Isotope Calculation Gadgets: A Series of Software for Isotope-Tracing Experiments in Garuda Platform


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
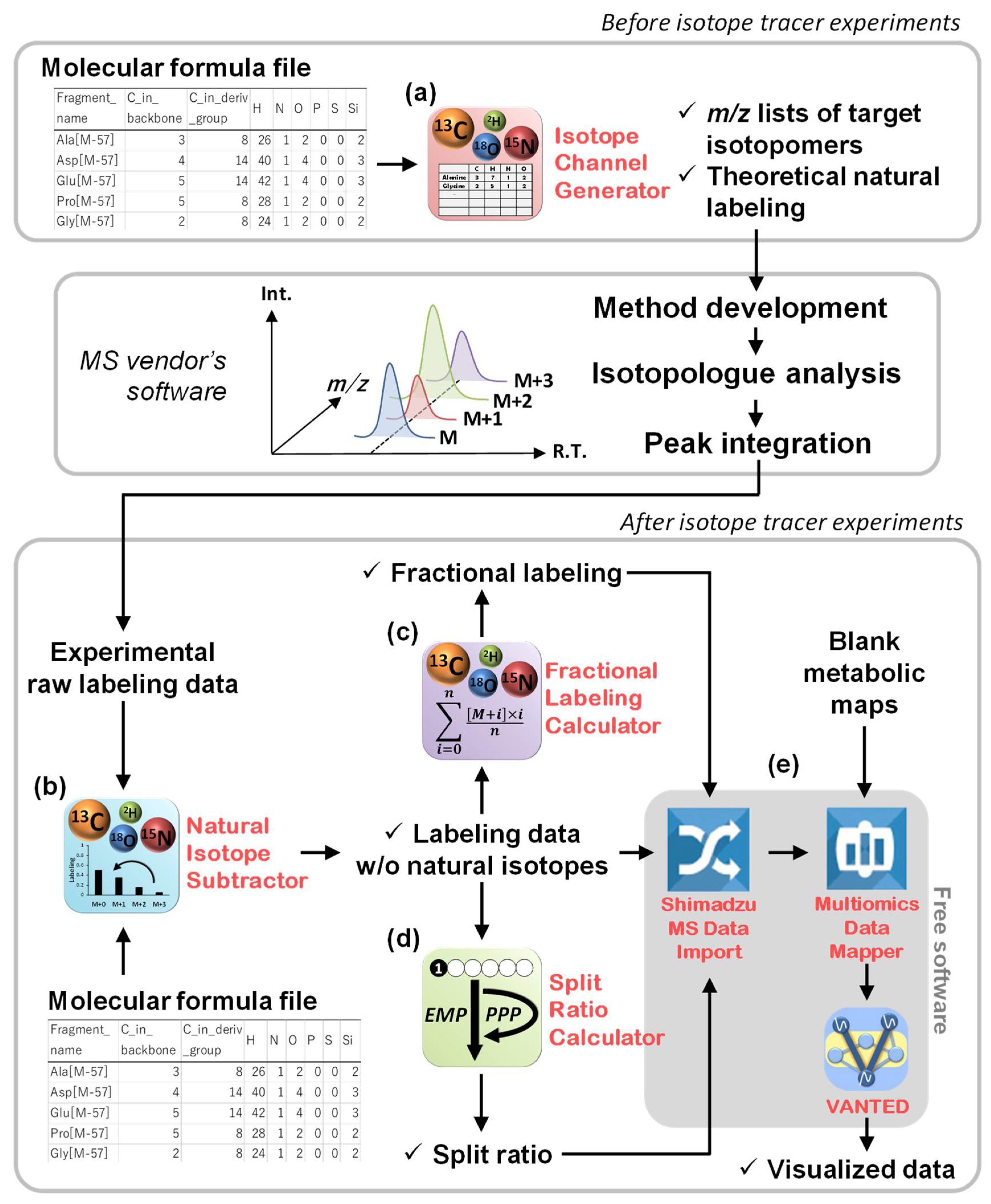
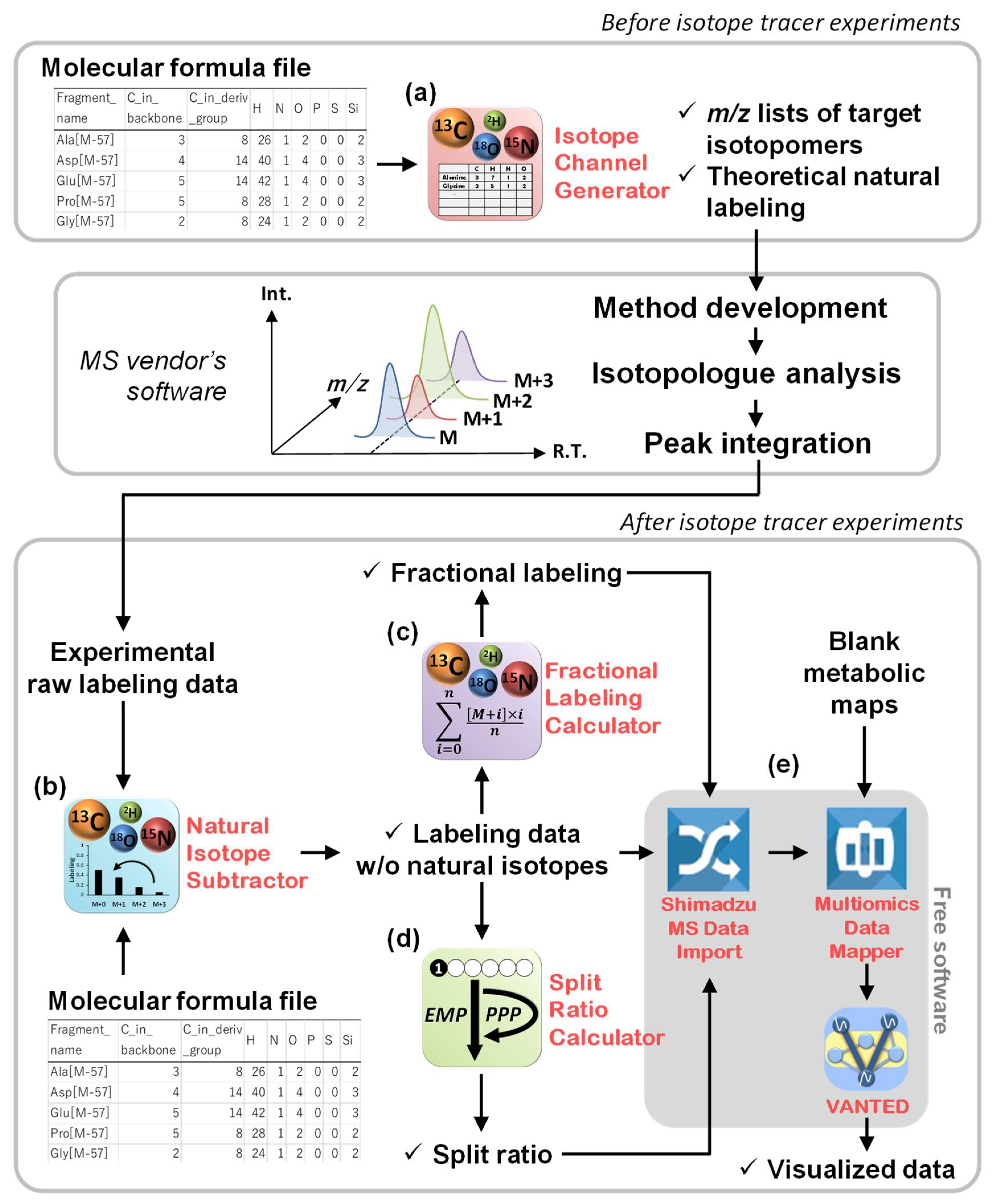
2.1. Overview of This Study
2.2. Channel Creation by the Isotope Channel Generator
2.3. Correction of the Effects of Naturally Occurring Isotopes by the Natural Isotope Subtractor
2.4. Data Interpretation Using the Fractional Labeling Calculator
2.5. Data Interpretation Using the Split Ratio Calculator
2.6. Data Mapping
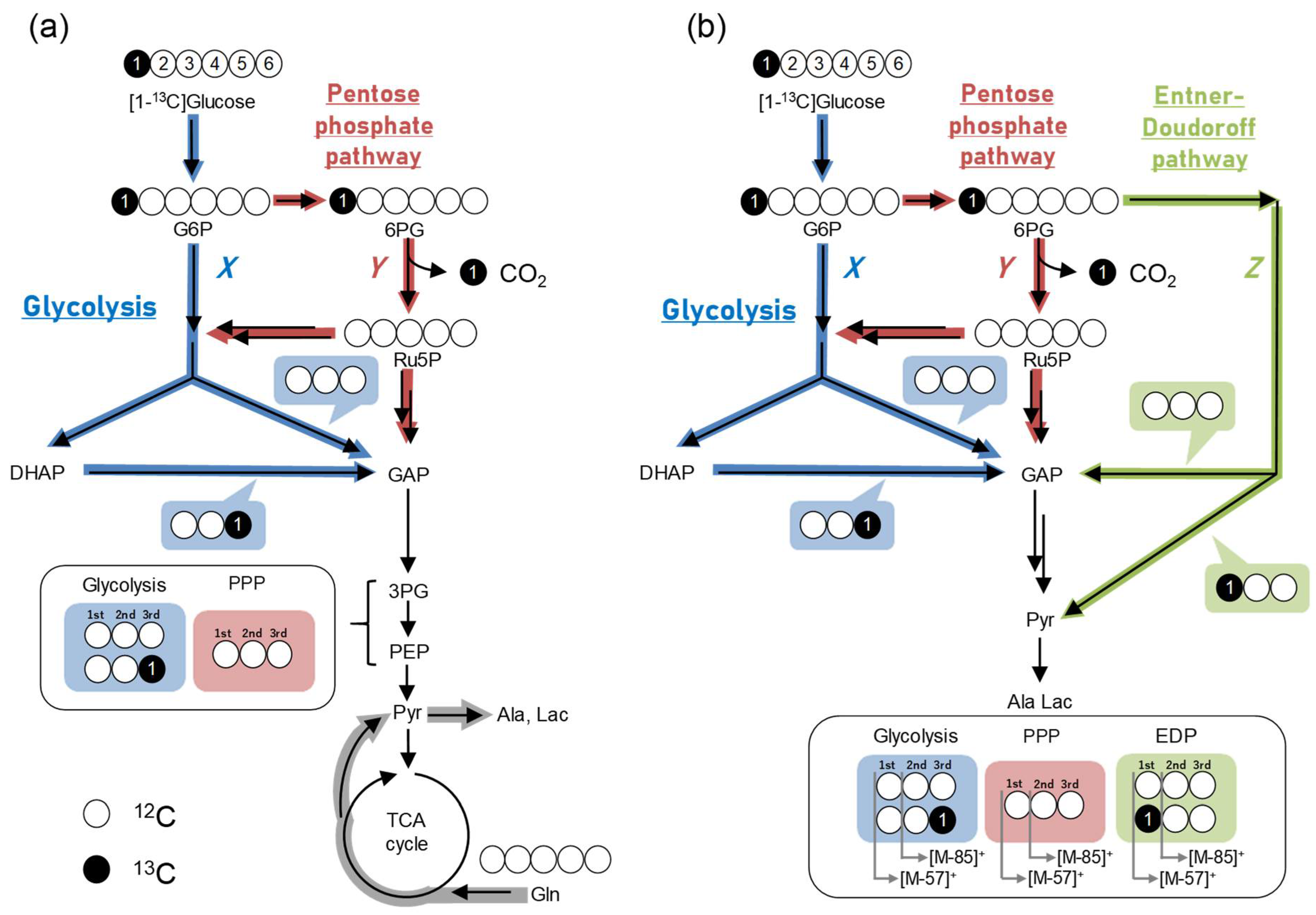
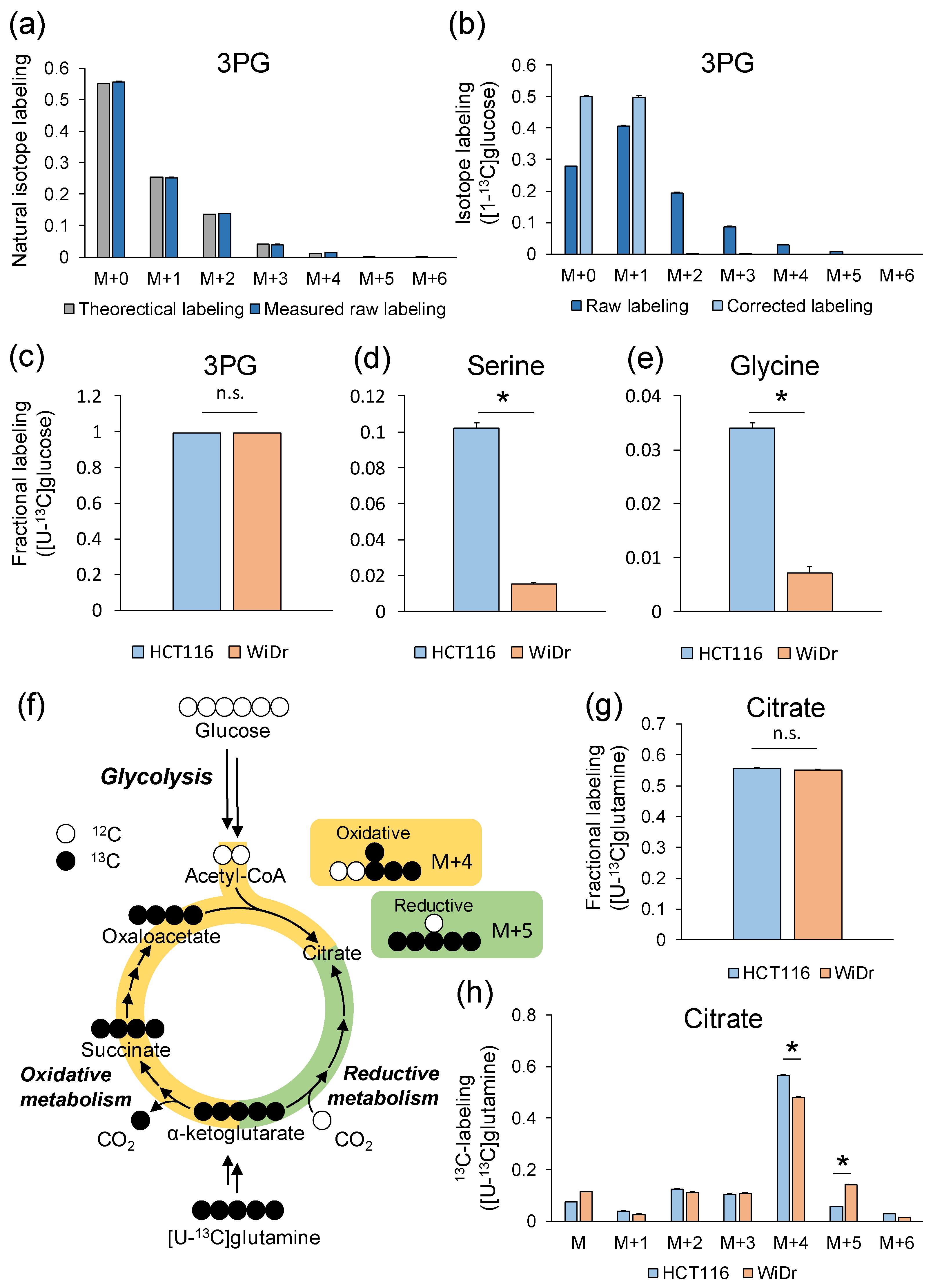
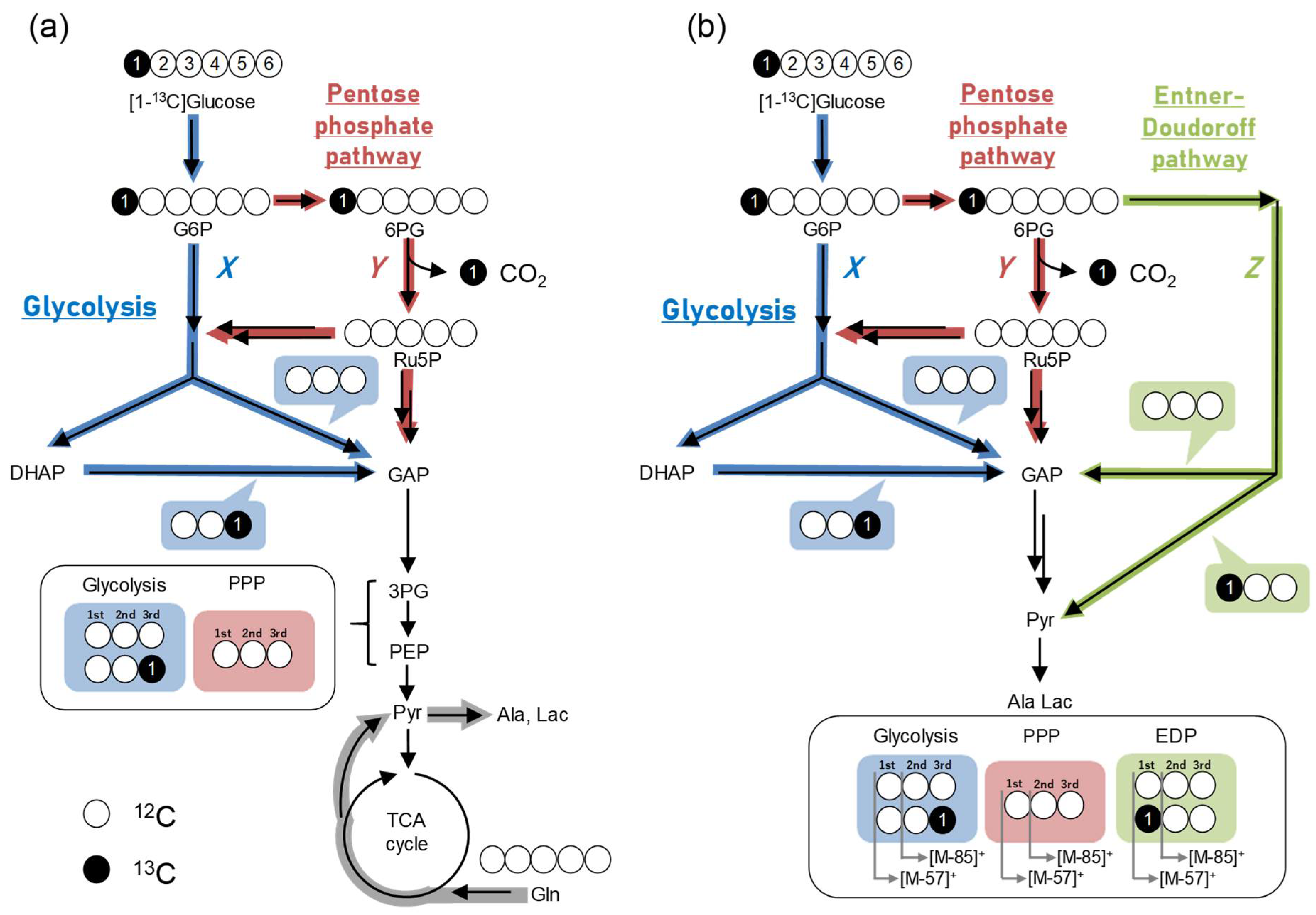
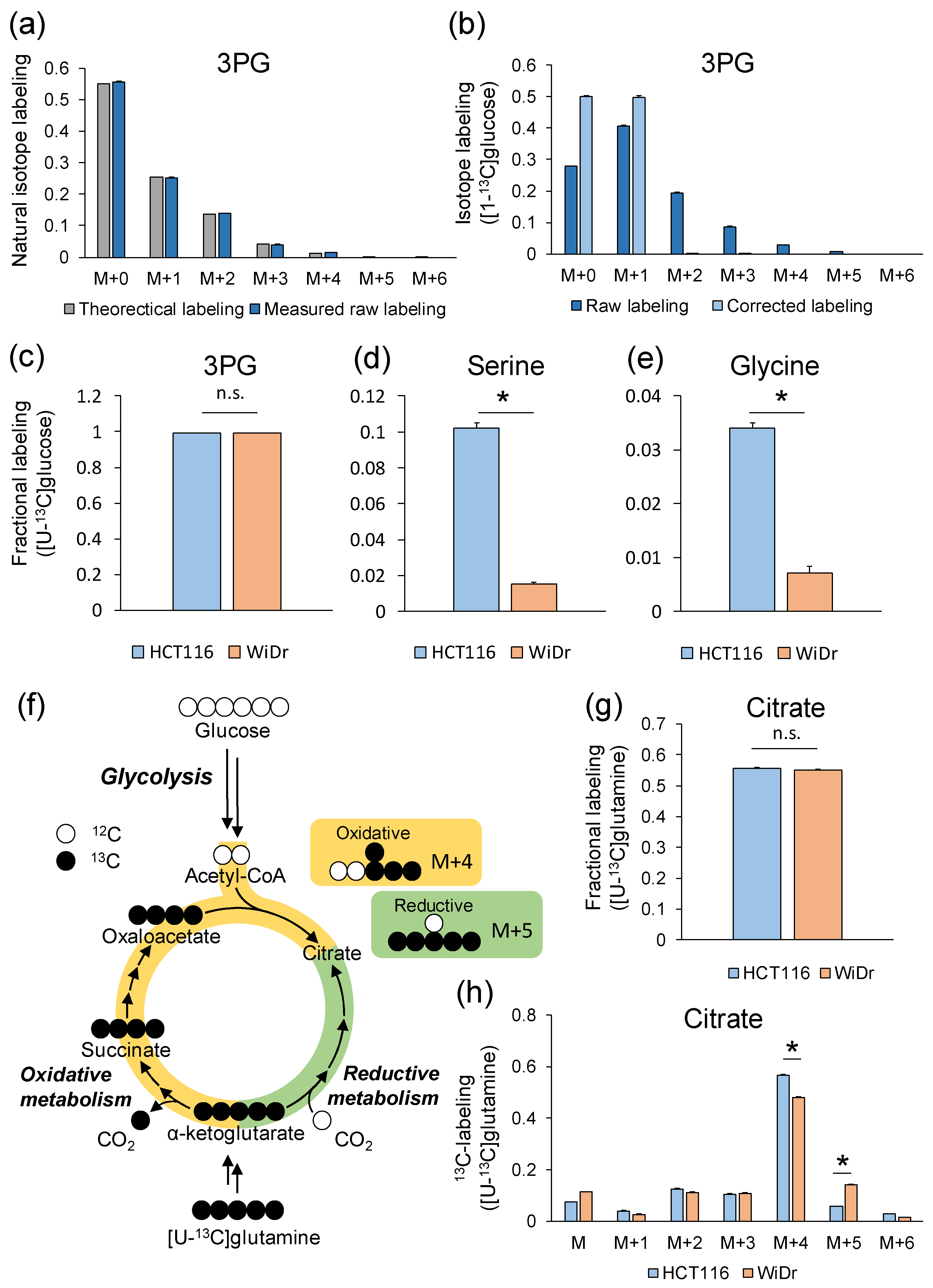
2.7. Example of Analysis Using GC–MS
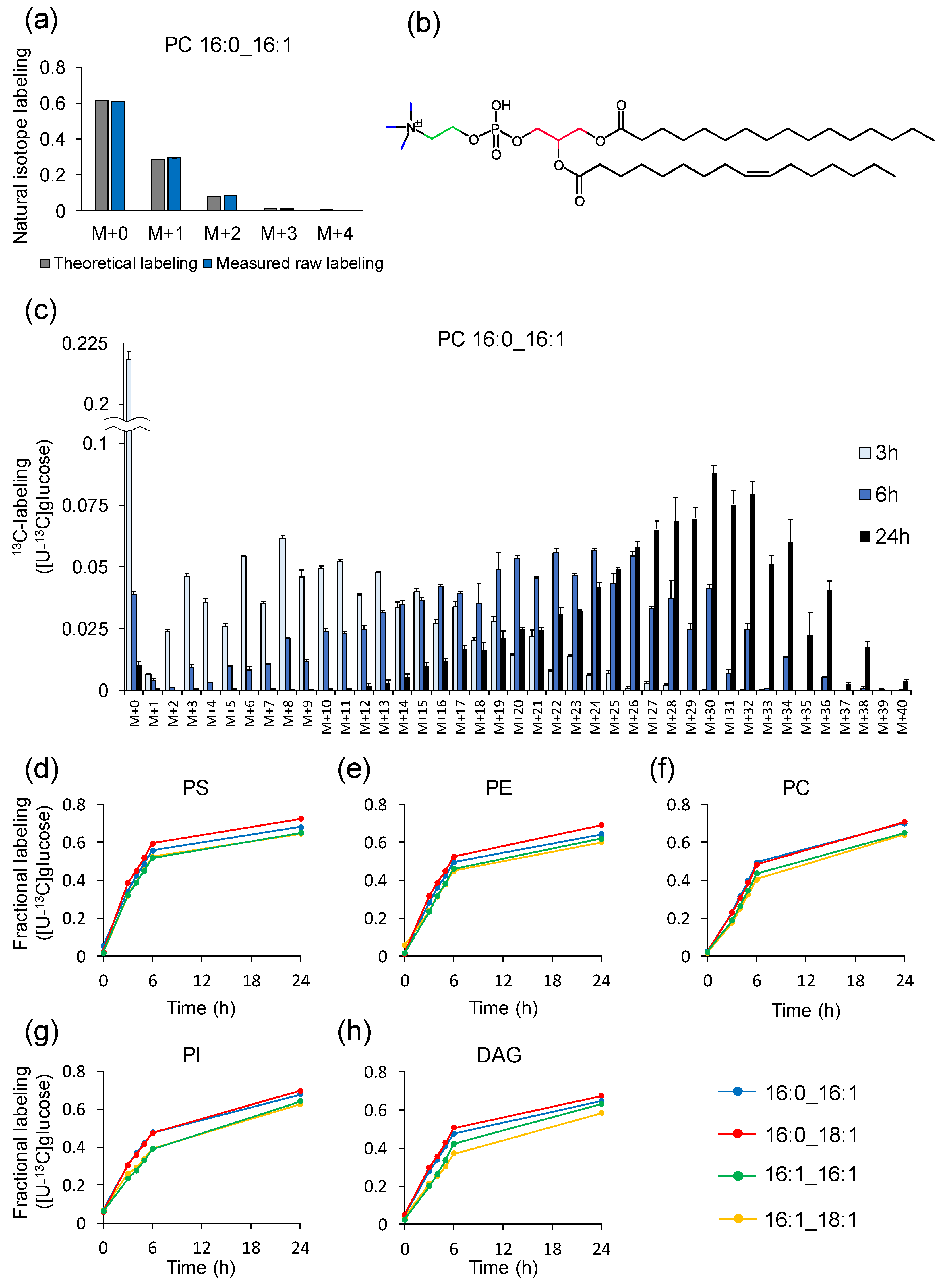
2.8. Example of Analysis Using LC–MS
3. Materials and Methods
3.1. Software Development
3.2. 13C-Tracing Culture of Cancer Cell Lines
3.3. 13C-Labeling Analysis of Water-Soluble Metabolites Using GC–MS
3.4. 13C-Tracing Culture of a Yeast Strain
3.5. 13C-Labeling Analysis of Lipids Using LC-QTOF/MS
