


A Novel UHPLC-MS/MS Based Method for Isomeric Separation and Quantitative Determination of Cyanogenic Glycosides in American Elderberry

 , ,
, , 
Abstract

1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. North American Elderberry Samples
2.3. Sample Preparation
2.4. UHPLC-MS/MS Analysis
2.5. Method Validation
2.6. Statistical Analysis
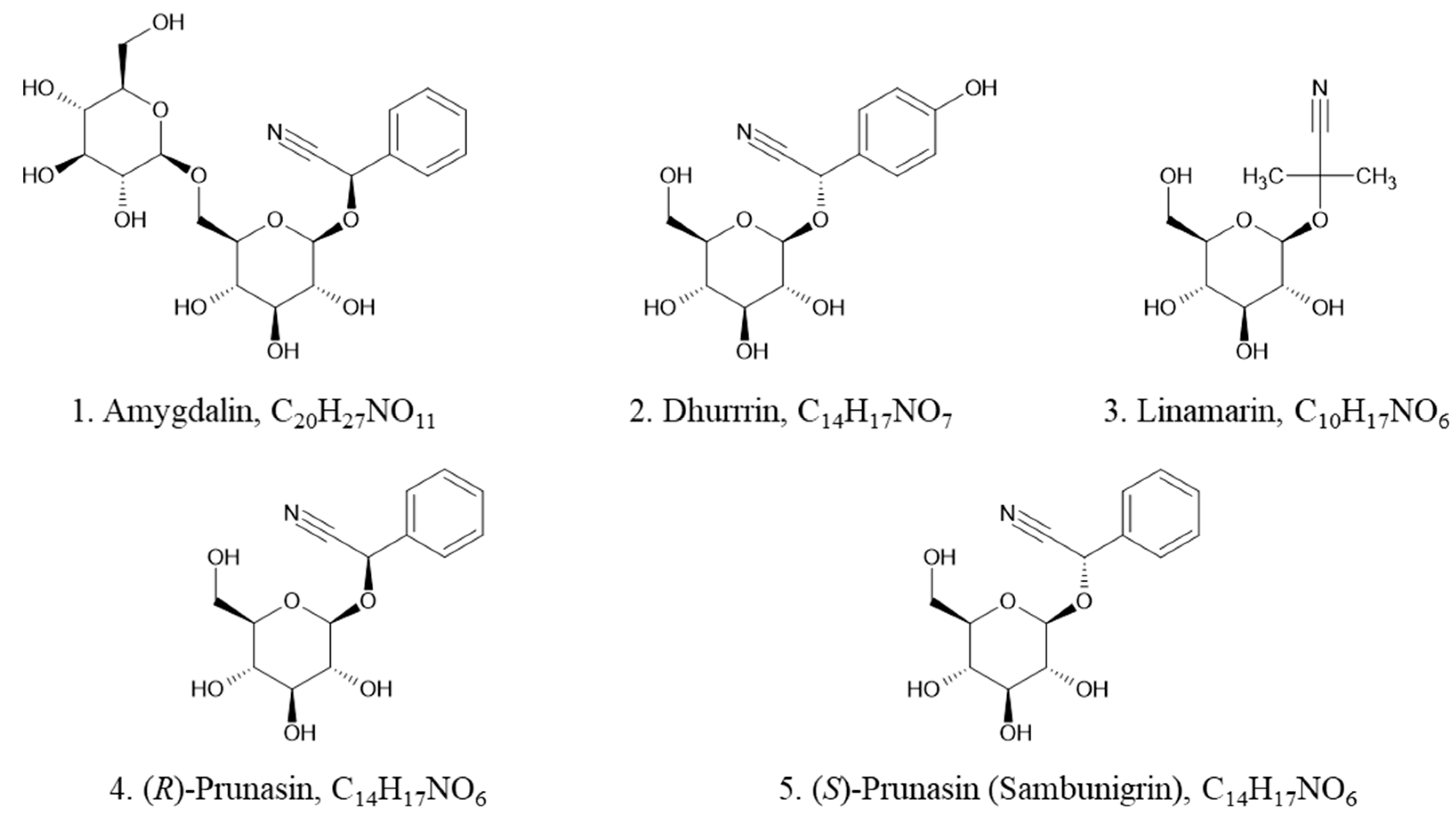
3. Results
3.1. Optimization of Chromatographic Conditions
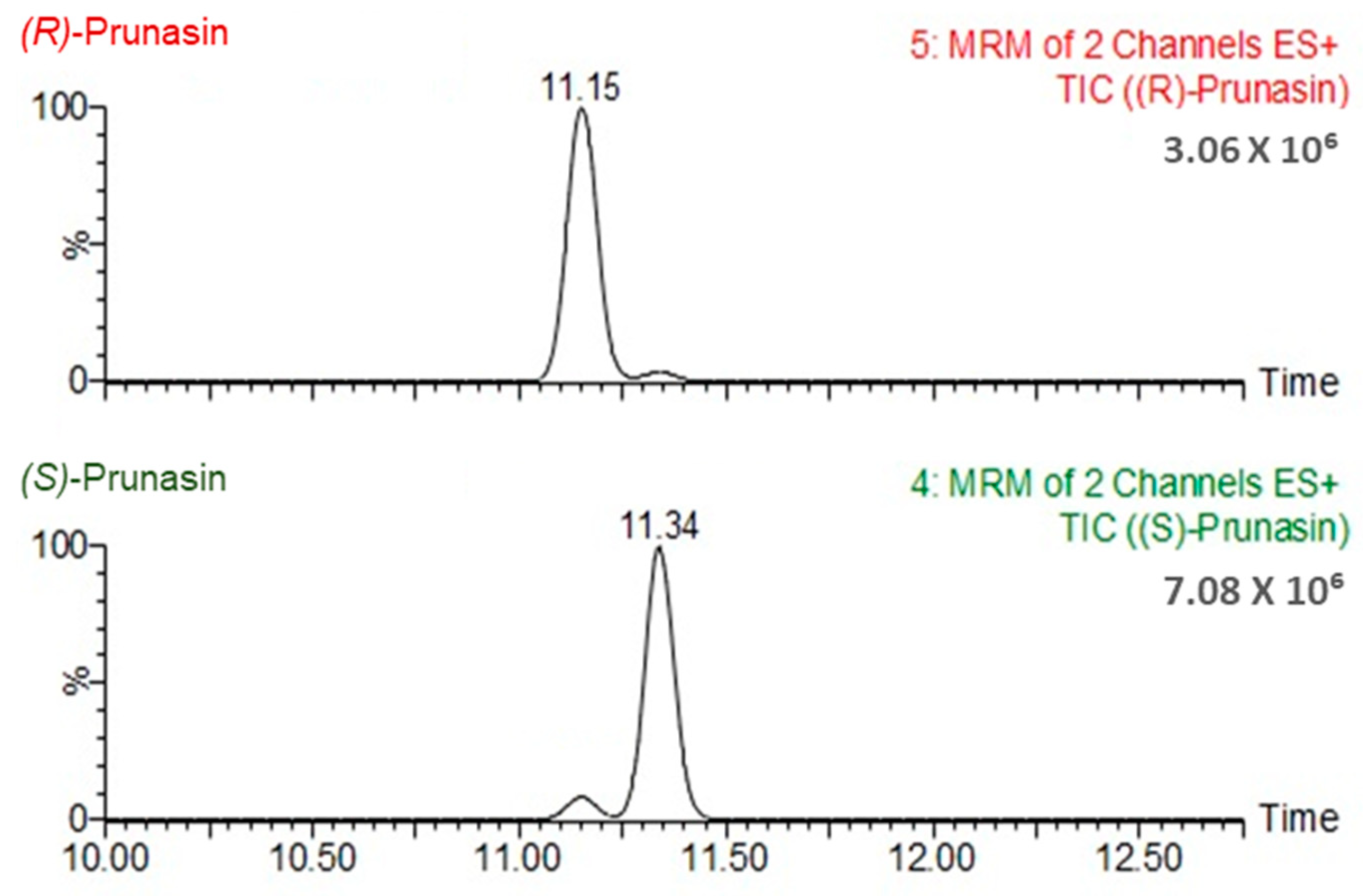
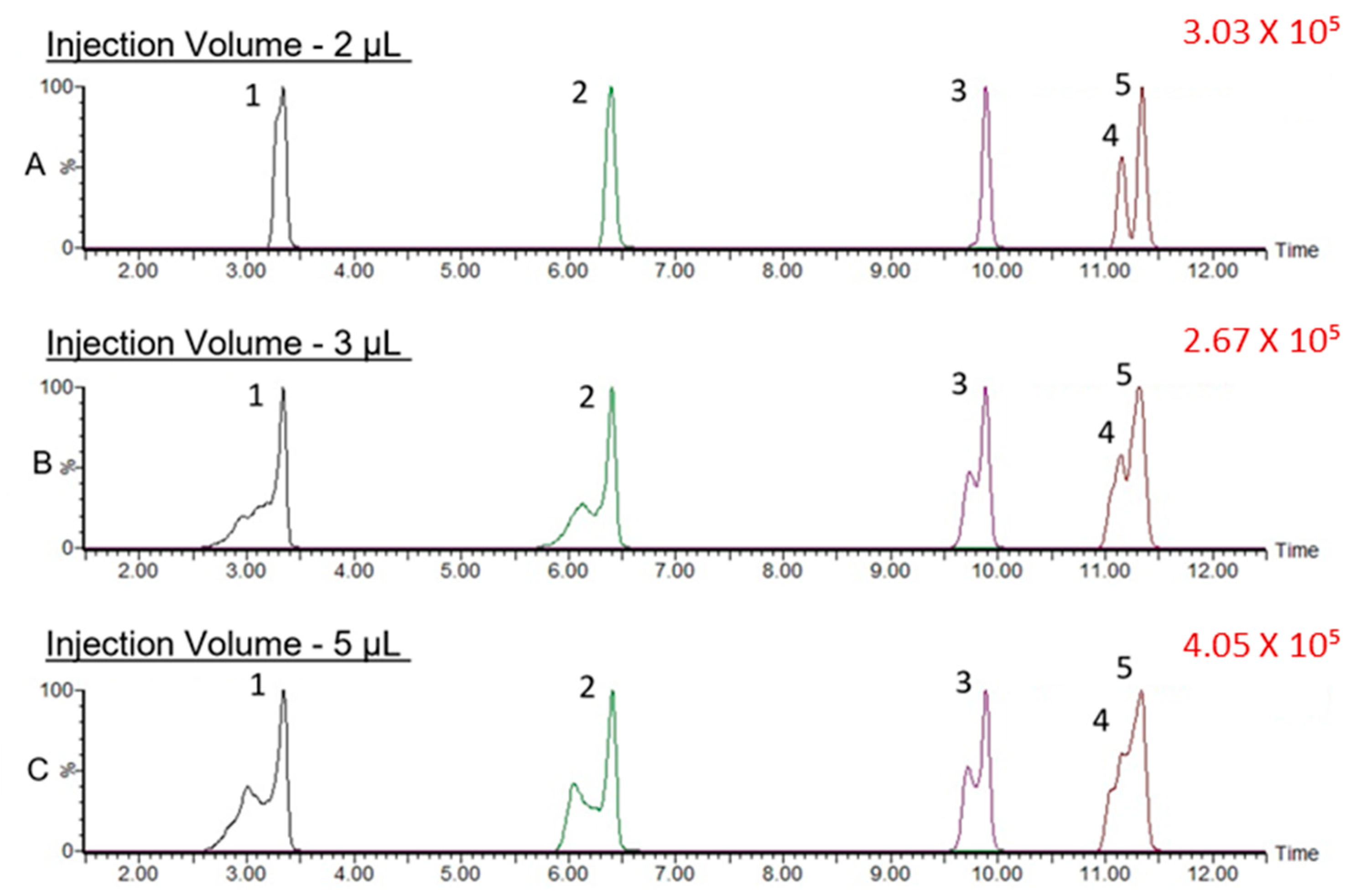
3.1.1. Impact of the Gradient Elution Program and Injection Volume on the Separation of Prunasin Isomers
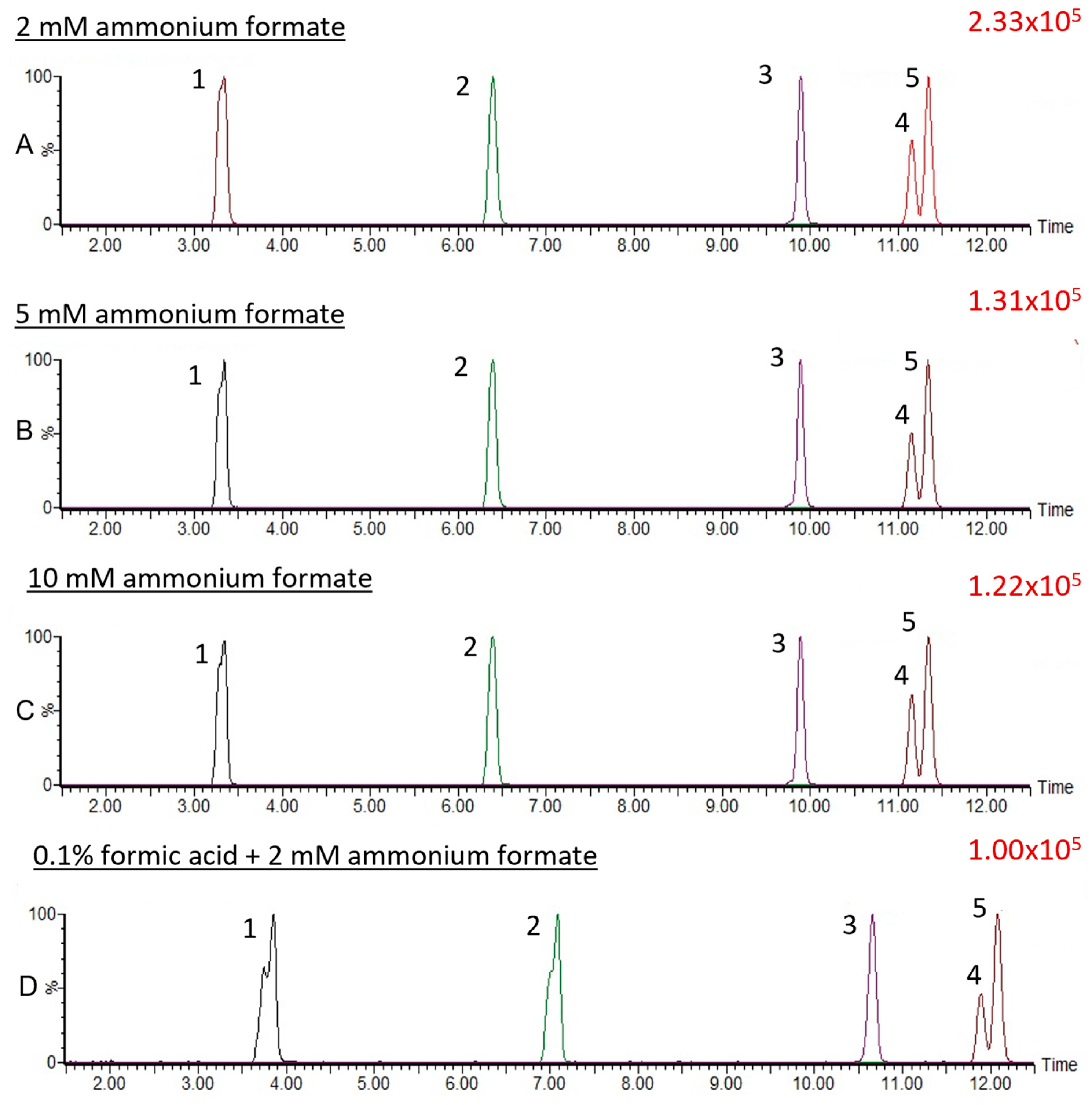
3.1.2. Effect of Mobile Phase Additives on MRM Transitions
3.2. Optimization of Mass Spectrometric Conditions
3.3. Method Validation
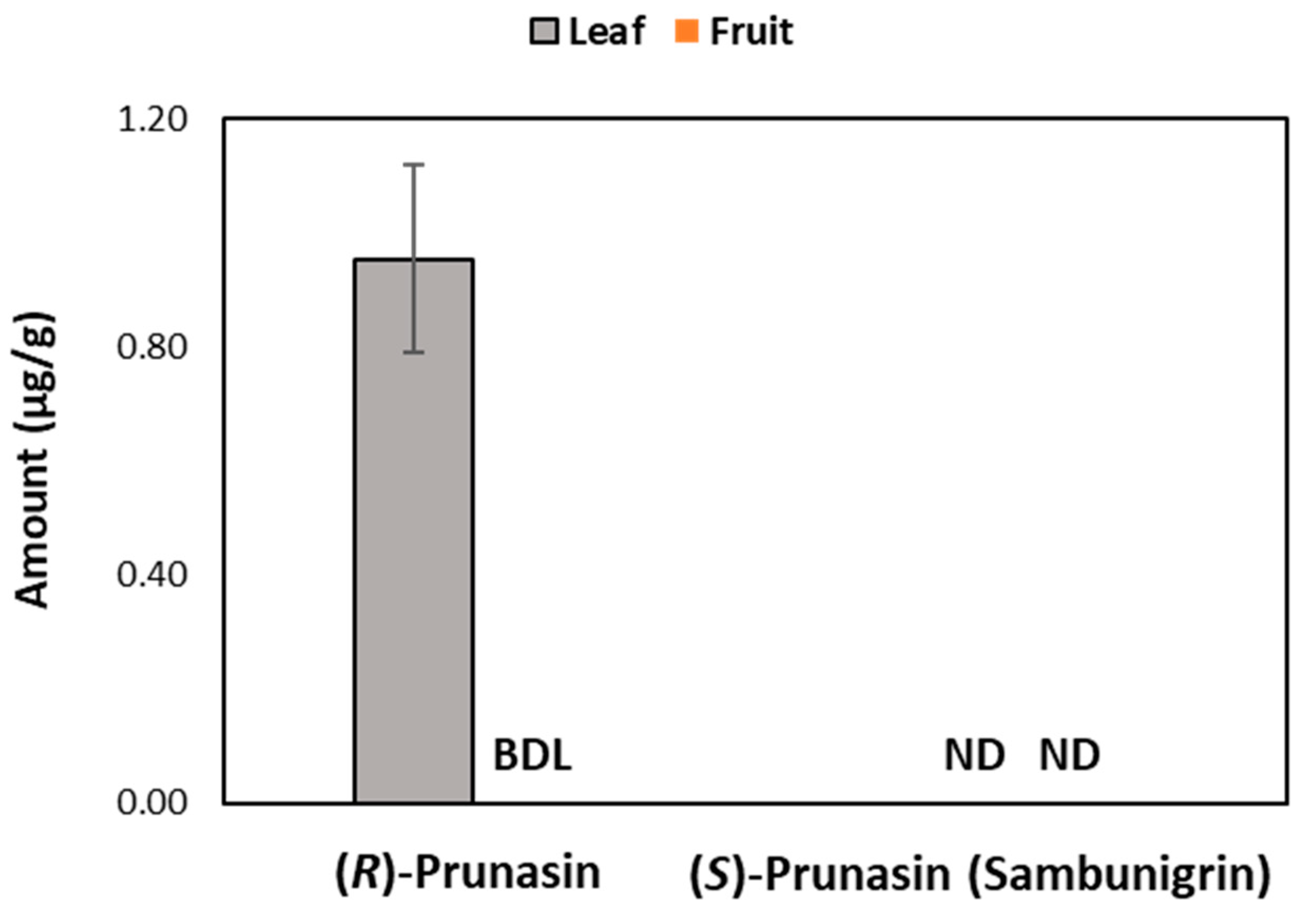
3.4. (R)-prunasin and Sambunigrin in Elderberry Fruit and Leaf Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ganjewala, D. Advances in cyanogenic glycosides biosynthesis and analyses in plants: A review. Acta Biol. Szeged. 2010, 54, 1–14. [Google Scholar]
