
Application of Mitochondrial and Oxidative Stress Biomarkers in the Evaluation of Neurocognitive Prognosis Following Acute Carbon Monoxide Poisoning



Abstract
:1. Introduction
2. Results
2.1. Characteristics of the Study Population
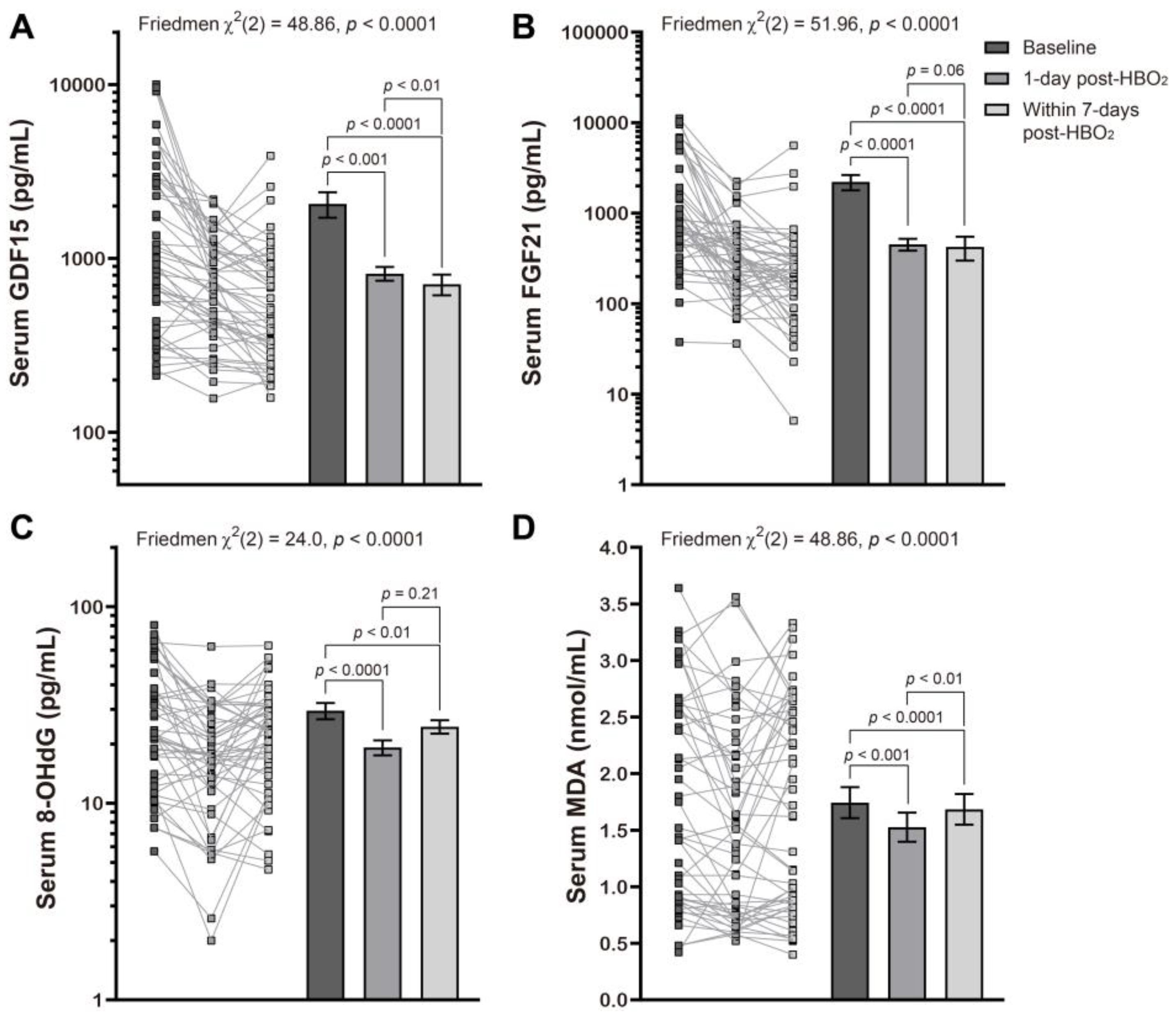
2.2. Changes in Stress Biomarkers Following Hyperbaric Oxygen Therapy
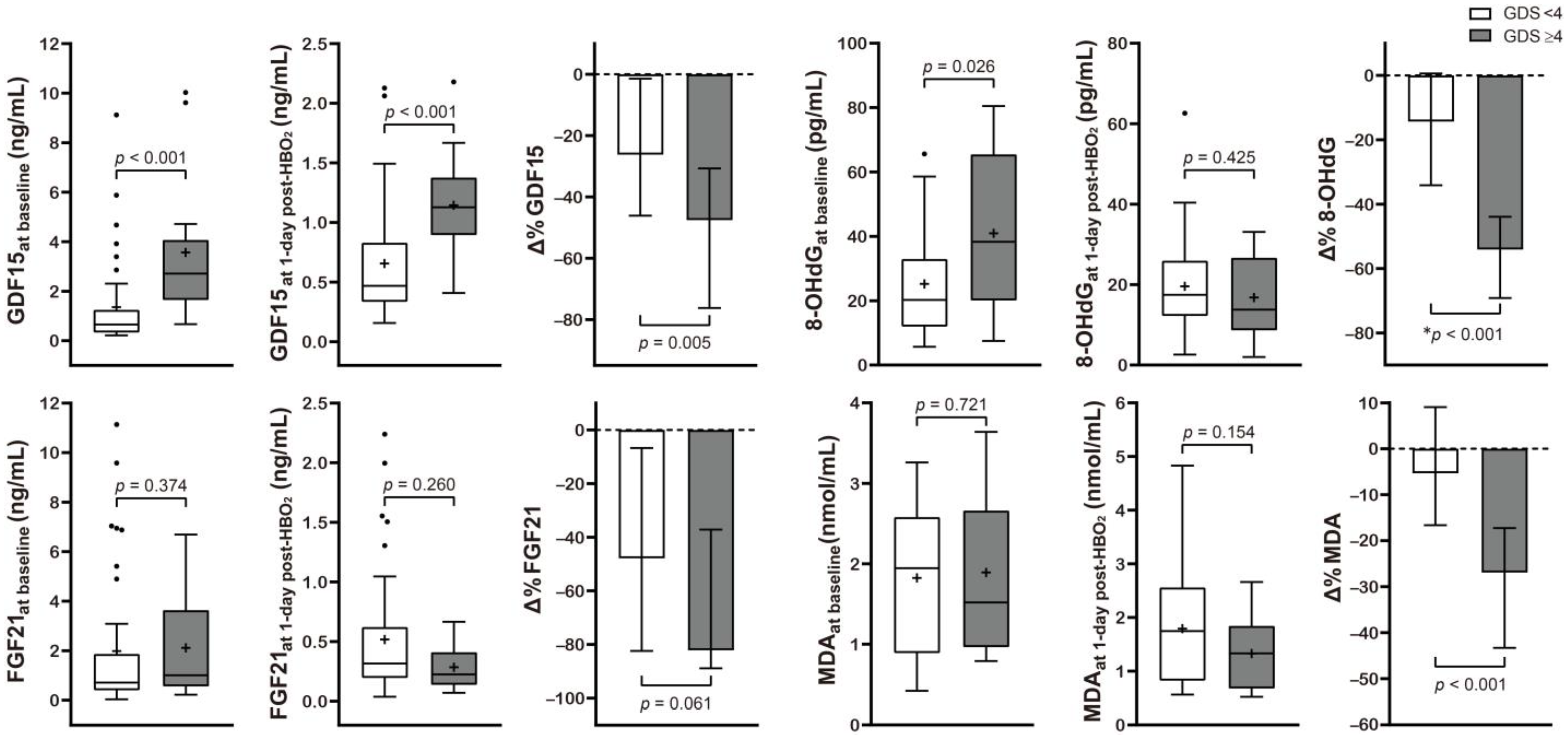
2.3. Stratification of the Serum Mitochondrial and Oxidative Stress Biomarkers Based on Neurocognitive Outcome Following CO Poisoning
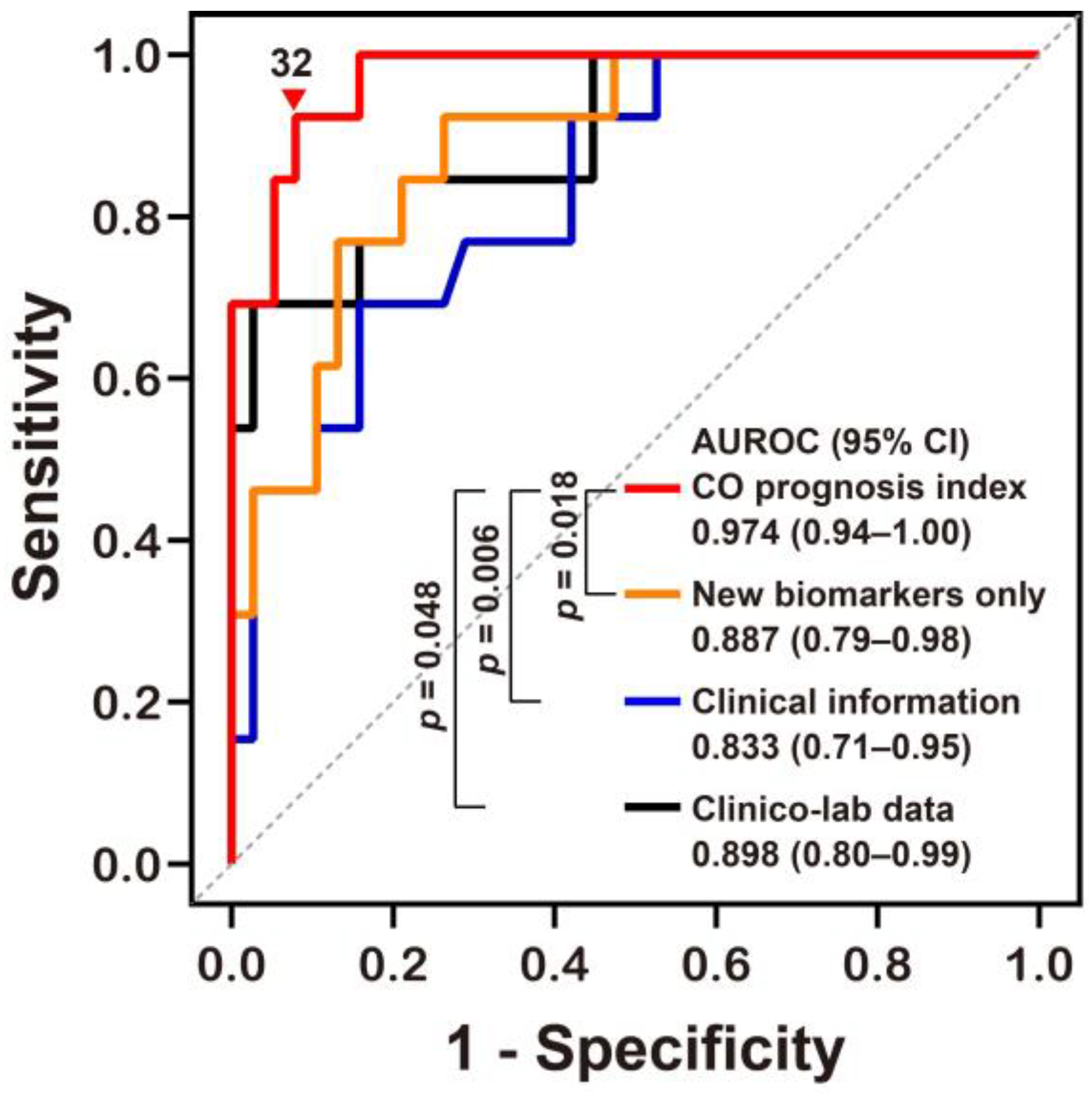
2.4. Potential Predictive Models for Neurocognitive Outcome Prognoses
3. Discussion
4. Materials and Methods
4.1. Study Design and Setting
4.2. Biomarkers of Mitochondrial and Oxidative Stress
4.3. Study Variables and Definitions
4.4. Study Outcomes
4.5. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rose, J.J.; Wang, L.; Xu, Q.; McTiernan, C.F.; Shiva, S.; Tejero, J.; Gladwin, M.T. Carbon monoxide poisoning: Pathogenesis, management, and future directions of therapy. Am. J. Respir. Crit. Care Med. 2017, 195, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Hampson, N.B.; Weaver, L.K. Carbon monoxide poisoning: A new incidence for an old disease. Undersea Hyperb Med. 2007, 34, 163–168. [Google Scholar] [PubMed]
- Hampson, N.B. U.S. Mortality due to carbon monoxide poisoning, 1999-2014. Accidental and intentional deaths. Ann. Am. Thor. Soc. 2016, 13, 1768–1774. [Google Scholar] [CrossRef]
- Weaver, L.K.; Hopkins, R.O.; Chan, K.J.; Churchill, S.; Elliott, C.G.; Clemmer, T.P.; Orme, J.F., Jr.; Thomas, F.O.; Morris, A.H. Hyperbaric oxygen for acute carbon monoxide poisoning. N. Engl. J. Med. 2002, 347, 1057–1067. [Google Scholar] [CrossRef]
- Goldbaum, L.R.; Orellano, T.; Dergal, E. Mechanism of the toxic action of carbon monoxide. Ann. Clin. Lab. Sci 1976, 6, 372–376. [Google Scholar]