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Buescher, J.M.; Antoniewicz, M.R.; Boros, L.G.; Burgess, S.C.; Brunengraber, H.; Clish, C.B.; DeBerardinis, R.J.; Feron, O.; Frezza, C.; Ghesquiere, B.; et al. A roadmap for interpreting 13C metabolite labeling patterns from cells. Curr. Opin. Biotechnol. 2015, 34, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.; Chen, L.; Rabinowitz, J.D. Metabolomics and Isotope Tracing. Cell 2018, 173, 822–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, F.; Maeda, K.; Taniguchi, T.; Kondo, Y.; Yatabe, F.; Okahashi, N.; Shimizu, H. mfapy: An open-source Python package for 13C-based metabolic flux analysis. Metab. Eng. Commun. 2021, 13, e00177. [Google Scholar] [CrossRef] [PubMed]
- Dunstan, R.H. Calculation of the relative frequencies of stable isotope labelled and unlabelled molecules in a gas chromatographic peak using gas chromatography/mass spectrometry. Biomed. Environ. Mass Spectrom. 1988, 15, 473–478. [Google Scholar] [CrossRef]
- Carreer, W.; Flight, R.; Moseley, H. A Computational Framework for High-Throughput Isotopic Natural Abundance Correction of Omics-Level Ultra-High Resolution FT-MS Datasets. Metabolites 2013, 3, 853–866. [Google Scholar] [CrossRef] [Green Version]