- Møller, B.L. Functional diversifications of cyanogenic glucosides. Curr. Opin. Plant Biol. 2010, 13, 337–346. [Google Scholar] [CrossRef]
- Panter, K.E. Cyanogenic glycoside–containing plants. In Veterinary Toxicology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 935–940. [Google Scholar]
- Yulvianti, M.; Zidorn, C. Chemical diversity of plant cyanogenic glycosides: An overview of reported natural products. Molecules 2021, 26, 719. [Google Scholar] [CrossRef]
- Mensah, M.A. Cyanogenic Glycosides as Food Toxins. In Analysis of Naturally Occurring Food Toxins of Plant Origin; CRC Press: Boca Raton, FL, USA, 2022; pp. 25–52. [Google Scholar]
- Cressey, P.; Reeve, J. Metabolism of cyanogenic glycosides: A review. Food Chem. Toxicol. 2019, 125, 225–232. [Google Scholar] [CrossRef]
- Liu, D.; He, X.-Q.; Wu, D.-T.; Li, H.-B.; Feng, Y.-B.; Zou, L.; Gan, R.-Y. Elderberry (Sambucus nigra L.): Bioactive compounds, health functions, and applications. J. Agric. Food Chem. 2022, 70, 4202–4220. [Google Scholar] [CrossRef]
- Buhrmester, R.A.; Ebinger, J.E.; Seigler, D.S. Sambunigrin and cyanogenic variability in populations of Sambucus canadensis L.(Caprifoliaceae). Biochem. Syst. Ecol. 2000, 28, 689–695. [Google Scholar] [CrossRef]
