
MAVEN2: An Updated Open-Source Mass Spectrometry Exploration Platform


Abstract
:
1. Introduction
2. Materials and Methods
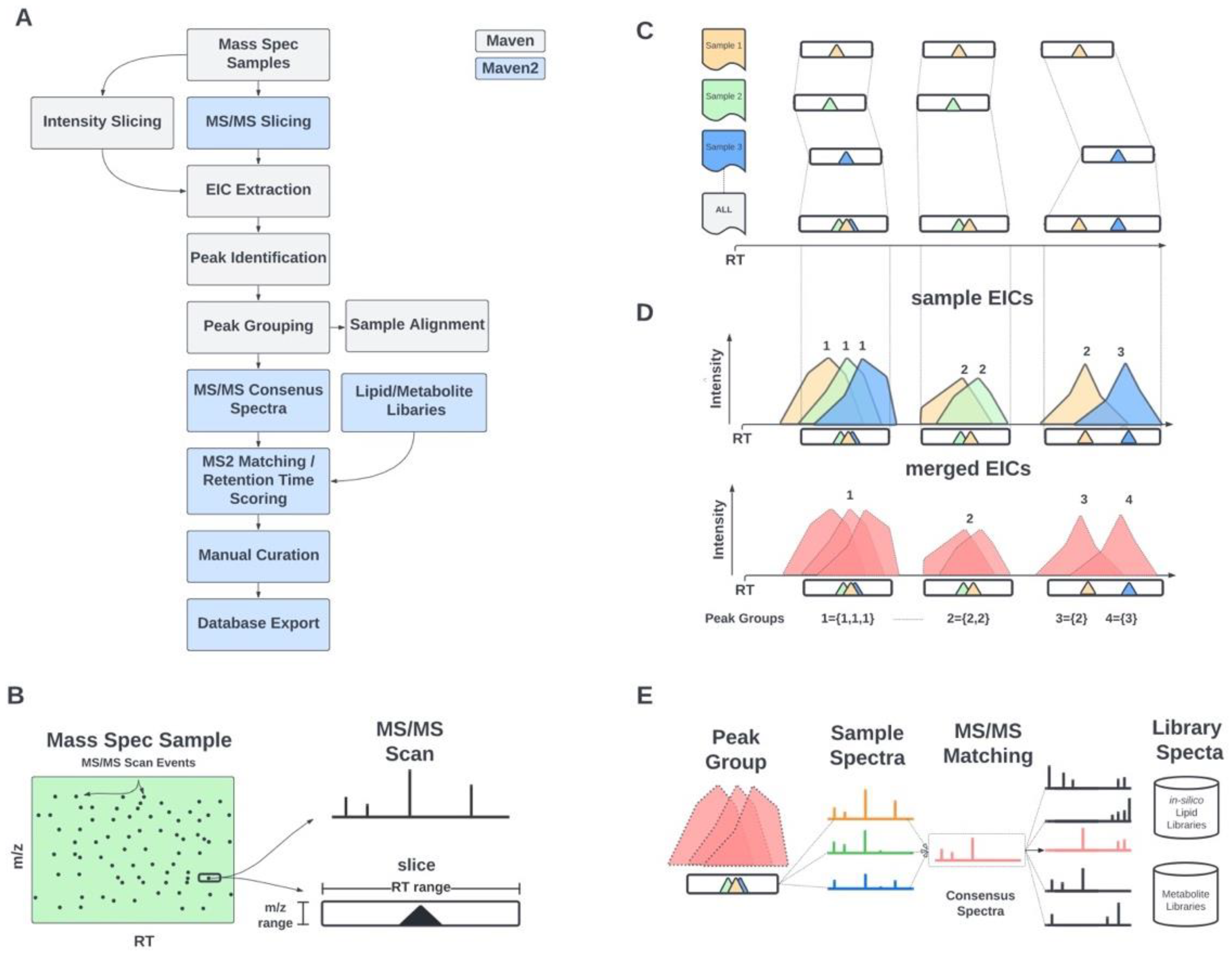
2.1. Informatic Methods
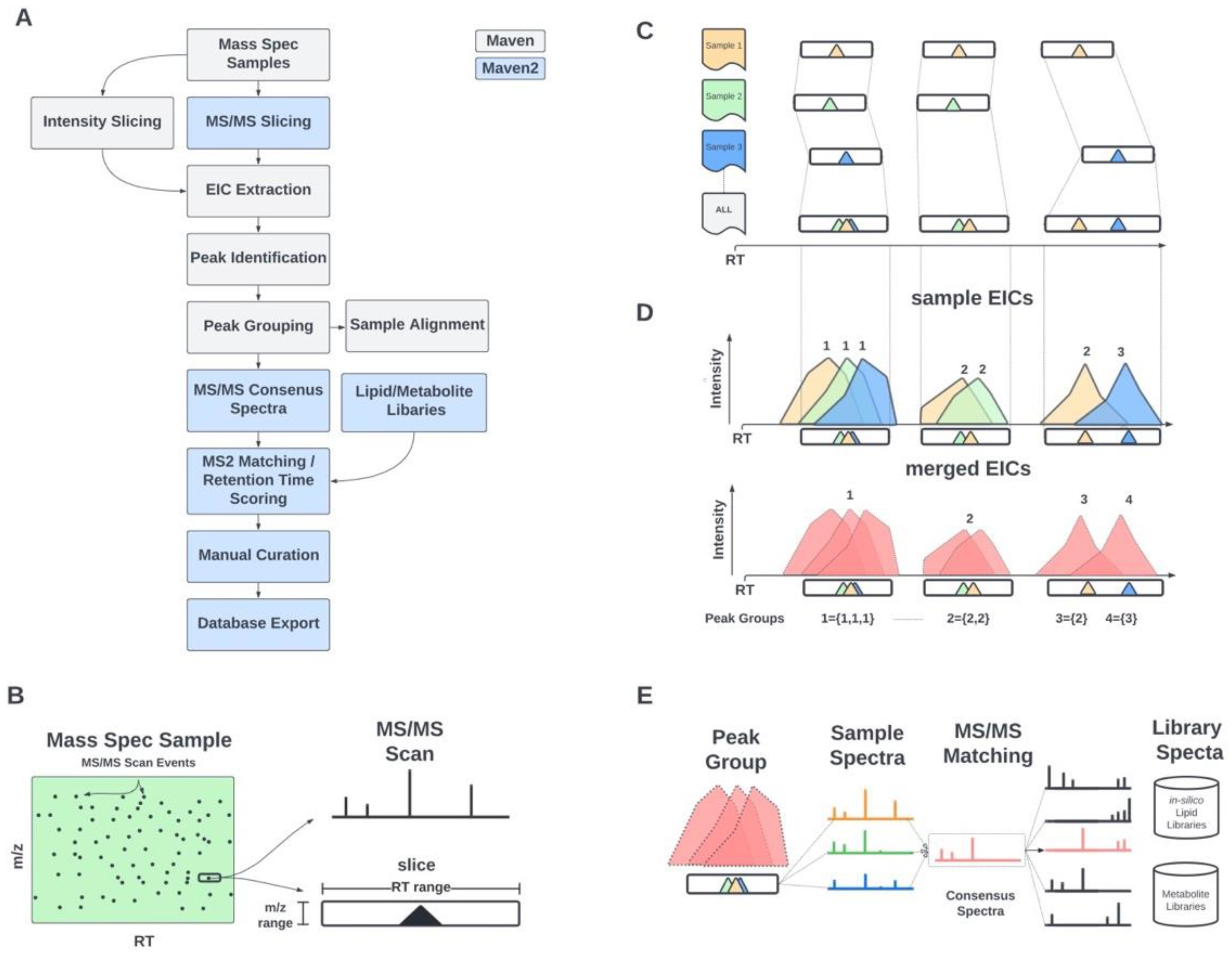
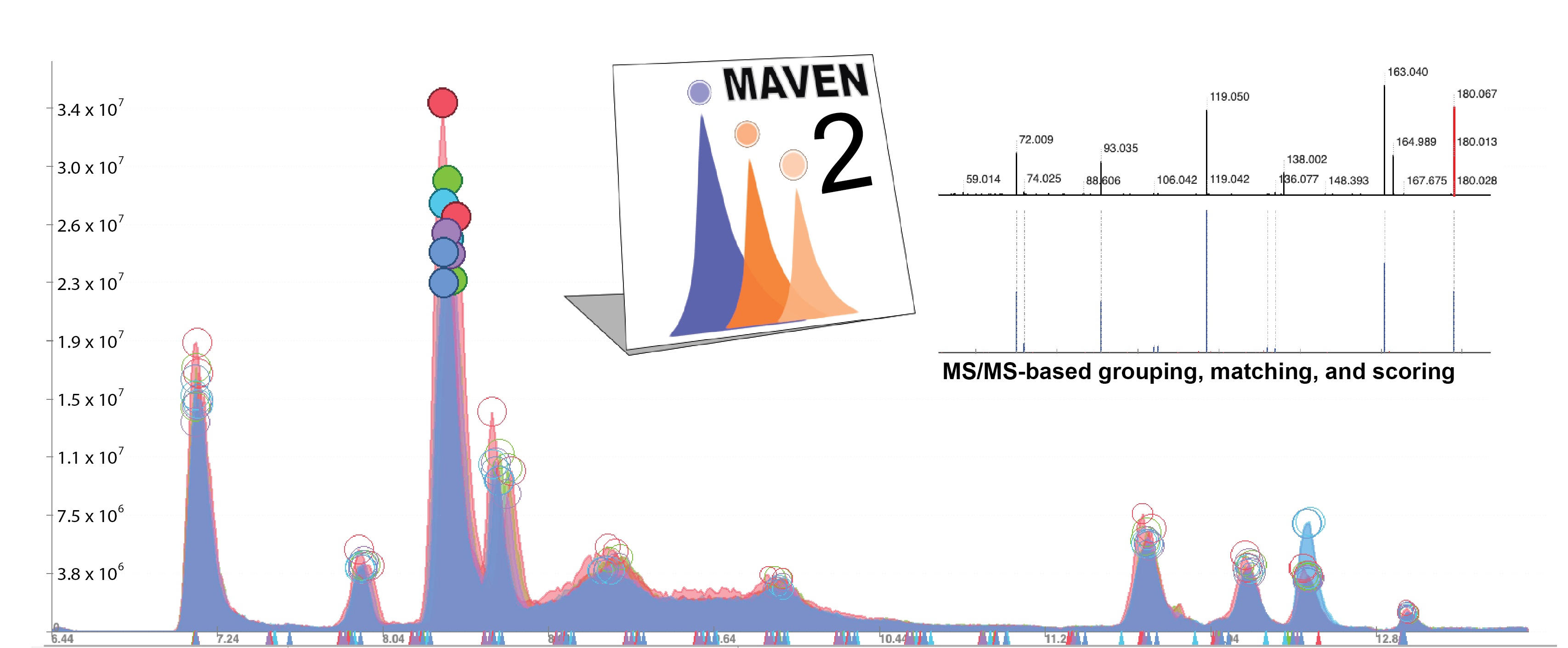
2.1.1. MS/MS-Based Peak Grouping and Compound Identification
2.1.2. MS/MS-Based Spectral Matching and Scoring Approaches
2.1.3. Lipidomics Search Compound Comparison Approach
