
Bile Acid Composition and Transcriptome Analysis of the Liver and Small Intestine in Different Species

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Experimental Animals and Samples
2.3. Metabolome Processing and Analyses
2.4. RNA Extraction and Sequencing
2.5. Transcriptome Analysis
3. Results
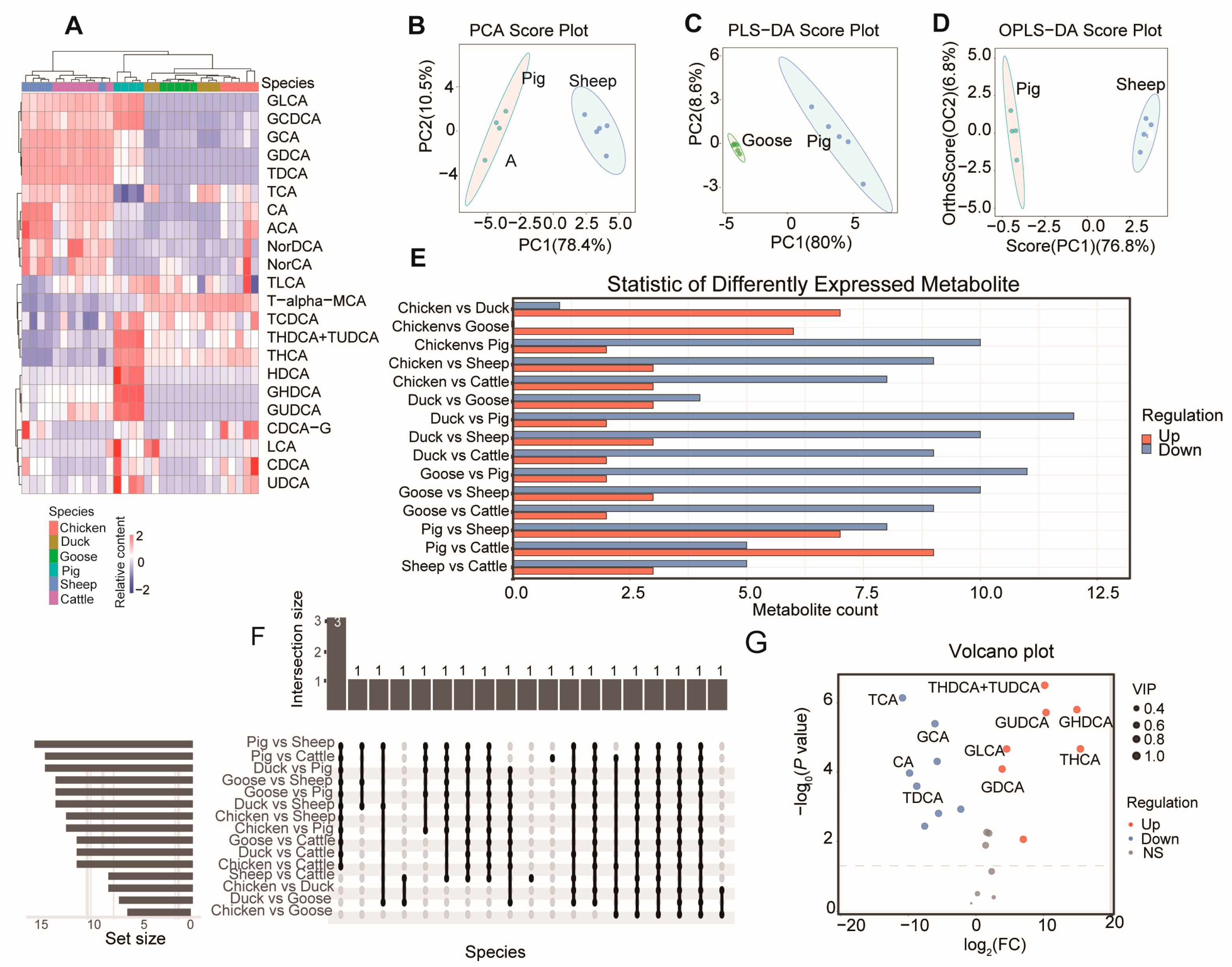
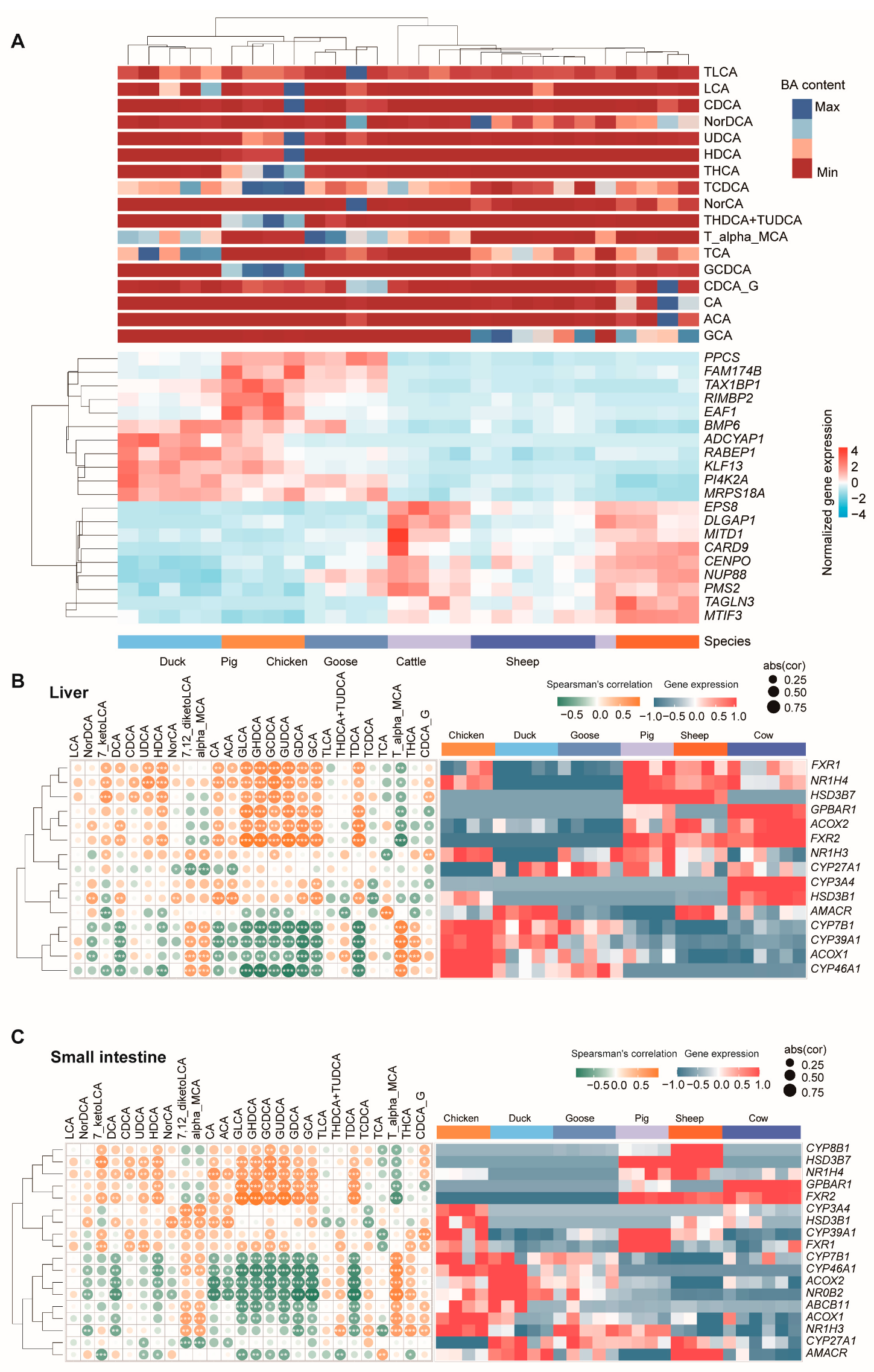
3.1. Analysis of the Bile Acid Composition of Different Species
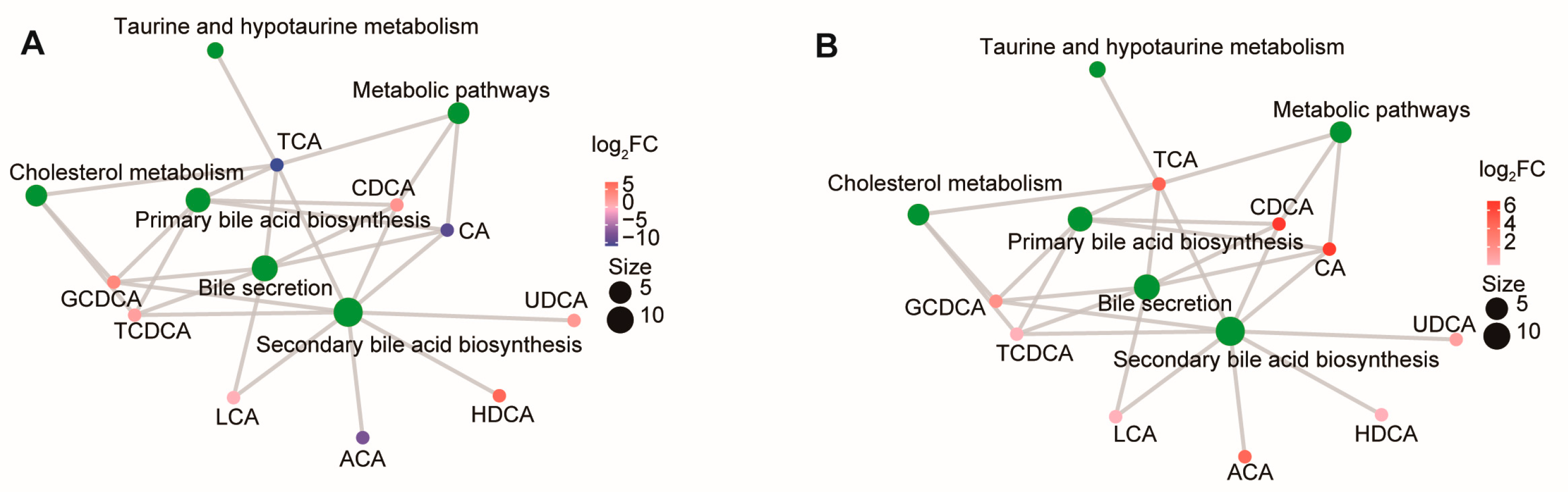
3.2. Differentiated BA Metabolites and KEGG Pathway
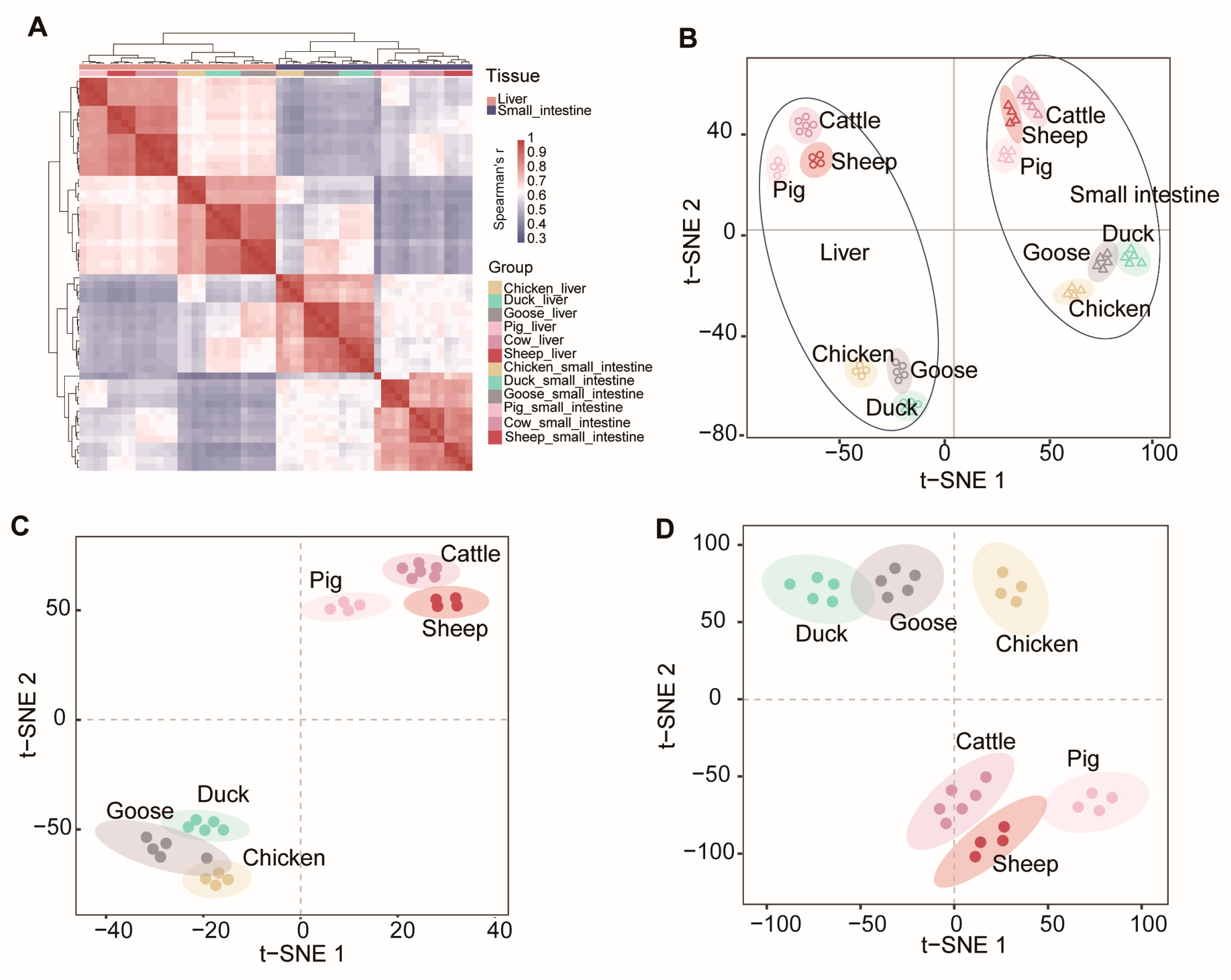
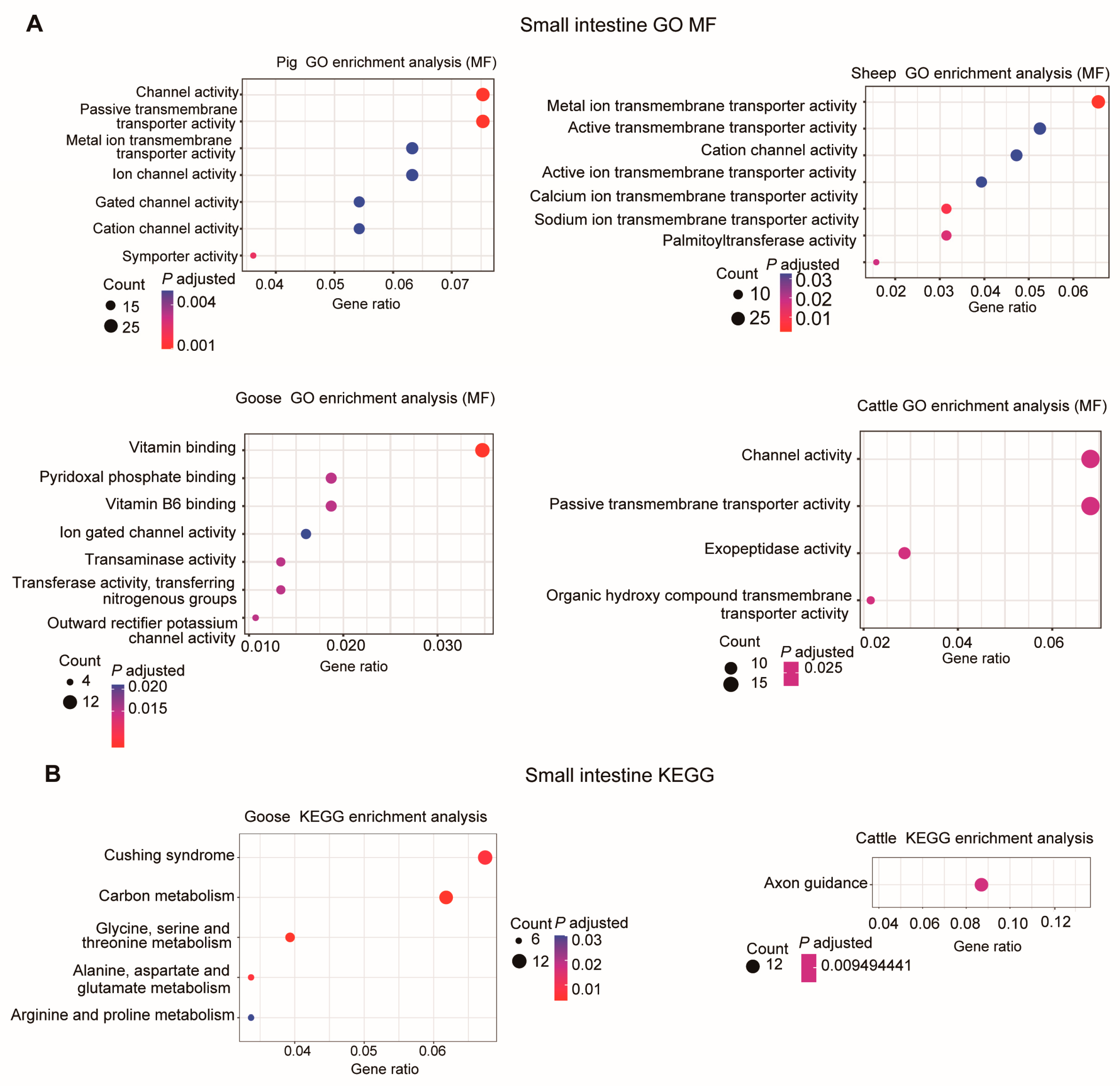
3.3. Analysis of the Transcriptome of the Liver and Small Intestine among Different Species
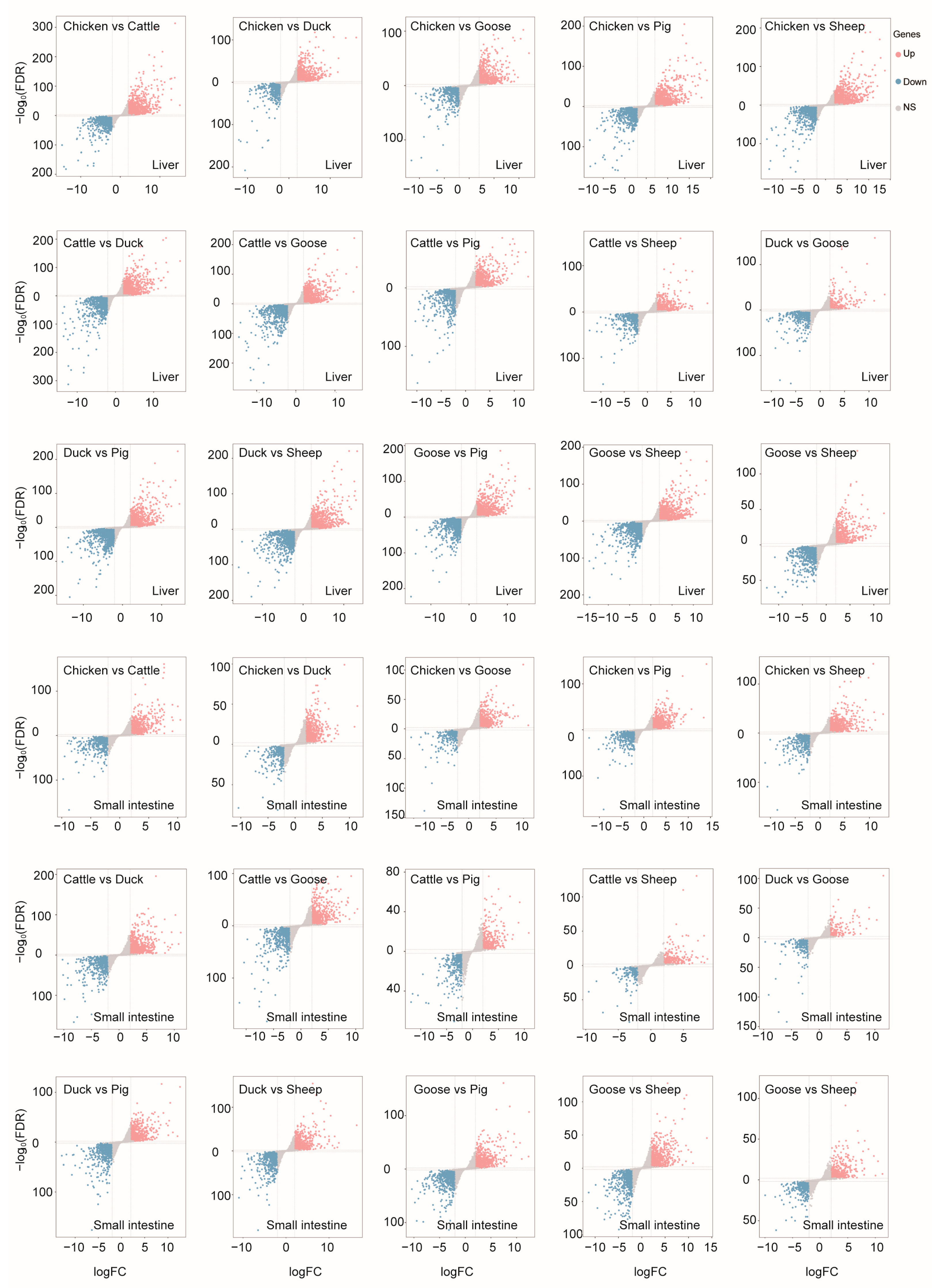
3.4. Differentially Expressed Genes in Two Tissues among Six Species
3.5. Correlation Analysis between Gene Expression and BA Content
4. Discussion
5. Limitations of the Study
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Łuczykowski, K.; Warmuzińska, N.; Bojko, B. Current approaches to the analysis of bile and the determination of bile acids in various biological matrices as supportive tools to traditional diagnostic testing for liver dysfunction and biliary diseases. TrAC Trends Anal. Chem. 2021, 142, 116307. [Google Scholar] [CrossRef]
- Farina, A.; Dumonceau, J.M.; Delhaye, M.; Frossard, J.L.; Hadengue, A.; Hochstrasser, D.F.; Lescuyer, P. A step further in the analysis of human bile proteome. J. Proteome Res. 2011, 10, 2047–2063. [Google Scholar] [CrossRef] [PubMed]