- Senica, M.; Stampar, F.; Veberic, R.; Mikulic-Petkovsek, M. The higher the better? Differences in phenolics and cyanogenic glycosides in Sambucus nigra leaves, flowers and berries from different altitudes. J. Sci. Food Agric. 2017, 97, 2623–2632. [Google Scholar] [CrossRef]
- Appenteng, M.K.; Krueger, R.; Johnson, M.C.; Ingold, H.; Bell, R.; Thomas, A.L.; Greenlief, C.M. Cyanogenic glycoside analysis in American elderberry. Molecules 2021, 26, 1384. [Google Scholar] [CrossRef]
- Senica, M.; Stampar, F.; Veberic, R.; Mikulic-Petkovsek, M. Processed elderberry (Sambucus nigra L.) products: A beneficial or harmful food alternative? LWT Food Sci. Technol. 2016, 72, 182–188. [Google Scholar] [CrossRef]
- Chassagne, D.; Crouzet, J.C.; Bayonove, C.L.; Baumes, R.L. Identification and quantification of passion fruit cyanogenic glycosides. J. Agric. Food Chem. 1996, 44, 3817–3820. [Google Scholar] [CrossRef]
- Sornyotha, S.; Kyu, K.L.; Ratanakhanokchai, K. Purification and detection of linamarin from cassava root cortex by high performance liquid chromatography. Food Chem. 2007, 104, 1750–1754. [Google Scholar] [CrossRef]