2.1.4. Generation of In Silico Lipid Spectral Library
2.2. Experimental Methods
2.2.1. Construction of Polar Metabolites Spectral Library
2.2.2. Generation of Metabolomics Datasets
2.2.3. Generation of Lipidomics Datasets
2.2.4. Metabolomics LC/MS Methods
2.2.5. Lipidomics LC/MS Methods
2.2.6. External Lipidomics Datasets
3. Results and Discussion
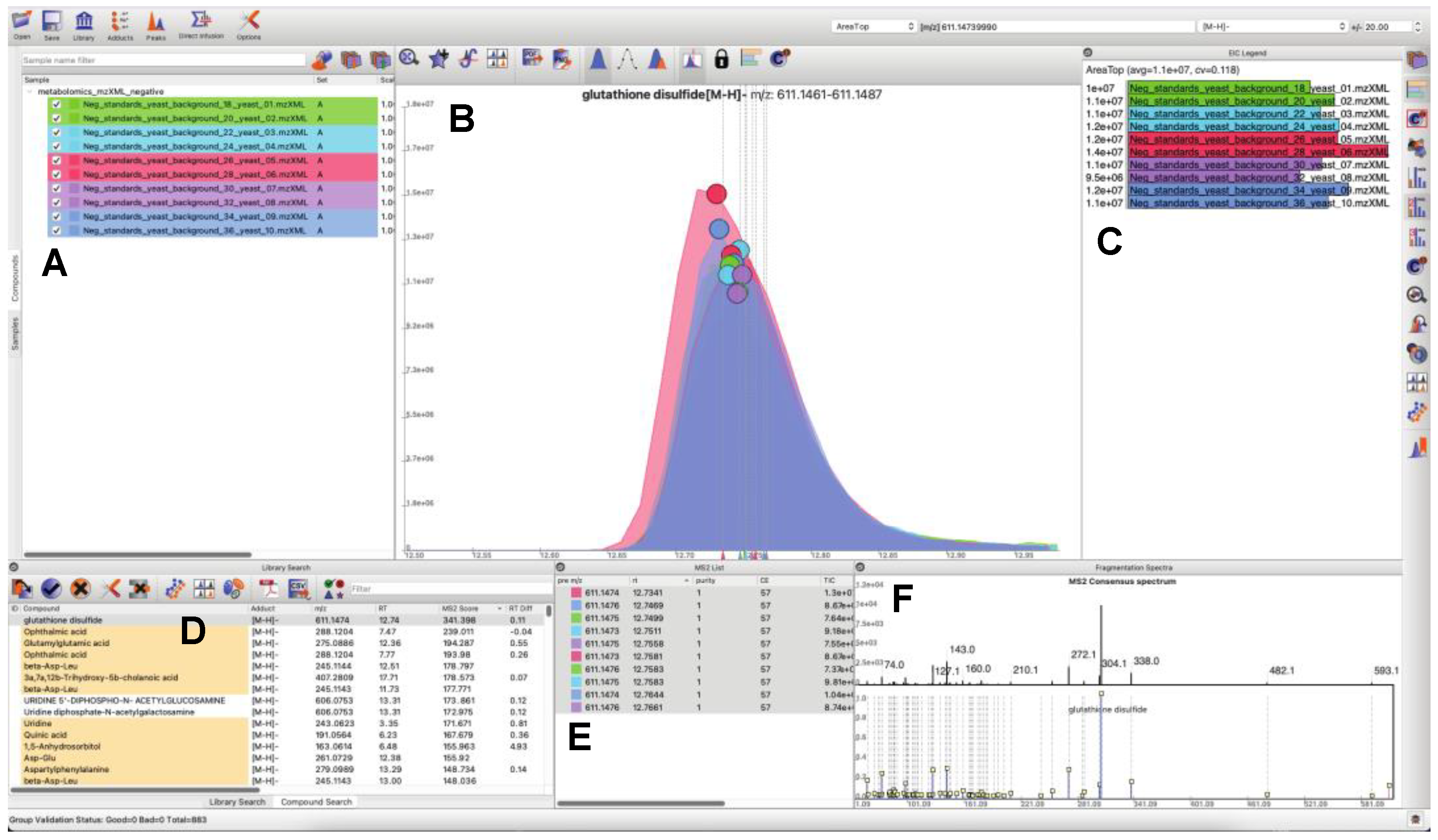
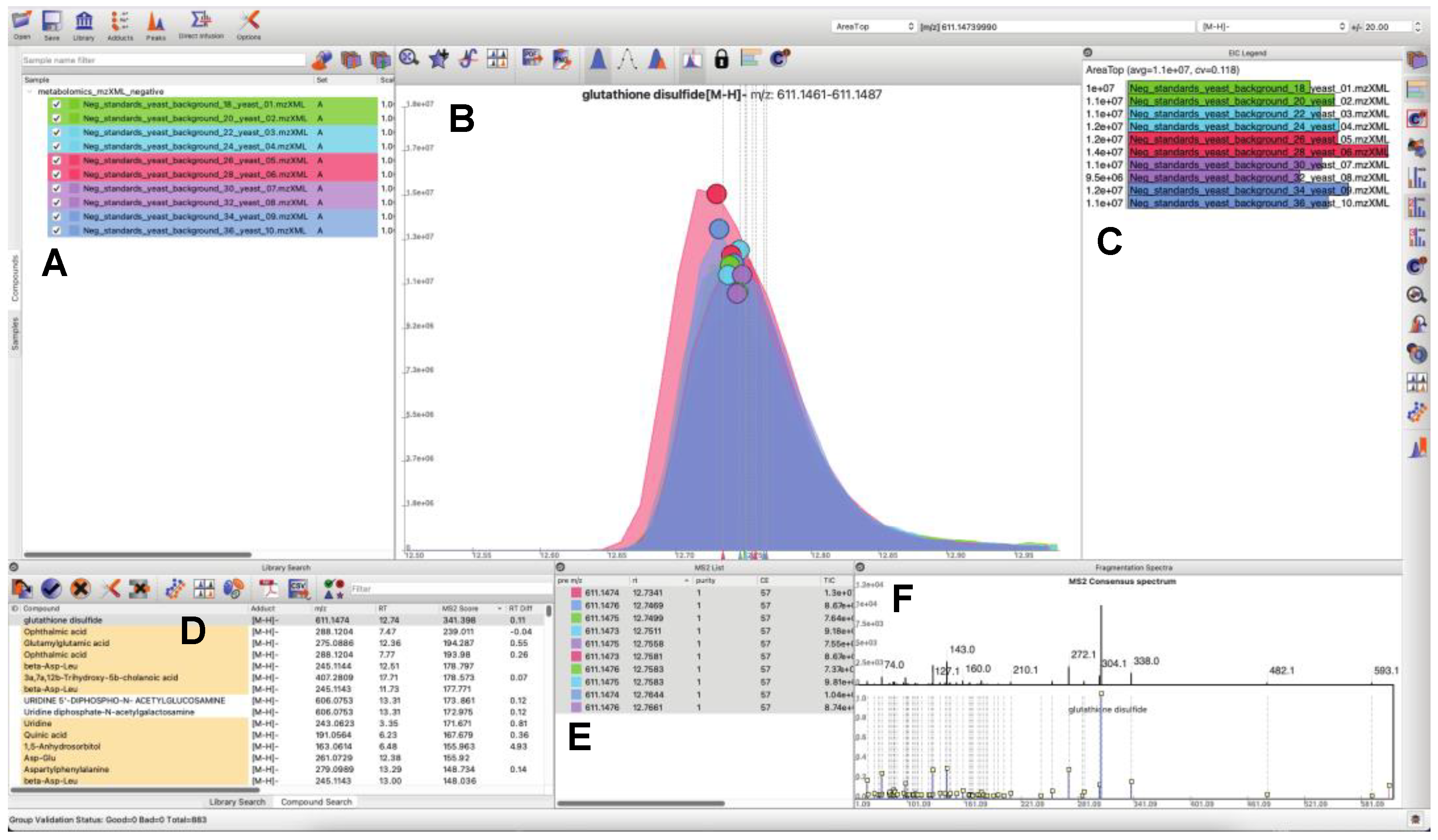
3.1. Updated GUI Facilitates Investigation of Large LC-MS/MS Datasets
- Users can load and search multiple fragmentation libraries simultaneously.
- Users can browse through individual compounds—automatically extracting EICs, displaying location of MS/MS events, and browse through all fragmentation spectra, which are sortable by retention time and spectral purity throughout the loaded dataset.
- Peaks in the raw data may be manually selected to trigger a targeted search of the peak of interest (in real time a consensus MS/MS spectrum is calculated, spectral libraries are scanned, and MS/MS similarity scores are computed). Spectral similarities are displayed in a dedicated MS/MS widget.
- Peaks may be quantified a number of different ways, or manually integrated. Manually integrated peak groups can be saved to a special “Bookmarks” table.
