mass spectrometer. Anal. Chem. 2002, 74, 941–949. [Google Scholar] [CrossRef]
- Stahlman, M.; Ejsing, C.S.; Tarasov, K.; Perman, J.; Boren, J.; Ekroos, K. High-throughput shotgun lipidomics by quadrupole time-of-flight mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 2664–2672. [Google Scholar] [CrossRef]
- Ejsing, C.S.; Duchoslav, E.; Sampaio, J.; Simons, K.; Bonner, R.; Thiele, C.; Ekroos, K.; Shevchenko, A. Automated identification and quantification of glycerophospholipid molecular species by multiple precursor ion scanning. Anal. Chem. 2006, 78, 6202–6214. [Google Scholar] [CrossRef]
- Quehenberger, O.; Dennis, E.A. The human plasma lipidome. N. Engl. J. Med. 2011, 365, 1812–1823. [Google Scholar] [CrossRef]
- Graessler, J.; Schwudke, D.; Schwarz, P.E.; Herzog, R.; Shevchenko, A.; Bornstein, S.R. Top-down lipidomics reveals ether lipid deficiency in blood plasma of hypertensive patients. PLoS One 2009, 4, e6261. [Google Scholar]
- Oresic, M.; Simell, S.; Sysi-Aho, M.; Nanto-Salonen, K.; Seppanen-Laakso, T.; Parikka, V.; Katajamaa, M.; Hekkala, A.; Mattila, I.; Keskinen, P.; et al. Dysregulation of lipid and amino acid metabolism precedes islet autoimmunity in children who later progress to type 1 diabetes. J. Exp. Med. 2008, 205, 2975–2984. [Google Scholar] [CrossRef]
- McAnoy, A.M.; Wu, C.C.; Murphy, R.C. Direct qualitative analysis of triacylglycerols by electrospray mass spectrometry using a linear ion trap. J. Am. Soc. Mass Spectrom. 2005, 16, 1498–1509. [Google Scholar] [CrossRef]
- Trimpin, S.; Tan, B.; Bohrer, B.C.; O'Dell, D.K.; Merenbloom, S.I.; Pazos, M.X.; Clemmer, D.E.; Walker, J.M. Profiling of phospholipids and related lipid structures using multidimensional ion mobility spectrometry-mass spectrometry. Int. J. Mass Spectrom. 2009, 287, 58–69. [Google Scholar] [CrossRef]
- Stahlman, M.; Pham, H.T.; Adiels, M.; Mitchell, T.W.; Blanksby, S.J.; Fagerberg, B.; Ekroos, K.; Boren, J. Clinical dyslipidaemia is associated with changes in the lipid composition and inflammatory properties of apolipoprotein-B-containing lipoproteins from women with type 2 diabetes. Diabetologia 2012. [Google Scholar]
Supplementary Files
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Simons, B.; Kauhanen, D.; Sylvänne, T.; Tarasov, K.; Duchoslav, E.; Ekroos, K. Shotgun Lipidomics by Sequential Precursor Ion Fragmentation on a Hybrid Quadrupole Time-of-Flight Mass Spectrometer. Metabolites 2012, 2, 195-213. https://doi.org/10.3390/metabo2010195
Simons B, Kauhanen D, Sylvänne T, Tarasov K, Duchoslav E, Ekroos K. Shotgun Lipidomics by Sequential Precursor Ion Fragmentation on a Hybrid Quadrupole Time-of-Flight Mass Spectrometer. Metabolites. 2012; 2(1):195-213. https://doi.org/10.3390/metabo2010195
Chicago/Turabian StyleSimons, Brigitte, Dimple Kauhanen, Tuulia Sylvänne, Kirill Tarasov, Eva Duchoslav, and Kim Ekroos. 2012. "Shotgun Lipidomics by Sequential Precursor Ion Fragmentation on a Hybrid Quadrupole Time-of-Flight Mass Spectrometer" Metabolites 2, no. 1: 195-213. https://doi.org/10.3390/metabo2010195
APA StyleSimons, B., Kauhanen, D., Sylvänne, T., Tarasov, K., Duchoslav, E., & Ekroos, K. (2012). Shotgun Lipidomics by Sequential Precursor Ion Fragmentation on a Hybrid Quadrupole Time-of-Flight Mass Spectrometer. Metabolites, 2(1), 195-213. https://doi.org/10.3390/metabo2010195