- Zhong, Y.; Xu, T.; Chen, Q.; Li, K.; Zhang, Z.; Song, H.; Wang, M.; Wu, X.; Lu, B. Development and validation of eight cyanogenic glucosides via ultra-high-performance liquid chromatography-tandem mass spectrometry in agri-food. Food Chem. 2020, 331, 127305. [Google Scholar] [CrossRef]
- Li, C.; Chu, S.; Tan, S.; Yin, X.; Jiang, Y.; Dai, X.; Gong, X.; Fang, X.; Tian, D. Towards higher sensitivity of mass spectrometry: A perspective from the mass analyzers. Front. Chem. 2021, 9, 1148. [Google Scholar] [CrossRef]
- Franks, T.; Hayasaka, Y.; Choimes, S.; Van Heeswijck, R. Cyanogenic glucosides in grapevine: Polymorphism, identification and developmental patterns. Phytochemistry 2005, 66, 165–173. [Google Scholar] [CrossRef]
- Food and Drug Administration. Guidance for Industry-Bioanalytical Method Validation; FDA: Silver Spring, MD, USA, 2018.
- Q2(R1) Validation of Analytical Procedures: Text and Methodology Guidance for Industry; FDA: Silver Spring, MD, USA, 2021.
- Harischandra, N.R.; Pallavi, M.; Bheemanna, M.; PavanKumar, K.; Reddy, V.C.S.; Udaykumar, N.R.; Paramasivam, M.; Yadav, S. Simultaneous determination of 79 pesticides in pigeonpea grains using GC–MS/MS and LC–MS/MS. Food Chem. 2021, 347, 128986. [Google Scholar] [CrossRef]