- Jungreuthmayer, C.; Neubauer, S.; Mairinger, T.; Zanghellini, J.; Hann, S. ICT: Isotope correction toolbox. Bioinformatics 2016, 32, 154–156. [Google Scholar] [CrossRef] [Green Version]
- Selivanov, V.A.; Benito, A.; Miranda, A.; Aguilar, E.; Polat, I.H.; Centelles, J.J.; Jayaraman, A.; Lee, P.W.N.; Marin, S.; Cascante, M. MIDcor, an R-program for deciphering mass interferences in mass spectra of metabolites enriched in stable isotopes. BMC Bioinform. 2017, 18, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, P.; Kohler, C.; Ellmann, L.; Kuerner, P.; Spang, R.; Oefner, P.J.; Dettmer, K. Correcting for natural isotope abundance and tracer impurity in MS-, MS/MS- and high-resolution-multiple-tracer-data from stable isotope labeling experiments with IsoCorrectoR. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Millard, P.; Delépine, B.; Guionnet, M.; Heuillet, M.; Bellvert, F.; Létisse, F. IsoCor: Isotope correction for high-resolution MS labeling experiments. Bioinformatics 2019, 35, 4484–4487. [Google Scholar] [CrossRef]
- Kumar, A.; Mitchener, J.; King, Z.A.; Metallo, C.M. Escher-Trace: A web application for pathway-based visualization of stable isotope tracing data. BMC Bioinform. 2020, 21, 1–10. [Google Scholar] [CrossRef]