- Brown, S.D.; Piantadosi, C.A. In vivo binding of carbon monoxide to cytochrome c oxidase in rat brain. J. Appl. Physiol. 1990, 68, 604–610. [Google Scholar] [CrossRef]
- Brown, S.D.; Piantadosi, C.A. Recovery of energy metabolism in rat brain after carbon monoxide hypoxia. J. Clin. Investig. 1992, 89, 666–672. [Google Scholar] [CrossRef]
- Gnaiger, E.; Lassnig, B.; Kuznetsov, A.; Rieger, G.; Margreiter, R. Mitochondrial oxygen affinity, respiratory flux control and excess capacity of cytochrome c oxidase. J. Exp. Biol. 1998, 201, 1129–1139. [Google Scholar] [CrossRef]
- Wald, G.; Allen, D.W. The equilibrium between cytochrome oxidase and carbon monoxide. J. Gen. Physiol 1957, 40, 593–608. [Google Scholar] [CrossRef] [Green Version]
- Fineschi, V.; Agricola, E.; Baroldi, G.; Bruni, G.; Cerretani, D.; Mondillo, S.; Parolini, M.; Turillazzi, E. Myocardial findings in fatal carbon monoxide poisoning: A human and experimental morphometric study. Int. J. Legal Med. 2000, 113, 276–282. [Google Scholar] [CrossRef]
- Lo Iacono, L.; Boczkowski, J.; Zini, R.; Salouage, I.; Berdeaux, A.; Motterlini, R.; Morin, D. A carbon monoxide-releasing molecule (corm-3) uncouples mitochondrial respiration and modulates the production of reactive oxygen species. Free Radic. Biol. Med. 2011, 50, 1556–1564. [Google Scholar] [CrossRef] [Green Version]
- Thom, S.R.; Bhopale, V.M.; Han, S.T.; Clark, J.M.; Hardy, K.R. Intravascular neutrophil activation due to carbon monoxide poisoning. Am. J. Respir. Crit. Care Med. 2006, 174, 1239–1248. [Google Scholar] [CrossRef] [PubMed]
- Piantadosi, C.A.; Zhang, J.; Levin, E.D.; Folz, R.J.; Schmechel, D.E. Apoptosis and delayed neuronal damage after carbon monoxide poisoning in the rat. Exp. Neurol. 1997, 147, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.; Chang, J.S.; Kong, I.D.; Baik, S.K.; Kim, M.Y.; Park, K.S. Multidimensional biomarker analysis including mitochondrial stress indicators for nonalcoholic fatty liver disease. Gut Liver 2022, 16. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.L.; Liang, C.; Sue, C.M. A comparison of current serum biomarkers as diagnostic indicators of mitochondrial diseases. Neurology 2016, 86, 2010–2015. [Google Scholar] [CrossRef] [PubMed]
- Chow, W.S.; Xu, A.; Woo, Y.C.; Tso, A.W.; Cheung, S.C.; Fong, C.H.; Tse, H.F.; Chau, M.T.; Cheung, B.M.; Lam, K.S. Serum fibroblast growth factor-21 levels are associated with carotid atherosclerosis independent of established cardiovascular risk factors. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2454–2459. [Google Scholar] [CrossRef] [Green Version]
- Adela, R.; Banerjee, S.K. Gdf-15 as a target and biomarker for diabetes and cardiovascular diseases: A translational prospective. J. Diabetes Res. 2015, 2015, 490842. [Google Scholar] [CrossRef]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, C. 8-hydroxy-2’ -deoxyguanosine (8-ohdg): A critical biomarker of oxidative stress and carcinogenesis. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2009, 27, 120–139. [Google Scholar] [CrossRef] [Green Version]
- Khoubnasabjafari, M.; Ansarin, K.; Jouyban, A. Reliability of malondialdehyde as a biomarker of oxidative stress in psychological disorders. Bioimpacts 2015, 5, 123–127. [Google Scholar] [CrossRef]
- Chang, J.S.; Chang, E.; Lee, Y.; Cha, Y.S.; Cha, S.K.; Cho, W.G.; Jeong, Y.; Kim, H.; Park, K.S. Hyperbaric oxygen exposure attenuates circulating stress biomarkers: A pilot interventional study. Int. J. Environ. Res. Public Health 2020, 17, 7853. [Google Scholar] [CrossRef]