- Peak groups may be tagged with user-configurable labels. Peak groups may take on an unlimited number of labels, and peak group results tables may be filtered based on label contents for efficient navigation and visualization.
- Internal infrastructure has been improved to save projects, sample information, and search results into SQLite databases. This makes it possible to interact with project files outside of MAVEN2, through both SQLite database visualization programs and programmatically via many mainstream programming languages.
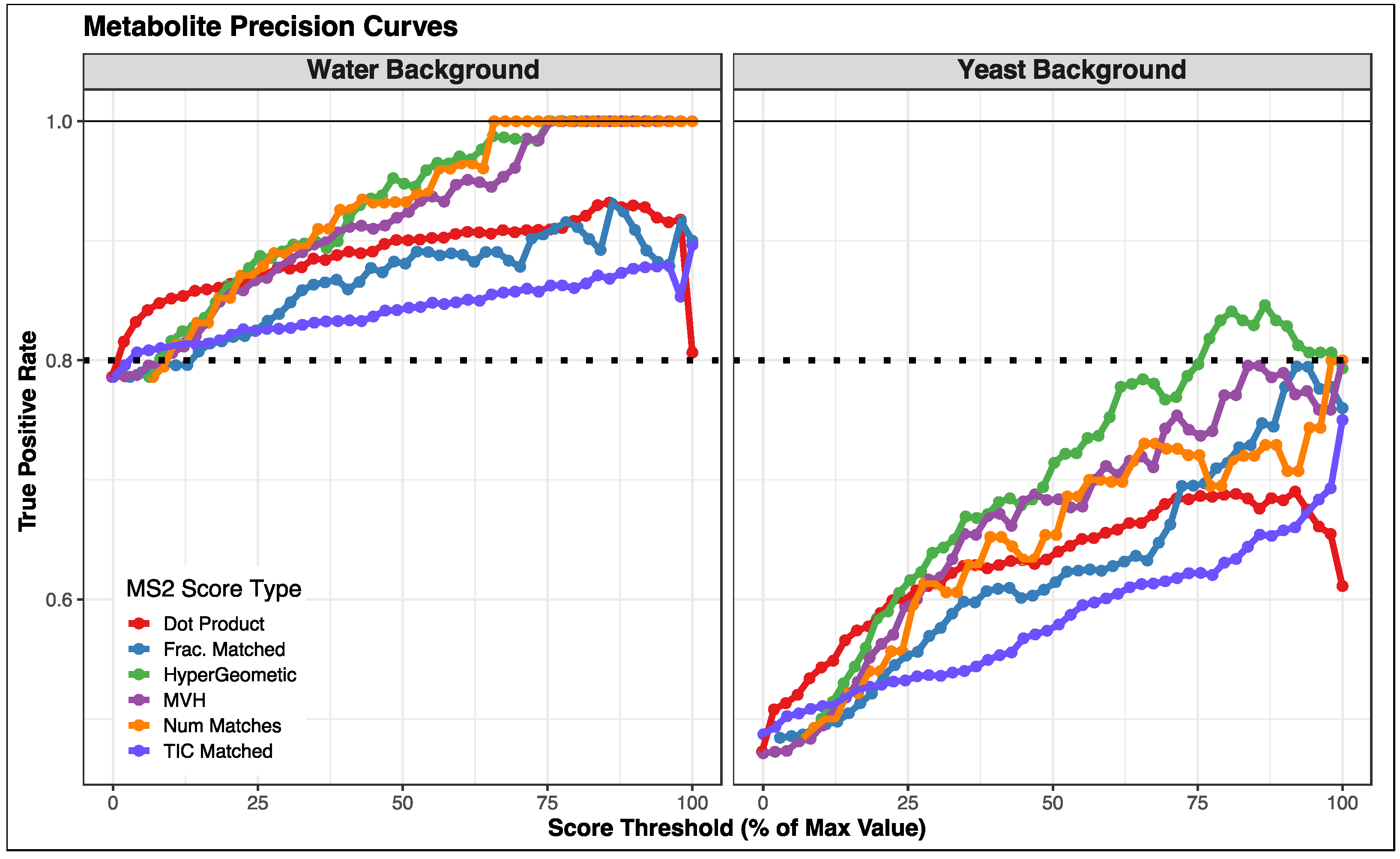
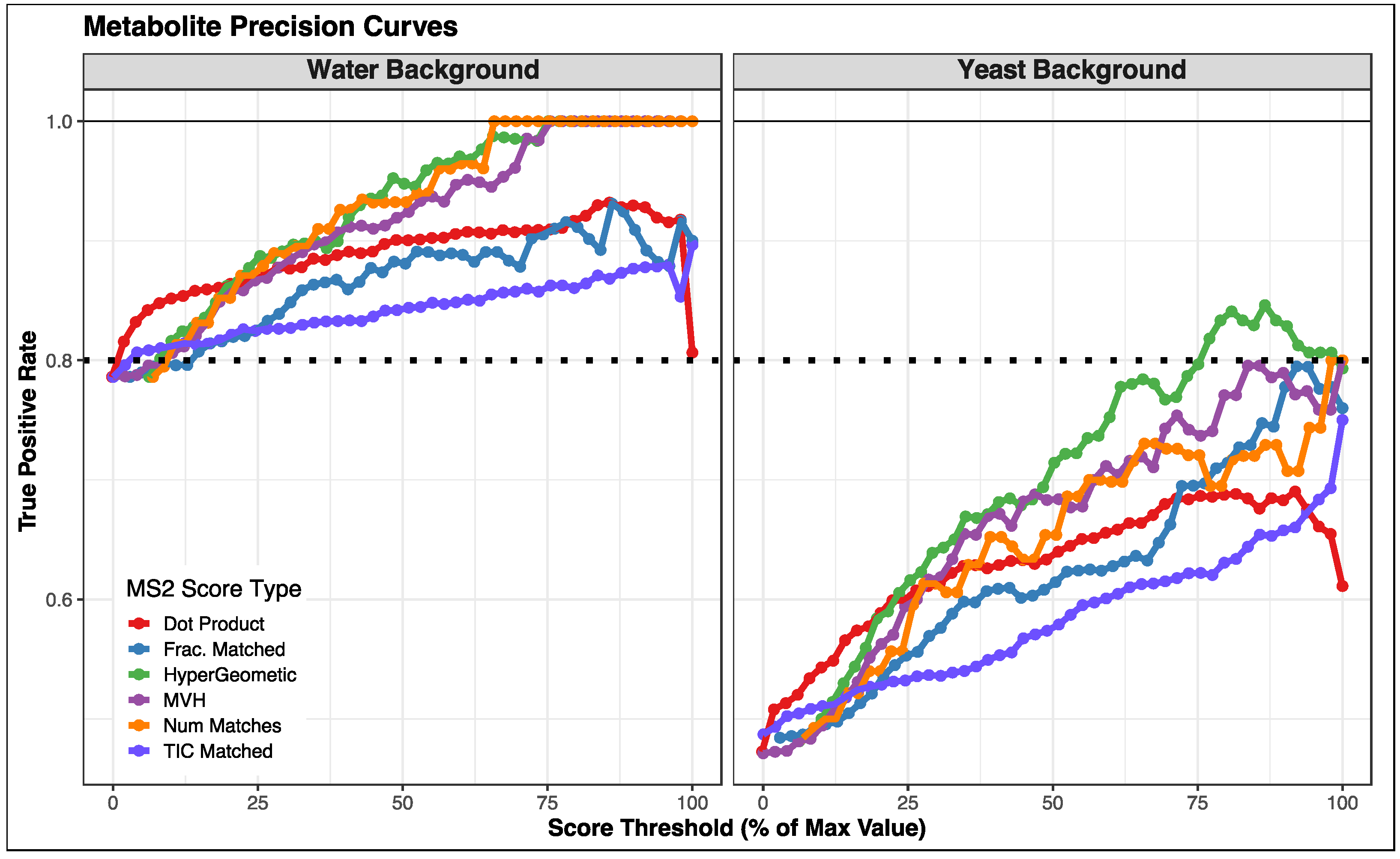
3.2. Comparison of Fragmentation Matching Scoring Methods
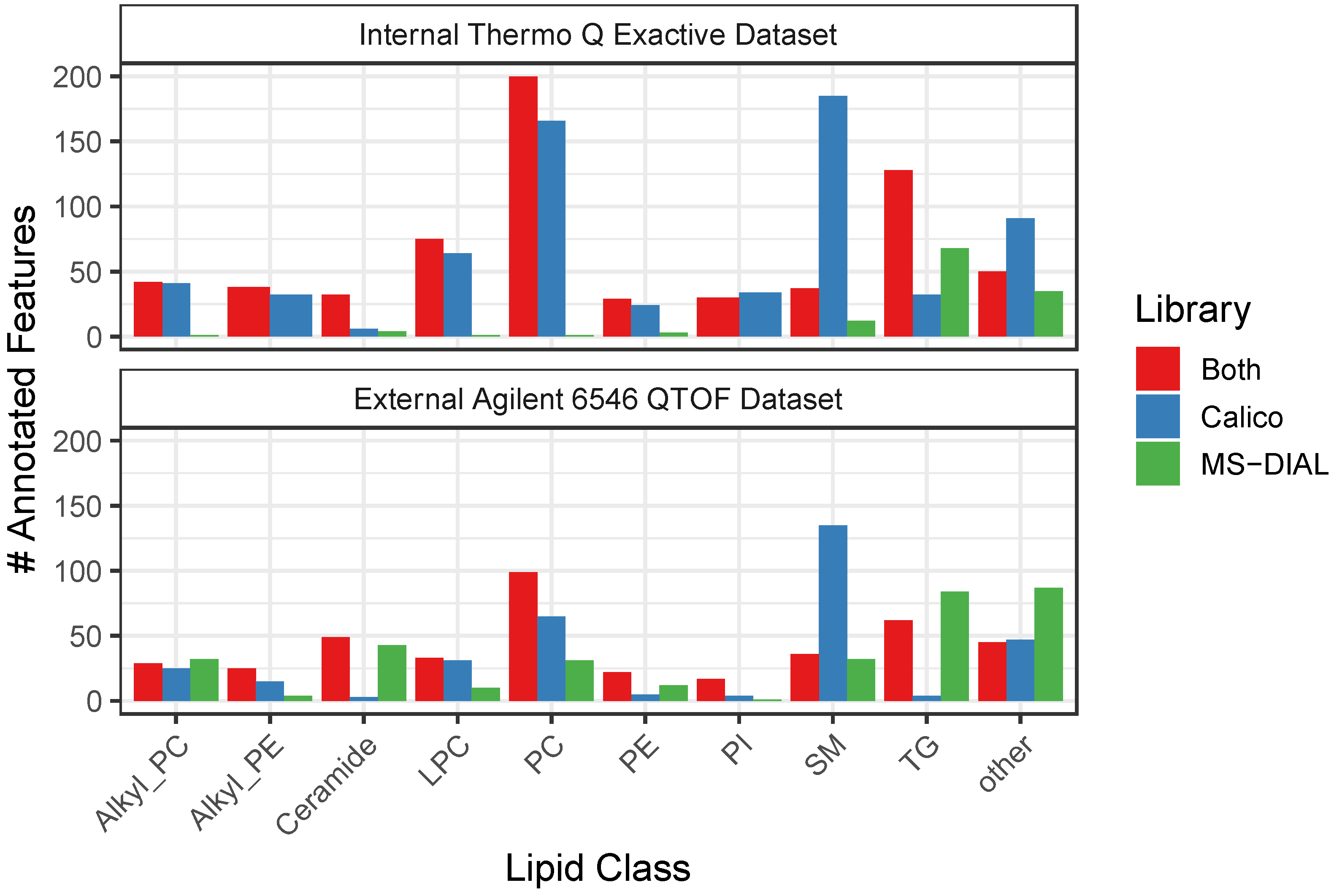
3.3. CalicoLipids Libraries Return Unique Lipid Identifications
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Doerr, A. Targeting with PRM. Nat. Methods 2012, 9, 950. [Google Scholar] [CrossRef] [PubMed]
- Junot, C.; Fenaille, F.; Colsch, B.; Bécher, F. High resolution mass spectrometry based techniques at the crossroads of metabolic pathways. Mass Spectrom. Rev. 2014, 33, 471–500. [Google Scholar] [CrossRef] [PubMed]
- Melamud, E.; Vastag, L.; Rabinowitz, J.D. Metabolomic analysis and visualization engine for LC-MS data. Anal. Chem. 2010, 82, 9818–9826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrawal, S.; Kumar, S.; Sehgal, R.; George, S.; Gupta, R.; Poddar, S.; Jha, A.; Pathak, S. El-MAVEN: A Fast, Robust, and User-Friendly Mass Spectrometry Data Processing Engine for Metabolomics. Methods Mol. Biol. 2019, 1978, 301–321. [Google Scholar]
- Stein, S.E.; Scott, D.R. Optimization and testing of mass spectral library search algorithms for compound identification. J. Am. Soc. Mass Spectrom. 1994, 5, 859–866. [Google Scholar] [CrossRef] [Green Version]
- Griss, J. Spectral library searching in proteomics. Proteomics 2016, 16, 729–740. [Google Scholar] [CrossRef]
- Kind, T.; Liu, K.-H.; Lee, D.Y.; DeFelice, B.; Meissen, J.K.; Fiehn, O. LipidBlast in silico tandem mass spectrometry database for lipid identification. Nat. Methods 2013, 10, 755–758. [Google Scholar] [CrossRef] [Green Version]
- Cajka, T.; Fiehn, O. LC–MS-Based Lipidomics and Automated Identification of Lipids Using the LipidBlast In-Silico MS/MS Library. In Lipidomics: Methods and Protocols; Bhattacharya, S.K., Ed.; Springer: New York, NY, USA, 2017; pp. 149–170. [Google Scholar]
- Wang, M.; Wang, C.; Han, X. Selection of internal standards for accurate quantification of complex lipid species in biological extracts by electrospray ionization mass spectrometry—What, how and why? Mass Spectrom. Rev. 2017, 36, 693–714. [Google Scholar] [CrossRef] [Green Version]