- Shansky, Y.; Bespyatykh, J. Bile Acids: Physiological Activity and Perspectives of Using in Clinical and Laboratory Diagnostics. Molecules 2022, 27, 7830. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Li, B.; Lin, D.; Miao, Y.R.; Luo, T.; Yue, T.; Luo, Q.; Guo, A.Y.; Zhang, Z. scLiverDB: A Database of Human and Mouse Liver Transcriptome Landscapes at Single-Cell Resolution. Small Methods 2023, 7, e2201421. [Google Scholar] [CrossRef] [PubMed]
- Jiao, T.-Y.; Ma, Y.-D.; Guo, X.-Z.; Ye, Y.-F.; Xie, C. Bile acid and receptors: Biology and drug discovery for nonalcoholic fatty liver disease. Acta Pharmacol. Sin. 2022, 43, 1103–1119. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Bambha, K. Bile acid receptors and nonalcoholic fatty liver disease. World J. Hepatol. 2015, 7, 2811–2818. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chiang, J.Y.L. Bile acid-based therapies for non-alcoholic steatohepatitis and alcoholic liver disease. Hepatobiliary Surg. Nutr. 2020, 9, 152–169. [Google Scholar] [CrossRef] [PubMed]
- Haslewood, G.A. Bile salt evolution. J. Lipid Res. 1967, 8, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F.; Hagey, L.R.; Krasowski, M.D. Bile salts of vertebrates: Structural variation and possible evolutionary significance. J. Lipid. Res. 2010, 51, 226–246. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Cheng, F.; Li, W.; Yu, Q.; Ma, C.; Zou, Y.; Xu, T.; Liu, S.; Zhang, S.; Wang, Q. Enhancement of anti-inflammatory effect of cattle bile by fermentation and its inhibition of neuroinflammation on microglia by inhibiting NLRP3 inflammasome. J. Biosci. Bioeng. 2022, 133, 146–154. [Google Scholar] [CrossRef]
- GB/T5915-2020; Compound Feed for Piglets and Growing and Fattening Pigs. State Administration for Market Regulation. Standardization Administration of the People’s Republic of China: Beijing, China, 2020.
