
Evaluating Cefoperazone-Induced Gut Metabolic Functional Changes in MR1-Deficient Mice

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
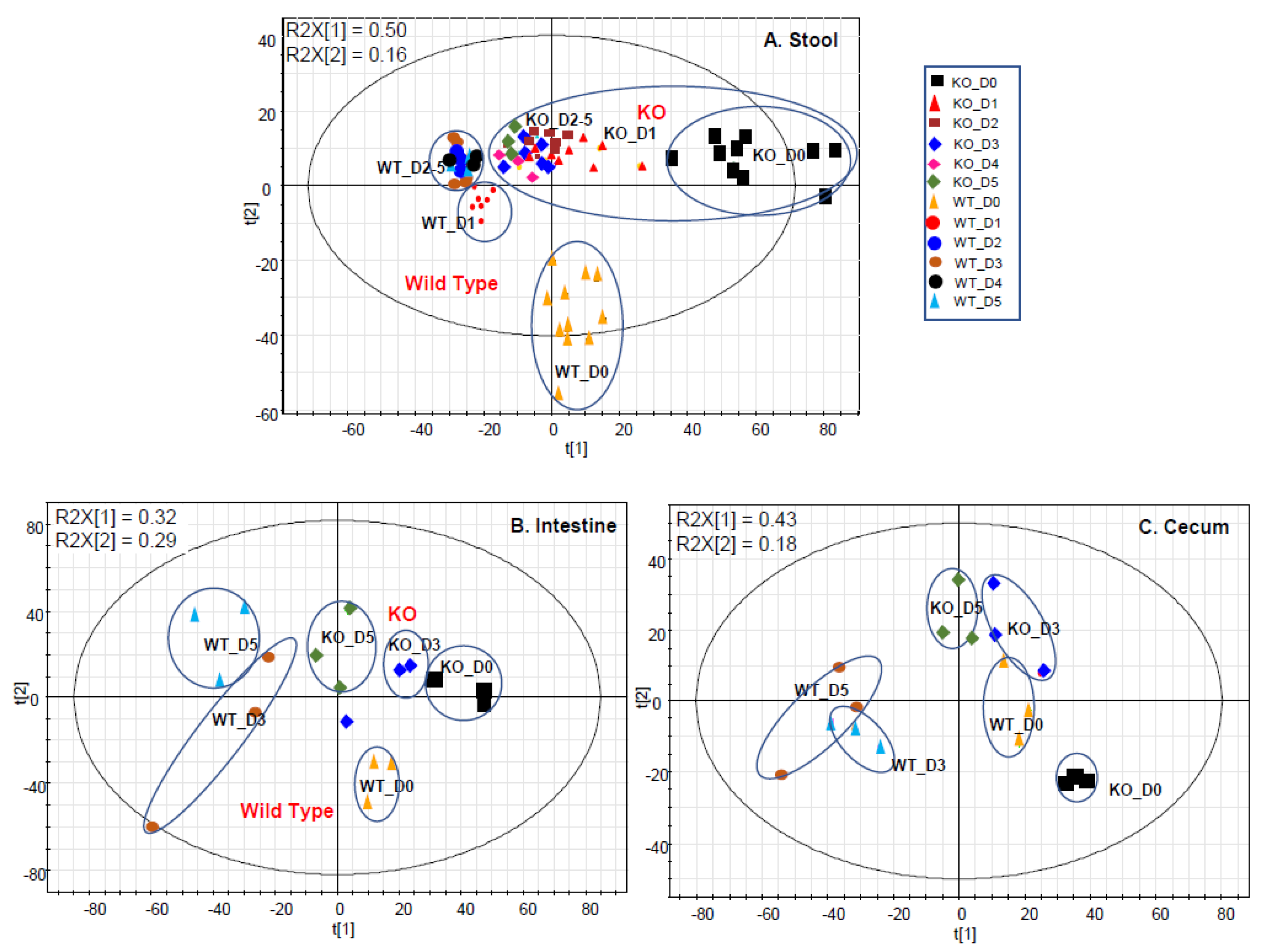
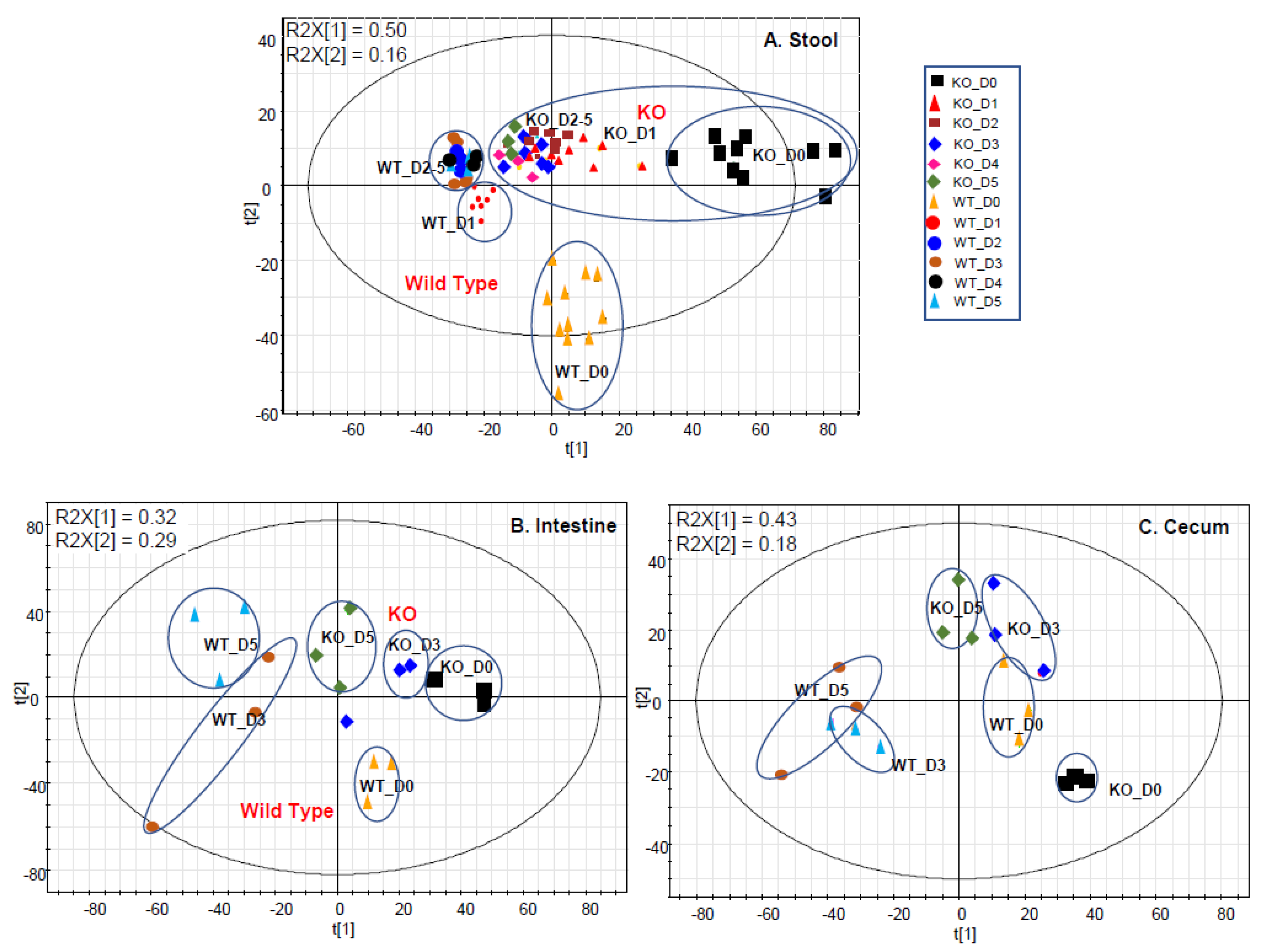
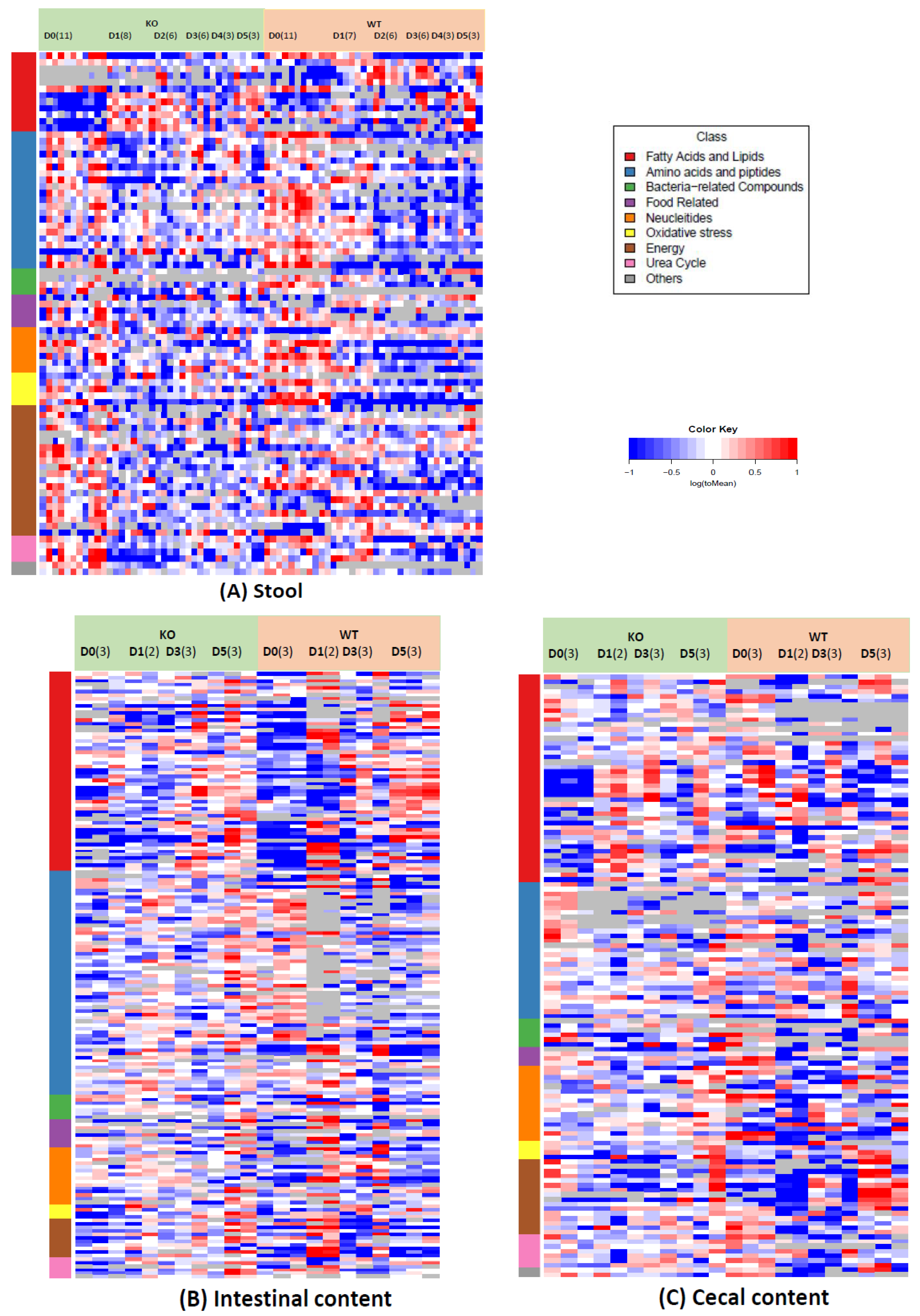
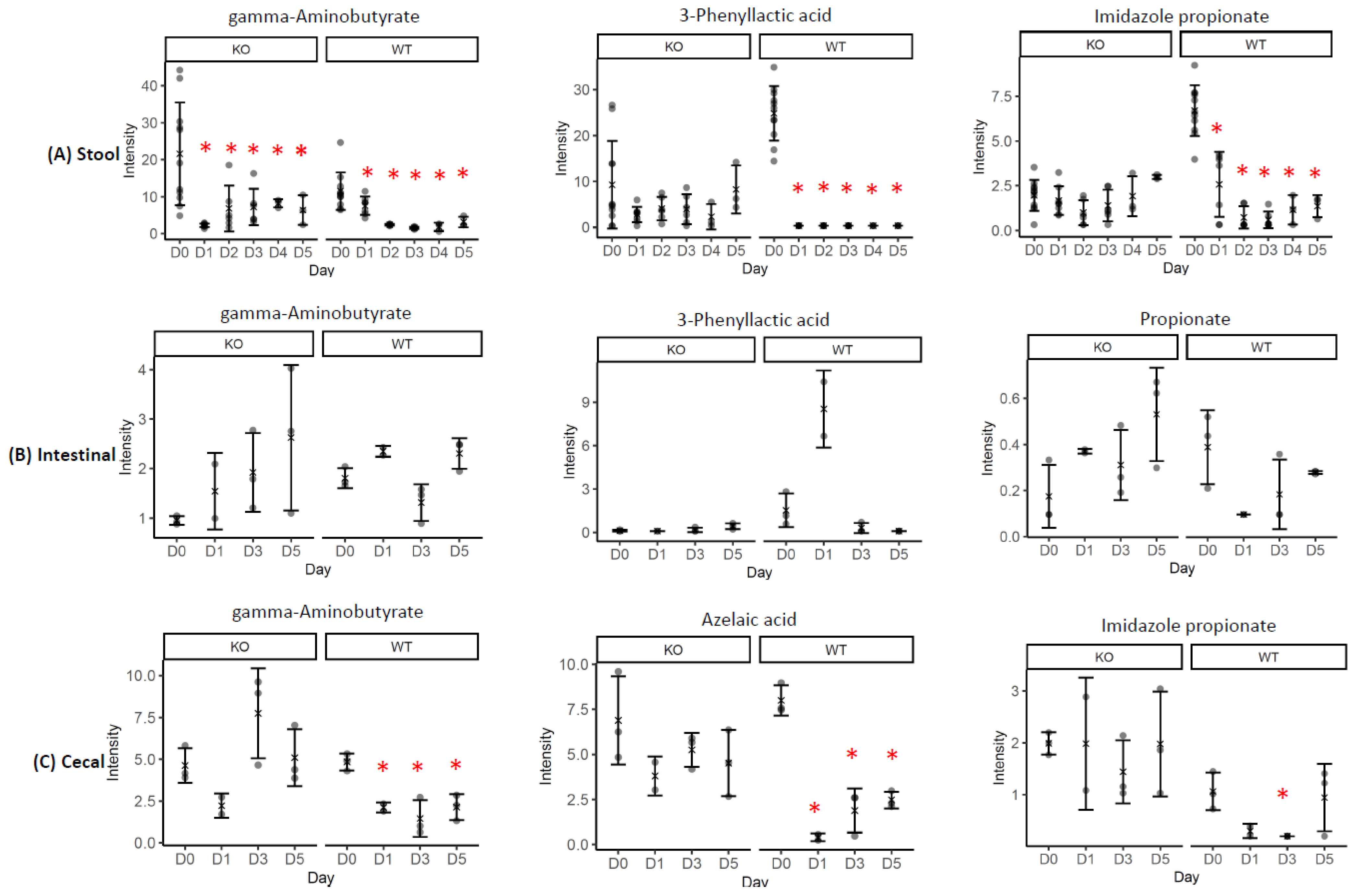
2.1. Gut Metabolome Alteration by Cef Treatments
2.2. Correlations of the GABA or Riboflavin to Gut Metabolites
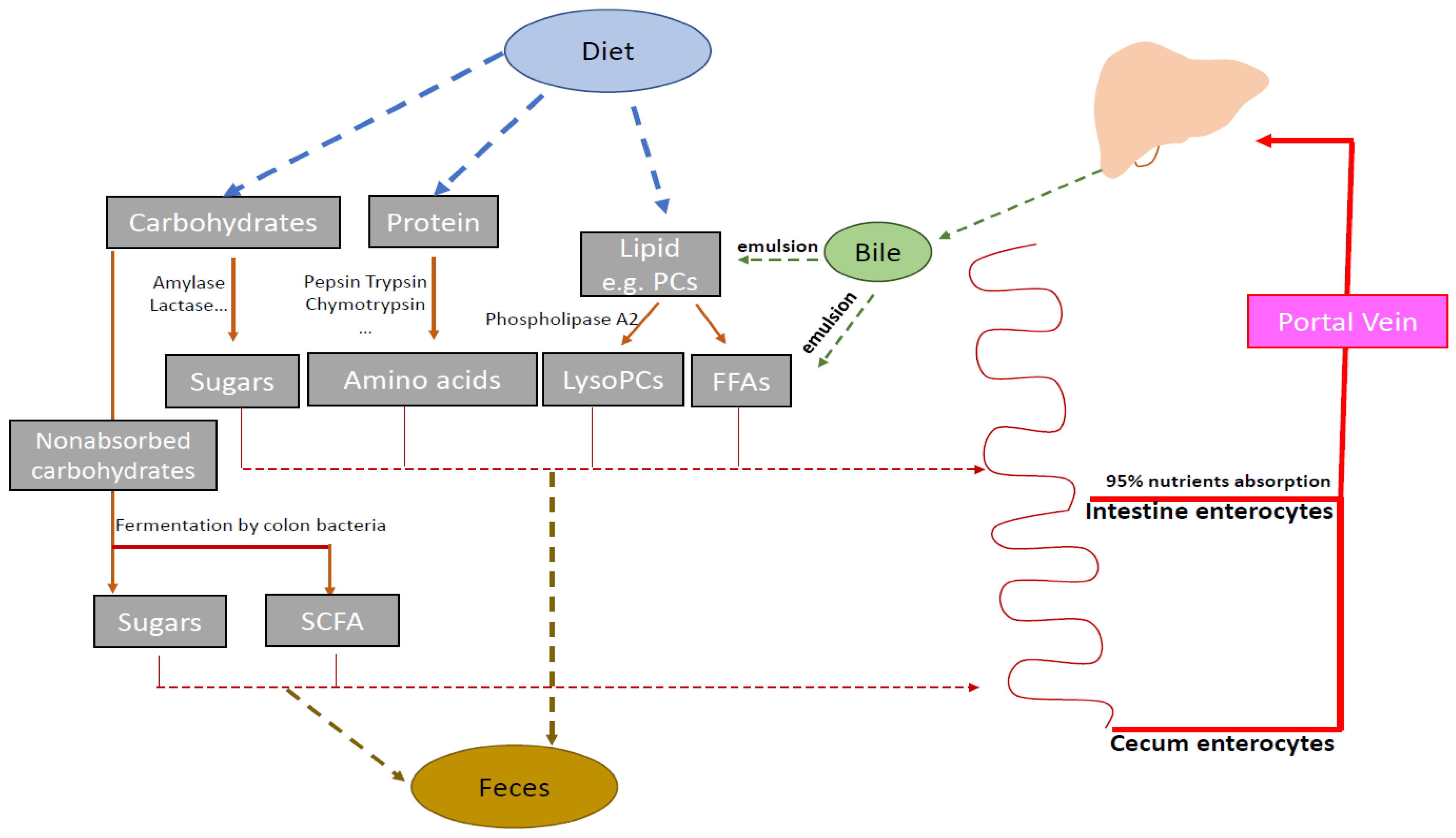
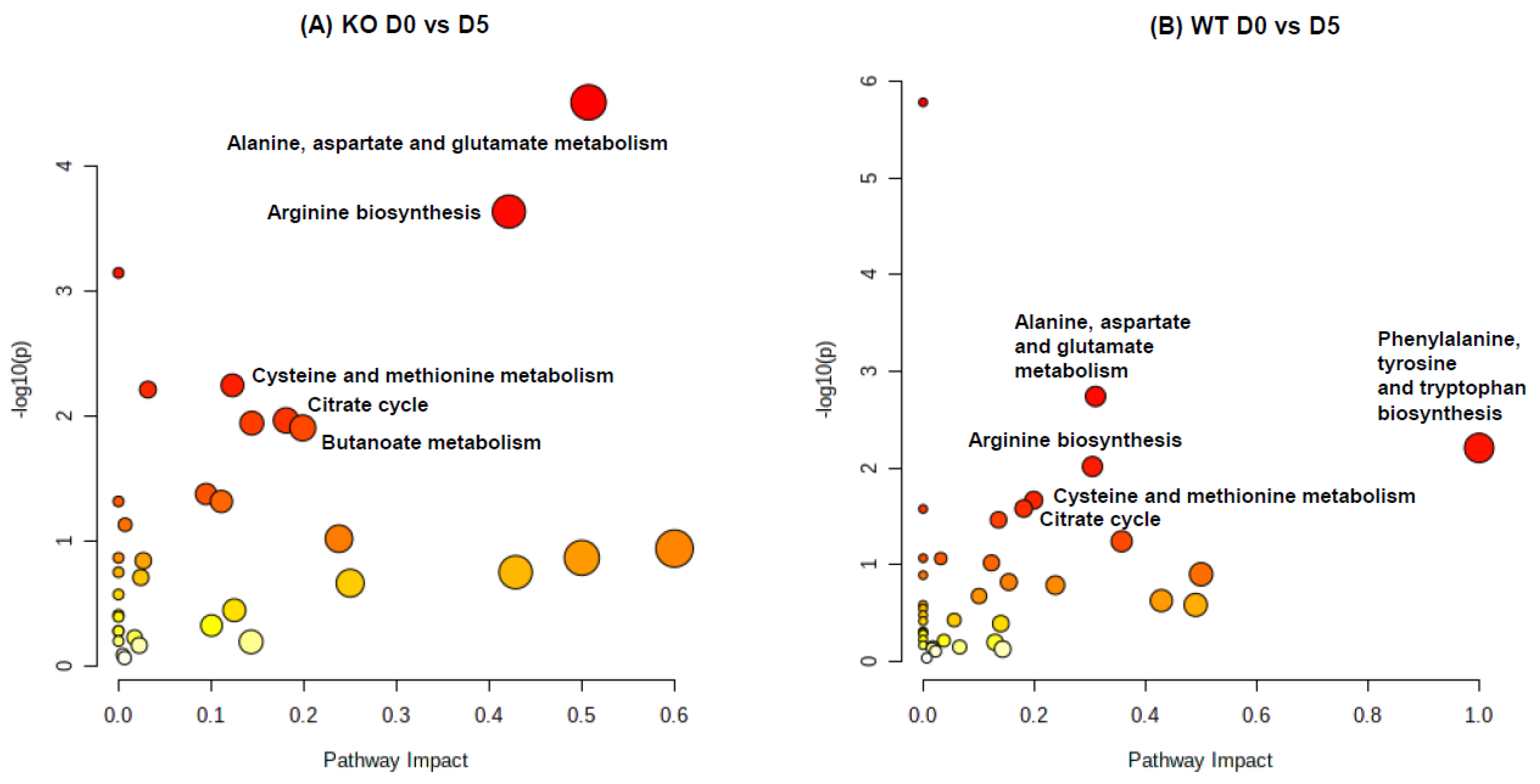
2.3. Pathway Impact Analysis
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Animal Care and Treatment
4.3. Quality Control in Metabolomics
4.4. Open Metabolic Profiling by UPLC/QTof-MS
4.5. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guarner, F.; Malagelada, J.R. Gut flora in health and disease. Lancet 2003, 361, 512–519. [Google Scholar] [CrossRef]
- Sokol, H.; Seksik, P.; Rigottier-Gois, L.; Lay, C.; Lepage, P.; Podglajen, I.; Marteau, P.; Dore, J. Specificities of the fecal microbiota in inflammatory bowel disease. Inflamm. Bowel Dis. 2006, 12, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Thompson-Chagoyan, O.C.; Maldonado, J.; Gil, A. Colonization and impact of disease and other factors on intestinal microbiota. Dig. Dis. Sci. 2007, 52, 2069–2077. [Google Scholar] [CrossRef]