- Ly, L.D.; Xu, S.; Choi, S.K.; Ha, C.M.; Thoudam, T.; Cha, S.K.; Wiederkehr, A.; Wollheim, C.B.; Lee, I.K.; Park, K.S. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp. Mol. Med. 2017, 49, e291. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Integrated stress response stimulates fgf21 expression: Systemic enhancer of longevity. Cell Signal 2017, 40, 10–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montero, R.; Yubero, D.; Villarroya, J.; Henares, D.; Jou, C.; Rodriguez, M.A.; Ramos, F.; Nascimento, A.; Ortez, C.I.; Campistol, J.; et al. Gdf-15 is elevated in children with mitochondrial diseases and is induced by mitochondrial dysfunction. PLoS ONE 2016, 11, e0148709. [Google Scholar] [CrossRef] [Green Version]
- Yatsuga, S.; Fujita, Y.; Ishii, A.; Fukumoto, Y.; Arahata, H.; Kakuma, T.; Kojima, T.; Ito, M.; Tanaka, M.; Saiki, R.; et al. Growth differentiation factor 15 as a useful biomarker for mitochondrial disorders. Ann. Neurol. 2015, 78, 814–823. [Google Scholar] [CrossRef] [Green Version]
- Morovat, A.; Weerasinghe, G.; Nesbitt, V.; Hofer, M.; Agnew, T.; Quaghebeur, G.; Sergeant, K.; Fratter, C.; Guha, N.; Mirzazadeh, M.; et al. Use of fgf-21 as a biomarker of mitochondrial disease in clinical practice. J. Clin. Med. 2017, 6, 80. [Google Scholar] [CrossRef] [Green Version]
- Chau, M.D.; Gao, J.; Yang, Q.; Wu, Z.; Gromada, J. Fibroblast growth factor 21 regulates energy metabolism by activating the ampk-sirt1-pgc-1alpha pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 12553–12558. [Google Scholar] [CrossRef] [Green Version]
- Planavila, A.; Redondo-Angulo, I.; Ribas, F.; Garrabou, G.; Casademont, J.; Giralt, M.; Villarroya, F. Fibroblast growth factor 21 protects the heart from oxidative stress. Cardiovasc. Res. 2015, 106, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Thiessen, S.E.; Vanhorebeek, I.; Derese, I.; Gunst, J.; Van den Berghe, G. Fgf21 response to critical illness: Effect of blood glucose control and relation with cellular stress and survival. J. Clin. Endocrinol. Metab. 2015, 100, E1319–E1327. [Google Scholar] [CrossRef] [Green Version]