- Trapp, A.L.; Taylor, R.F. Methods of euthanasia in poultry and food-producing animals. Vet. Clin. N. Am. Food Anim. Pract. 1986, 2, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Wen, X.; Meng, Q.; Wu, W.; Everaert, N.; Xie, J.; Zhang, H. Alteration in bile acids profile in Large White pigs during chronic heat exposure. J. Therm. Biol. 2019, 84, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Smith, M.D.; Mische, L.; Harrington, E.; Fitzgerald, K.C.; Martin, K.; Kim, S.; Reyes, A.A.; Gonzalez-Cardona, J.; Volsko, C.; et al. Bile acid metabolism is altered in multiple sclerosis and supplementation ameliorates neuroinflammation. J. Clin. Invest. 2020, 130, 3467–3482. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; An, Z.; Shi, C.; Li, P.; Liu, L. A sensitive and efficient method for simultaneous profiling of bile acids and fatty acids by UPLC-MS/MS. J. Pharm. Biomed. Anal. 2020, 178, 112815. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Shu, T.; Liu, G.; Mei, H.; Zhu, X.; Huang, X.; Zhang, L.; Jiang, Z. Quantitative profiling of 19 bile acids in rat plasma, liver, bile and different intestinal section contents to investigate bile acid homeostasis and the application of temporal variation of endogenous bile acids. J. Steroid Biochem. Mol. Biol. 2017, 172, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Thévenot, E.A.; Roux, A.; Xu, Y.; Ezan, E.; Junot, C. Analysis of the Human Adult Urinary Metabolome Variations with Age, Body Mass Index, and Gender by Implementing a Comprehensive Workflow for Univariate and OPLS Statistical Analyses. J. Proteome Res. 2015, 14, 3322–3335. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010, 38, 1767–1771. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef] [PubMed]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Gu, Y.; Zhou, X.; Jin, L.; Guan, J.; Liu, R.; Li, J.; Long, K.; Tian, S.; Che, T.; et al. Comparative transcriptomics of 5 high-altitude vertebrates and their low-altitude relatives. Gigascience 2017, 6, 1–9. [Google Scholar] [CrossRef]
- Brawand, D.; Soumillon, M.; Necsulea, A.; Julien, P.; Csárdi, G.; Harrigan, P.; Weier, M.; Liechti, A.; Aximu-Petri, A.; Kircher, M.; et al. The evolution of gene expression levels in mammalian organs. Nature 2011, 478, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Waxman, D.J.; Chang, T.K. An isocratic high-performance liquid chromatographic assay for CYP7A1-catalyzed cholesterol 7 alpha-hydroxylation. Methods Mol. Biol. 1998, 107, 169–173. [Google Scholar] [CrossRef]
- Dubrac, S.; Lear, S.R.; Ananthanarayanan, M.; Balasubramaniyan, N.; Bollineni, J.; Shefer, S.; Hyogo, H.; Cohen, D.E.; Blanche, P.J.; Krauss, R.M.; et al. Role of CYP27A in cholesterol and bile acid metabolism. J. Lipid Res. 2005, 46, 76–85. [Google Scholar] [CrossRef]
- Fan, L.; Joseph, J.F.; Durairaj, P.; Parr, M.K.; Bureik, M. Conversion of chenodeoxycholic acid to cholic acid by human CYP8B1. Biol. Chem. 2019, 400, 625–628. [Google Scholar] [CrossRef]
- Hosamani, S.; Chakraborty, S. Cholesterol Allosterically Modulates the Structure and Dynamics of the Taurocholate Export Pump (ABCB11). J. Phys. Chem. Lett. 2024, 15, 7901–7908. [Google Scholar] [CrossRef]
- Ticho, A.L.; Malhotra, P.; Dudeja, P.K.; Gill, R.K.; Alrefai, W.A. Intestinal Absorption of Bile Acids in Health and Disease. Compr. Physiol. 2019, 10, 21–56. [Google Scholar] [CrossRef]