- Sommer, F.; Anderson, J.M.; Bharti, R.; Raes, J.; Rosenstiel, P. The resilience of the intestinal microbiota influences health and disease. Nat. Rev. Microbiol. 2017, 15, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Moya, A.; Ferrer, M. Functional Redundancy-Induced Stability of Gut Microbiota Subjected to Disturbance. Trends Microbiol. 2016, 24, 402–413. [Google Scholar] [CrossRef]
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The impact of the gut microbiota on human health: An integrative view. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef] [Green Version]
- Larabi, A.; Barnich, N.; Nguyen, H.T.T. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy 2020, 16, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Backhed, F.; Fulton, L.; Gordon, J.I. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 2008, 3, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Swann, J.R.; Tuohy, K.M.; Lindfors, P.; Brown, D.T.; Gibson, G.R.; Wilson, I.D.; Sidaway, J.; Nicholson, J.K.; Holmes, E. Variation in antibiotic-induced microbial recolonization impacts on the host metabolic phenotypes of rats. J. Proteome. Res. 2011, 10, 3590–3603. [Google Scholar] [CrossRef] [Green Version]
- Yap, I.K.; Li, J.V.; Saric, J.; Martin, F.P.; Davies, H.; Wang, Y.; Wilson, I.D.; Nicholson, J.K.; Utzinger, J.; Marchesi, J.R.; et al. Metabonomic and microbiological analysis of the dynamic effect of vancomycin-induced gut microbiota modification in the mouse. J. Proteome. Res. 2008, 7, 3718–3728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackie, R.I.; Sghir, A.; Gaskins, H.R. Developmental microbial ecology of the neonatal gastrointestinal tract. Am. J. Clin. Nutr. 1999, 69, 1035S–1045S. [Google Scholar] [CrossRef]
- Angelucci, F.; Cechova, K.; Amlerova, J.; Hort, J. Antibiotics, gut microbiota, and Alzheimer’s disease. J. Neuroinflammation 2019, 16, 108. [Google Scholar] [CrossRef] [PubMed]
- Koenigsknecht, M.J.; Theriot, C.M.; Bergin, I.L.; Schumacher, C.A.; Schloss, P.D.; Young, V.B. Dynamics and establishment of Clostridium difficile infection in the murine gastrointestinal tract. Infect. Immun. 2015, 83, 934–941. [Google Scholar] [CrossRef] [Green Version]