- Ost, M.; Coleman, V.; Voigt, A.; van Schothorst, E.M.; Keipert, S.; van der Stelt, I.; Ringel, S.; Graja, A.; Ambrosi, T.; Kipp, A.P.; et al. Muscle mitochondrial stress adaptation operates independently of endogenous fgf21 action. Mol. Metab. 2016, 5, 79–90. [Google Scholar] [CrossRef]
- Kang, S.G.; Choi, M.J.; Jung, S.B.; Chung, H.K.; Chang, J.Y.; Kim, J.T.; Kang, Y.E.; Lee, J.H.; Hong, H.J.; Jun, S.M.; et al. Differential roles of gdf15 and fgf21 in systemic metabolic adaptation to the mitochondrial integrated stress response. iScience 2021, 24, 102181. [Google Scholar] [CrossRef]
- Gregorevic, P.; Lynch, G.S.; Williams, D.A. Hyperbaric oxygen modulates antioxidant enzyme activity in rat skeletal muscles. Eur. J. Appl. Physiol. 2001, 86, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Thom, S.R. Antagonism of carbon monoxide-mediated brain lipid peroxidation by hyperbaric oxygen. Toxicol. Appl. Pharmacol. 1990, 105, 340–344. [Google Scholar] [CrossRef]
- Thom, S.R.; Mendiguren, I.; Hardy, K.; Bolotin, T.; Fisher, D.; Nebolon, M.; Kilpatrick, L. Inhibition of human neutrophil beta2-integrin-dependent adherence by hyperbaric O2. Am. J. Physiol. 1997, 272, C770–C777. [Google Scholar] [CrossRef] [PubMed]
- Thom, S.R.; Bhopale, V.M.; Fisher, D. Hyperbaric oxygen reduces delayed immune-mediated neuropathology in experimental carbon monoxide toxicity. Toxicol. Appl. Pharmacol. 2006, 213, 152–159. [Google Scholar] [CrossRef]
- Dave, K.R.; Prado, R.; Busto, R.; Raval, A.P.; Bradley, W.G.; Torbati, D.; Pérez-Pinzón, M.A. Hyperbaric oxygen therapy protects against mitochondrial dysfunction and delays onset of motor neuron disease in wobbler mice. Neuroscience 2003, 120, 113–120. [Google Scholar] [CrossRef]
- Suzuki, J. Endurance performance is enhanced by intermittent hyperbaric exposure via up-regulation of proteins involved in mitochondrial biogenesis in mice. Physiol. Rep. 2017, 5, e13349. [Google Scholar] [CrossRef]