- Ayewoh, E.N.; Czuba, L.C.; Nguyen, T.T.; Swaan, P.W. S-acylation status of bile acid transporter hASBT regulates its function, metabolic stability, membrane expression, and phosphorylation state. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183510. [Google Scholar] [CrossRef]
- Gan, L.; Pan, S.; Cui, J.; Bai, J.; Jiang, P.; He, Y. Functional analysis of the correlation between ABCB11 gene mutation and primary intrahepatic stone. Mol. Med. Rep. 2019, 19, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Kosters, A.; Frijters, R.J.; Schaap, F.G.; Vink, E.; Plösch, T.; Ottenhoff, R.; Jirsa, M.; De Cuyper, I.M.; Kuipers, F.; Groen, A.K. Relation between hepatic expression of ATP-binding cassette transporters G5 and G8 and biliary cholesterol secretion in mice. J. Hepatol. 2003, 38, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Fang, C.; Zhao, R.H.; Zou, L.; Miao, H.; Zhao, Y.Y. Bile acid metabolism in health and ageing-related diseases. Biochem. Pharmacol. 2024, 225, 116313. [Google Scholar] [CrossRef] [PubMed]
- Fleishman, J.S.; Kumar, S. Bile acid metabolism and signaling in health and disease: Molecular mechanisms and therapeutic targets. Signal Transduct. Target. Ther. 2024, 9, 97. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Y. [An overview of bile acid synthesis and its physiological and pathological functions]. Yi Chuan 2019, 41, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Browning, M.G.; Pessoa, B.M.; Khoraki, J.; Campos, G.M. Changes in Bile Acid Metabolism, Transport, and Signaling as Central Drivers for Metabolic Improvements After Bariatric Surgery. Curr. Obes. Rep. 