- Theriot, C.M.; Koenigsknecht, M.J.; Carlson, P.E., Jr.; Hatton, G.E.; Nelson, A.M.; Li, B.; Huffnagle, G.B.; Li, J.Z.; Young, V.B. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat. Commun. 2014, 5, 3114. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Foss, E.D.; Zhang, I.; Hastie, J.L.; Giordano, N.P.; Gasparyan, L.; VinhNguyen, L.P.; Schubert, A.M.; Prasad, D.; McMichael, H.L.; et al. Microbiota of MR1 deficient mice confer resistance against Clostridium difficile infection. PLoS ONE 2019, 14, e0223025. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Cao, Z.; Smith, A.D.; Carlson, P.E., Jr.; Coryell, M.; Chen, H.; Beger, R.D. Bile Acid Profile and its Changes in Response to Cefoperazone Treatment in MR1 Deficient Mice. Metabolites 2020, 10, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Schnackenberg, L.K.; Khare, S.; Yang, X.; Greenhaw, J.; Salminen, W.; Mendrick, D.L.; Beger, R.D. Evaluating effects of penicillin treatment on the metabolome of rats. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2013, 932, 134–143. [Google Scholar] [CrossRef]
- Gherardin, N.A.; Keller, A.N.; Woolley, R.E.; Le Nours, J.; Ritchie, D.S.; Neeson, P.J.; Birkinshaw, R.W.; Eckle, S.B.G.; Waddington, J.N.; Liu, L.; et al. Diversity of T Cells Restricted by the MHC Class I-Related Molecule MR1 Facilitates Differential Antigen Recognition. Immunity 2016, 44, 32–45. [Google Scholar] [CrossRef] [Green Version]