- Kind, T.; Fiehn, O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinform. 2007, 8, 105. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Seitzer, P.M.; Searle, B.C. Incorporating In-Source Fragment Information Improves Metabolite Identification Accuracy in Untargeted LC–MS Data Sets. J. Proteome Res. 2019, 18, 791–796. [Google Scholar] [CrossRef]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; Vander Gheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods. 2015, 12, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, R.; Ishikawa, M. Precise and global identification of phospholipid molecular species by an Orbitrap mass spectrometer and automated search engine Lipid Search. J. Chromatogr. A 2010, 1217, 4229–4239. [Google Scholar] [CrossRef] [PubMed]
- Herzog, R.; Schuhmann, K.; Schwudke, D.; Sampaio, J.L.; Bornstein, S.R.; Schroeder, M.; Shevchenko, A. LipidXplorer: A software for consensual cross-platform lipidomics. PLoS ONE 2012, 7, e29851. [Google Scholar] [CrossRef] [Green Version]
- Comstock, K.; Ding, C.; Stratton, T.; Wang, K.; Eiserberg, G. Rapid and Confident Metabolite Profiling and Identification Using Bench-Top Orbitrap Q Exactive and Compound Discoverer. 20889142189. Available online: http://208.89.142.189/content/dam/tfs/ATG/CMD/cmd-documents/sci-res/posters/ms/events/asms2014/PN-64125-Identification-Q-Exactive-ASMS2014-PN64125-EN.pdf (accessed on 6 July 2022).