2019, 8, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Yin, P.; Ma, Z.; Ma, Y.; Zhang, H.; Kong, H.; Zhu, Y. Characteristics of bile acids metabolism profile in the second and third trimesters of normal pregnancy. Metabolism 2019, 95, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.J.; Zhong, Y.S.; Weng, J.F.; Huang, H.H.; Hsieh, P.Y. Determination of bile acids in pig liver, pig kidney and bovine liver by gas chromatography-chemical ionization tandem mass spectrometry with total ion chromatograms and extraction ion chromatograms. J. Chromatogr. A 2011, 1218, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Kuang, J.; Wang, J.; Li, Y.; Li, M.; Zhao, M.; Ge, K.; Zheng, D.; Cheung, K.C.P.; Liao, B.; Wang, S.; et al. Hyodeoxycholic acid alleviates non-alcoholic fatty liver disease through modulating the gut-liver axis. Cell Metab. 2023, 35, 1752–1766.e1758. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; He, X.; Gao, X.; Liu, Q.; Zhao, Y.; Hong, Y.; Zhu, W.; Yan, J.; Li, Y.; Li, Y.; et al. Hyodeoxycholic acid ameliorates nonalcoholic fatty liver disease by inhibiting RAN-mediated PPARα nucleus-cytoplasm shuttling. Nat. Commun. 2023, 14, 5451. [Google Scholar] [CrossRef] [PubMed]
- Horvatits, T.; Drolz, A.; Roedl, K.; Rutter, K.; Ferlitsch, A.; Fauler, G.; Trauner, M.; Fuhrmann, V. Serum bile acids as marker for acute decompensation and acute-on-chronic liver failure in patients with non-cholestatic cirrhosis. Liver Int. 2017, 37, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, Z.; Huang, M.; Sun, X.; Liu, B.; Guo, Q.; Chang, Q.; Duan, Z. Taurocholic acid is an active promoting factor, not just a biomarker of progression of liver cirrhosis: Evidence from a human metabolomic study and in vitro experiments. BMC Gastroenterol. 2018, 18, 112. [Google Scholar] [CrossRef] [PubMed]