- Jeong, H.; Yu, Y.; Johansson, H.J.; Schroeder, F.C.; Lehtiö, J.; Vacanti, N.M. Correcting for naturally occurring mass isotopologue abundances in stable-isotope tracing experiments with polymid. Metabolites 2021, 11, 310. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.; Sauer, U. Metabolic flux profiling of Escherichia coli mutants in central carbon metabolism using GC-MS. Eur. J. Biochem. 2003, 270, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Hörl, M.; Schnidder, J.; Sauer, U.; Zamboni, N. Non-stationary 13C-metabolic flux ratio analysis. Biotechnol. Bioeng. 2013, 110, 3164–3176. [Google Scholar] [CrossRef] [PubMed]
- Kogadeeva, M.; Zamboni, N. SUMOFLUX: A Generalized Method for Targeted 13C Metabolic Flux Ratio Analysis. PLoS Comput. Biol. 2016, 12, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, F.; Maeda, K.; Okahashi, N. Computational data mining method for isotopomer analysis in the quantitative assessment of metabolic reprogramming. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Antoniewicz, M.R.; Kelleher, J.K.; Stephanopoulos, G. Accurate assessment of amino acid mass isotopomer distributions for metabolic flux analysis. Anal. Chem. 2007, 79, 7554–7559. [Google Scholar] [CrossRef]