- Pihlström, T.; Fernández-Alba, A.R.; Gamón, M.; Amate, C.; Poulsen, M.; Lippold, R.; Anastassiades, M. Analytical quality control and method validation procedures for pesticide residues analysis in food and feed. Sante 2017, 11813, 21–22. [Google Scholar]
- Kruve, A.; Kaupmees, K. Adduct formation in ESI/MS by mobile phase additives. J. Am. Soc. Mass Spectrom. 2017, 28, 887–894. [Google Scholar] [CrossRef]
- Johnson, M. LC-MS/MS Method Development for Quantification of Bioactive Compounds in Elderberry and Garlic Botanicals. Ph.D. Thesis, University of Missouri, Columbia, MO, USA, 2016. [Google Scholar]
- Gallart-Ayala, H.; Moyano, E.; Galceran, M. Recent advances in mass spectrometry analysis of phenolic endocrine disruptors and related compounds. Mass Spectrom. Rev. 2010, 29, 776–805. [Google Scholar] [CrossRef]
- Park, S.Y.; Jung, M.Y. UHPLC-ESI-MS/MS for the quantification of eight major Gingerols and Shogaols in ginger products: Effects of ionization polarity and mobile phase modifier on the sensitivity. J. Food Sci. 2016, 81, C2457–C2465. [Google Scholar] [CrossRef]