- Moazzam-Jazi, M.; Najd-Hassan-Bonab, L.; Masjoudi, S.; Tohidi, M.; Hedayati, M.; Azizi, F.; Daneshpour, M.S. Risk of type 2 diabetes and KCNJ11 gene polymorphisms: A nested case–control study and meta-analysis. Sci. Rep. 2022, 12, 20709. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; de Molliens, M.P.; Schneebeli, S.T.; Brewer, M.; Song, G.; Chatenet, D.; Braas, K.M.; May, V.; Li, J. Targeting the PAC1 Receptor for Neurological and Metabolic Disorders. Curr. Top. Med. Chem. 2019, 19, 1399–1417. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Gong, J.; Wu, L.; Lin, X.; Zhang, Y.; Lin, W.; Huang, H.; Zhu, C. ADCY3: The pivotal gene in classical ketogenic diet for the treatment of epilepsy. Front. Cell Neurosci. 2024, 18, 1305867. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.A.; Gomes, D.A.; Fiorotto, R.; Guerra, M.T.; Weerachayaphorn, J.; Bo, T.; Sessa, W.C.; Strazzabosco, M.; Nathanson, M.H. Molecular determinants of peri-apical targeting of inositol 1,4,5-trisphosphate receptor type 3 in cholangiocytes. Hepatol. Commun. 2022, 6, 2748–2764. [Google Scholar] [CrossRef] [PubMed]
- Meiring, S.; Meessen, E.C.E.; van Baar, A.C.G.; Holleman, F.; Nieuwdorp, M.; Olde Damink, S.W.; Schaap, F.G.; Vaz, F.M.; Groen, A.K.; Soeters, M.R.; et al. Duodenal mucosal resurfacing with a GLP-1 receptor agonist increases postprandial unconjugated bile acids in patients with insulin-dependent type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2022, 322, E132–E140. [Google Scholar] [CrossRef] [PubMed]