- Kjer-Nielsen, L.; Patel, O.; Corbett, A.J.; Le Nours, J.; Meehan, B.; Liu, L.; Bhati, M.; Chen, Z.; Kostenko, L.; Reantragoon, R.; et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature 2012, 491, 717–723. [Google Scholar] [CrossRef]
- Huang, S.; Martin, E.; Kim, S.; Yu, L.; Soudais, C.; Fremont, D.H.; Lantz, O.; Hansen, T.H. MR1 antigen presentation to mucosal-associated invariant T cells was highly conserved in evolution. Proc. Natl. Acad. Sci. USA 2009, 106, 8290–8295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.D.; Zhang, I.T.; Schubert, A.M.; Giordano, N.P.; Hastie, J.E.; Cowley, S.C.; Carlson, P.E. MAIT cells: Shaping the microbiome, contributing to Clostridium difficile infection. J. Immunol. 2017, 198, 216.3. [Google Scholar]
- Toivari, M.H.; Nygard, Y.; Penttila, M.; Ruohonen, L.; Wiebe, M.G. Microbial D-xylonate production. Appl. Microbiol. Biotechnol. 2012, 96, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Chen, Q.; Wang, X.; Chen, K.; Ouyang, P. The Biosynthesis of D-1,2,4-Butanetriol From d-Arabinose With an Engineered Escherichia coli. Front. Bioeng. Biotechnol. 2022, 10, 844517. [Google Scholar] [CrossRef] [PubMed]