- Dzuman, Z.; Jonatova, P.; Stranska-Zachariasova, M.; Prusova, N.; Brabenec, O.; Novakova, A.; Fenclova, M.; Hajslova, J. Development of a new LC-MS method for accurate and sensitive determination of 33 pyrrolizidine and 21 tropane alkaloids in plant-based food matrices. Anal. Bioanal. Chem. 2020, 412, 7155–7167. [Google Scholar] [CrossRef]
- Kasote, D.M.; Duncan, G.J.; Neacsu, M.; Russell, W.R. Rapid method for quantification of anthocyanidins and anthocyanins in human biological samples. Food Chem. 2019, 290, 56–63. [Google Scholar] [CrossRef]
- Rodríguez Madrera, R.; Suárez Valles, B. Analysis of cyanogenic compounds derived from mandelonitrile by ultrasound-assisted extraction and high-performance liquid chromatography in Rosaceae and Sambucus families. Molecules 2021, 26, 7563. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. No. | Compound | Rt (min) | Ion type | Transition (m/z) | Cone (V) | Collision (eV) |
|---|---|---|---|---|---|---|
| 1. | Linamarin | 3.34 | Quantifier | 265.40 > 180.01 | 4 | 8 |
| Qualifier | 265.40 > 162.92 | 4 | 10 | |||