- Funabashi, M.; Grove, T.L.; Wang, M.; Varma, Y.; McFadden, M.E.; Brown, L.C.; Guo, C.; Higginbottom, S.; Almo, S.C.; Fischbach, M.A. A metabolic pathway for bile acid dehydroxylation by the gut microbiome. Nature 2020, 582, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Luo, C.; Wu, J.; Deng, Y.; Mu, X.; Zhang, T.; Yang, X.; Liu, Q.; Li, Z.; Tang, S.; et al. Ion channels and transporters regulate nutrient absorption in health and disease. J. Cell Mol. Med. 2023, 27, 2631–2642. [Google Scholar] [CrossRef] [PubMed]
- Zou, P.; Wang, L. Dietary pattern and hepatic lipid metabolism. Liver Res. 2023, 7, 275–284. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, D.; Zheng, T.; Yang, M.; Huang, Z.; Wang, T.; Wang, Q.; Chen, B. Bile Acid Composition and Transcriptome Analysis of the Liver and Small Intestine in Different Species. Metabolites 2024, 14, 451. https://doi.org/10.3390/metabo14080451
Qi D, Zheng T, Yang M, Huang Z, Wang T, Wang Q, Chen B. Bile Acid Composition and Transcriptome Analysis of the Liver and Small Intestine in Different Species. Metabolites. 2024; 14(8):451. https://doi.org/10.3390/metabo14080451
Chicago/Turabian StyleQi, Dongming, Tingting Zheng, Maosen Yang, Zhiying Huang, Tao Wang, Qiang Wang, and Binlong Chen. 2024. "Bile Acid Composition and Transcriptome Analysis of the Liver and Small Intestine in Different Species" Metabolites 14, no. 8: 451. https://doi.org/10.3390/metabo14080451
APA StyleQi, D., Zheng, T., Yang, M., Huang, Z., Wang, T., Wang, Q., & Chen, B. (2024). Bile Acid Composition and Transcriptome Analysis of the Liver and Small Intestine in Different Species. Metabolites, 14(8), 451. https://doi.org/10.3390/metabo14080451