- Eberl, C.; Weiss, A.S.; Jochum, L.M.; Durai Raj, A.C.; Ring, D.; Hussain, S.; Herp, S.; Meng, C.; Kleigrewe, K.; Gigl, M.; et al. E. coli enhance colonization resistance against Salmonella Typhimurium by competing for galactitol, a context-dependent limiting carbon source. Cell Host Microbe 2021, 29, 1680–1692. [Google Scholar] [CrossRef]
- Osbelt, L.; Wende, M.; Almasi, E.; Derksen, E.; Muthukumarasamy, U.; Lesker, T.R.; Galvez, E.J.C.; Pils, M.C.; Schalk, E.; Chhatwal, P.; et al. Klebsiella oxytoca causes colonization resistance against multidrug-resistant K. pneumoniae in the gut via cooperative carbohydrate competition. Cell Host Microbe 2021, 29, 1663–1679. [Google Scholar] [CrossRef]
- Zhu, L.; Mack, C.; Wirtz, A.; Kranz, A.; Polen, T.; Baumgart, M.; Bott, M. Regulation of gamma-Aminobutyrate (GABA) Utilization in Corynebacterium glutamicum by the PucR-Type Transcriptional Regulator GabR and by Alternative Nitrogen and Carbon Sources. Front. Microbiol. 2020, 11, 544045. [Google Scholar] [CrossRef]