| 2. | Dhurrin | 6.40 | Quantifier | 329.45 > 131.90 | 8 | 10 |
| Qualifier | 329.45 > 84.80 | 8 | 26 | |||
| 3. | Amygdalin | 9.89 | Quantifier | 475.46 > 84.87 | 24 | 30 |
| Qualifier | 475.46 > 144.92 | 24 | 20 | |||
| 4. | (R)-Prunasin | 11.14 | Quantifier | 313.45 > 180.01 | 20 | 8 |
| Qualifier | 313.45 > 144.92 | 20 | 12 | |||
| 5. | (S)-Prunasin (Sambunigrin) | 11.34 | Quantifier | 313.45 > 180.01 | 20 | 8 |
| Qualifier | 313.45 > 144.92 | 20 | 12 |
| Sr. No. | Compound | R2 | LOD (ng mL−1) | LOQ (ng mL−1) | Repeatability (RSD) | Accuracy (%RE) | Matrix Effect (%ME) | |
|---|---|---|---|---|---|---|---|---|
| Leaf Tissue | Fruit Tissue | |||||||
| 1. | Linamarin | 0.999 | 0.0012 | 0.0037 | 8.35 | 93.9 | −23.6 | 1.2 |
| 2. | Dhurrin | 0.999 | 0.0012 | 0.0035 | 7.53 | 96.4 | 45.6 | 7.3 |
| 3. | Amygdalin | 0.998 | 0.0008 | 0.0023 | 4.81 | 86.4 | 32.4 | 9.2 |
| 4. | (R)-Prunasin | 0.997 | 0.0020 | 0.0061 | 5.37 | 100.4 | 8.3 | 13.1 |
| 5. | (S)-Prunasin (Sambunigrin) | 0.995 | 0.0009 | 0.0027 | 6.52 | 87.6 | 31.4 | 14.