- Choi, J.; Antoniewicz, M.R. Tandem mass spectrometry: A novel approach for metabolic flux analysis. Metab. Eng. 2011, 13, 225–233. [Google Scholar] [CrossRef]
- Rühl, M.; Rupp, B.; Nöh, K.; Wiechert, W.; Sauer, U.; Zamboni, N. Collisional fragmentation of central carbon metabolites in LC-MS/MS increases precision of 13C metabolic flux analysis. Biotechnol. Bioeng. 2012, 109, 763–771. [Google Scholar] [CrossRef]
- McCloskey, D.; Young, J.D.; Xu, S.; Palsson, B.O.; Feist, A.M. MID Max: LC-MS/MS Method for Measuring the Precursor and Product Mass Isotopomer Distributions of Metabolic Intermediates and Cofactors for Metabolic Flux Analysis Applications. Anal. Chem. 2016, 88, 1362–1370. [Google Scholar] [CrossRef] [Green Version]
- Okahashi, N.; Kawana, S.; Iida, J.; Shimizu, H.; Matsuda, F. GC-MS/MS survey of collision-induced dissociation of tert-butyldimethylsilyl-derivatized amino acids and its application to 13C-metabolic flux analysis of Escherichia coli central metabolism. Anal. Bioanal. Chem. 2016, 408, 6133–6140. [Google Scholar] [CrossRef]
- Ghosh, S.; Matsuoka, Y.; Asai, Y.; Hsin, K.Y.; Kitano, H. Software for systems biology: From tools to integrated platforms. Nat. Rev. Genet. 2011, 12, 821–832. [Google Scholar] [CrossRef] [PubMed]