- Aron, A.T.; Gentry, E.C.; McPhail, K.L.; Nothias, L.F.; Nothias-Esposito, M.; Bouslimani, A.; Petras, D.; Gauglitz, J.M.; Sikora, N.; Vargas, F.; et al. Reproducible molecular networking of untargeted mass spectrometry data using GNPS. Nat. Protoc. 2020, 15, 1954–1991. [Google Scholar] [CrossRef] [PubMed]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daly, R.; Rogers, S.; Wandy, J.; Jankevics, A.; Burgess, K.E.V.; Breitling, R. MetAssign: Probabilistic annotation of metabolites from LC–MS data using a Bayesian clustering approach. Bioinformatics 2014, 30, 2764–2771. [Google Scholar] [CrossRef] [Green Version]
- Tabb, D.L.; Fernando, C.G.; Chambers, M.C. MyriMatch: Highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J. Proteome Res. 2007, 6, 654–661. [Google Scholar] [CrossRef] [Green Version]
- Haug, K.; Cochrane, K.; Nainala, V.C.; Williams, M.; Chang, J.; Jayaseelan, K.V.; O’Donovan, C. MetaboLights: A resource evolving in response to the needs of its scientific community. Nucleic. Acids Res. 2020, 48, D440–D444. [Google Scholar] [CrossRef] [Green Version]
- Tsugawa, H.; Ikeda, K.; Takahashi, M.; Satoh, A.; Mori, Y.; Uchino, H.; Okahashi, N.; Yamada, Y.; Tada, I.; Bonini, P.; et al. A lipidome atlas in MS-DIAL 4. Nat. Biotechnol. 2020, 38, 1159–1163. [Google Scholar] [CrossRef]
- Clasquin, M.F.; Melamud, E.; Rabinowitz, J.D. LC-MS data processing with MAVEN: A metabolomic analysis and visualization engine. Curr. Protoc. Bioinform. 2012, 14. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Annotation Type | # Features (%) | # Compounds |
|---|---|---|
| All Features | 12,054 (100%) | |
| Unannotated | 9442 (78.30%) | |
| Identified only in CalicoLipids library | 1009 (8.40%) | 768 |
| Identified only in MS-DIAL library | 461 (3.80%) | 429 |
| Identified by both libraries | 1142 (9.50%) | |
| Identified by both libraries with same lipid class | 1078 (8.90%) | |
| Identified by both libraries with same lipid class and adduct | 994 (8.20%) | |
| Identified by both libraries with same summed composition and adduct | 990 (8.20%) | |
| Identified by both libraries as same compound (full agreement) | 512 (4.20%) | 253 |
| Identified by both libraries with different lipid class, adduct, summed composition, and compound (full disagreement) | 44 (0.4%) |
| Dataset | Library | Ionization Mode | # Features | # Features Identified (%) | # Compounds Identified |
|---|---|---|---|---|---|
| Internal Thermo Q Exactive Dataset | CalicoLipids | positive | 3412 | 995 (29.20%) | 741 |
| MS-DIAL | positive | 3412 | 525 (15.40%) | 442 | |
| CalicoLipids | negative | 1301 | 384 (29.5%) | 300 | |
| MS-DIAL | negative | 1301 | 304 (23.40%) | 263 | |
| External Agilent 6546 QTOF Dataset | CalicoLipids | positive | 4162 | 502 (12.10%) | 415 |
| MS-DIAL | positive | 4162 | 378 (9.10%) | 358 | |
| CalicoLipids | negative | 3179 | 270 (8.50%) | 245 | |
| MS-DIAL | negative | 3179 | 396 (12.50%) | 362 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seitzer, P.; Bennett, B.; Melamud, E. MAVEN2: An Updated Open-Source Mass Spectrometry Exploration Platform. Metabolites 2022, 12, 684. https://doi.org/10.3390/metabo12080684
Seitzer P, Bennett B, Melamud E. MAVEN2: An Updated Open-Source Mass Spectrometry Exploration Platform. Metabolites. 2022; 12(8):684. https://doi.org/10.3390/metabo12080684
Chicago/Turabian StyleSeitzer, Phillip, Bryson Bennett, and Eugene Melamud. 2022. "MAVEN2: An Updated Open-Source Mass Spectrometry Exploration Platform" Metabolites 12, no. 8: 684. https://doi.org/10.3390/metabo12080684
APA StyleSeitzer, P., Bennett, B., & Melamud, E. (2022). MAVEN2: An Updated Open-Source Mass Spectrometry Exploration Platform. Metabolites, 12(8), 684. https://doi.org/10.3390/metabo12080684