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasote, D.M.; Lei, Z.; Kranawetter, C.D.; Conway-Anderson, A.; Sumner, B.W.; Sumner, L.W. A Novel UHPLC-MS/MS Based Method for Isomeric Separation and Quantitative Determination of Cyanogenic Glycosides in American Elderberry. Metabolites 2024, 14, 360. https://doi.org/10.3390/metabo14070360
Kasote DM, Lei Z, Kranawetter CD, Conway-Anderson A, Sumner BW, Sumner LW. A Novel UHPLC-MS/MS Based Method for Isomeric Separation and Quantitative Determination of Cyanogenic Glycosides in American Elderberry. Metabolites. 2024; 14(7):360. https://doi.org/10.3390/metabo14070360
Chicago/Turabian StyleKasote, Deepak M., Zhentian Lei, Clayton D. Kranawetter, Ashley Conway-Anderson, Barbara W. Sumner, and Lloyd W. Sumner. 2024. "A Novel UHPLC-MS/MS Based Method for Isomeric Separation and Quantitative Determination of Cyanogenic Glycosides in American Elderberry" Metabolites 14, no. 7: 360. https://doi.org/10.3390/metabo14070360
APA StyleKasote, D. M., Lei, Z., Kranawetter, C. D., Conway-Anderson, A., Sumner, B. W., & Sumner, L. W. (2024). A Novel UHPLC-MS/MS Based Method for Isomeric Separation and Quantitative Determination of Cyanogenic Glycosides in American Elderberry. Metabolites, 14(7), 360. https://doi.org/10.3390/metabo14070360