- van Winden, W.A.; Wittmann, C.; Heinzle, E.; Heijnen, J.J. Correcting mass isotopomer distributions for naturally occurring isotopes. Biotechnol. Bioeng. 2002, 80, 477–479. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Nomura, Y.; Ishii, J.; Matsuda, F.; Kondo, A.; Shimizu, H. Heterologous expression of bacterial phosphoenol pyruvate carboxylase and Entner–Doudoroff pathway in Saccharomyces cerevisiae for improvement of isobutanol production. J. Biosci. Bioeng. 2017, 124, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Okahashi, N.; Matsuda, F.; Yoshikawa, K.; Shirai, T.; Matsumoto, Y.; Wada, M.; Shimizu, H. Metabolic engineering of isopropyl alcohol-producing Escherichia coli strains with 13C-metabolic flux analysis. Biotechnol. Bioeng. 2017, 114, 2782–2793. [Google Scholar] [CrossRef]
- Junker, B.H.; Klukas, C.; Schreiber, F. Vanted: A system for advanced data analysis and visualization in the context of biological networks. BMC Bioinform. 2006, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Okahashi, N.; Maeda, K.; Kawana, S.; Iida, J.; Shimizu, H.; Matsuda, F. Sugar phosphate analysis with baseline separation and soft ionization by gas chromatography-negative chemical ionization-mass spectrometry improves flux estimation of bidirectional reactions in cancer cells. Metab. Eng. 2019, 51, 43–49. [Google Scholar] [CrossRef]