- Yoon, M.Y.; Min, K.B.; Lee, K.M.; Yoon, Y.; Kim, Y.; Oh, Y.T.; Lee, K.; Chun, J.; Kim, B.Y.; Yoon, S.H.; et al. A single gene of a commensal microbe affects host susceptibility to enteric infection. Nat. Commun. 2016, 7, 11606. [Google Scholar] [CrossRef] [Green Version]
- Gika, H.G.; Zisi, C.; Theodoridis, G.; Wilson, I.D. Protocol for quality control in metabolic profiling of biological fluids by U(H)PLC-MS. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2016, 1008, 15–25. [Google Scholar] [CrossRef]
- Sun, J.; Von Tungeln, L.S.; Hines, W.; Beger, R.D. Identification of metabolite profiles of the catechol-O-methyl transferase inhibitor tolcapone in rat urine using LC/MS-based metabonomics analysis. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 2557–2565. [Google Scholar] [CrossRef]
- Sun, J.; Schnackenberg, L.K.; Hansen, D.K.; Beger, R.D. Study of valproic acid-induced endogenous and exogenous metabolite alterations using LC-MS-based metabolomics. Bioanalysis 2010, 2, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vazquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic. Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The Human Metabolome Database. Nucleic. Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing, 3.3.1 ed.; R Foundation for Statistical Computing: Vienna, Austria, 2016; Available online: http://www.R-project.org/ (accessed on 1 January 2022).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, J.; Cao, Z.; Smith, A.D.; Carlson Jr, P.E.; Coryell, M.; Chen, H.; Beger, R.D. Evaluating Cefoperazone-Induced Gut Metabolic Functional Changes in MR1-Deficient Mice. Metabolites 2022, 12, 380. https://doi.org/10.3390/metabo12050380
Sun J, Cao Z, Smith AD, Carlson Jr PE, Coryell M, Chen H, Beger RD. Evaluating Cefoperazone-Induced Gut Metabolic Functional Changes in MR1-Deficient Mice. Metabolites. 2022; 12(5):380. https://doi.org/10.3390/metabo12050380
Chicago/Turabian StyleSun, Jinchun, Zhijun Cao, Ashley D. Smith, Paul E. Carlson Jr, Michael Coryell, Huizhong Chen, and Richard D. Beger. 2022. "Evaluating Cefoperazone-Induced Gut Metabolic Functional Changes in MR1-Deficient Mice" Metabolites 12, no. 5: 380. https://doi.org/10.3390/metabo12050380
APA StyleSun, J., Cao, Z., Smith, A. D., Carlson Jr, P. E., Coryell, M., Chen, H., & Beger, R. D. (2022). Evaluating Cefoperazone-Induced Gut Metabolic Functional Changes in MR1-Deficient Mice. Metabolites, 12(5), 380. https://doi.org/10.3390/metabo12050380