- Araki, C.; Okahashi, N.; Maeda, K.; Shimizu, H.; Matsuda, F. Mass spectrometry-based method to study inhibitor-induced metabolic redirection in the central metabolism of cancer cells. Mass Spectrom. 2018, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012, 481, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Okahashi, N.; Ueda, M.; Yasuda, S.; Tsugawa, H.; Arita, M. Global profiling of gut microbiota-associated lipid metabolites in antibiotic-treated mice by LC-MS/MS-based analyses. STAR Protoc. 2021, 2, 100492. [Google Scholar] [CrossRef]
- Tsugawa, H.; Ikeda, K.; Takahashi, M.; Satoh, A.; Mori, Y.; Uchino, H.; Okahashi, N.; Yamada, Y.; Tada, I.; Bonini, P.; et al. A lipidome atlas in MS-DIAL 4. Nat. Biotechnol. 2020, 38, 1159–1163. [Google Scholar] [CrossRef]
- de Kroon, A.I.P.M. Metabolism of phosphatidylcholine and its implications for lipid acyl chain composition in Saccharomyces cerevisiae. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2007, 1771, 343–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Getz, G.S.; Jakovcic, S.; Heywood, J.; Frank, J.; Rabinowitz, M. A two-dimensional thin-layer chromatographic system for phospholipid separation the analysis of yeast phospholipids. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab. 1970, 218, 441–452. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okahashi, N.; Yamada, Y.; Iida, J.; Matsuda, F. Isotope Calculation Gadgets: A Series of Software for Isotope-Tracing Experiments in Garuda Platform. Metabolites 2022, 12, 646. https://doi.org/10.3390/metabo12070646
Okahashi N, Yamada Y, Iida J, Matsuda F. Isotope Calculation Gadgets: A Series of Software for Isotope-Tracing Experiments in Garuda Platform. Metabolites. 2022; 12(7):646. https://doi.org/10.3390/metabo12070646
Chicago/Turabian StyleOkahashi, Nobuyuki, Yuki Yamada, Junko Iida, and Fumio Matsuda. 2022. "Isotope Calculation Gadgets: A Series of Software for Isotope-Tracing Experiments in Garuda Platform" Metabolites 12, no. 7: 646. https://doi.org/10.3390/metabo12070646
APA StyleOkahashi, N., Yamada, Y., Iida, J., & Matsuda, F. (2022). Isotope Calculation Gadgets: A Series of Software for Isotope-Tracing Experiments in Garuda Platform. Metabolites, 12(7), 646. https://doi.org/10.3390/metabo12070646