- Takemura, A.; Ishihara, A. Mild hyperbaric oxygen inhibits growth-related decrease in muscle oxidative capacity of rats with metabolic syndrome. J. Atheroscler. Thromb. 2017, 24, 26–38. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.D.; Piantadosi, C.A. Reversal of carbon monoxide-cytochrome c oxidase binding by hyperbaric oxygen in vivo. Adv. Exp. Med. Biol. 1989, 248, 747–754. [Google Scholar]
- Rothfuss, A.; Radermacher, P.; Speit, G. Involvement of heme oxygenase-1 (ho-1) in the adaptive protection of human lymphocytes after hyperbaric oxygen (hbo) treatment. Carcinogenesis 2001, 22, 1979–1985. [Google Scholar] [CrossRef] [Green Version]
- Speit, G.; Dennog, C.; Eichhorn, U.; Rothfuss, A.; Kaina, B. Induction of heme oxygenase-1 and adaptive protection against the induction of DNA damage after hyperbaric oxygen treatment. Carcinogenesis 2000, 21, 1795–1799. [Google Scholar] [CrossRef] [Green Version]
- Godman, C.A.; Joshi, R.; Giardina, C.; Perdrizet, G.; Hightower, L.E. Hyperbaric oxygen treatment induces antioxidant gene expression. Ann. N. Y. Acad. Sci. 2010, 1197, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Shyu, W.C.; Lin, S.Z.; Saeki, K.; Kubosaki, A.; Matsumoto, Y.; Onodera, T.; Chiang, M.F.; Thajeb, P.; Li, H. Hyperbaric oxygen enhances the expression of prion protein and heat shock protein 70 in a mouse neuroblastoma cell line. Cell Mol. Neurobiol. 2004, 24, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Koide, S.; Kinoshita, Y.; Ito, N.; Kimura, J.; Yokoyama, K.; Karube, I. Determination of human serum 8-hydroxy-2’-deoxyguanosine (8-ohdg) by hplc-ecd combined with solid phase extraction (spe). J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2010, 878, 2163–2167. [Google Scholar] [CrossRef] [PubMed]
- Mas-Bargues, C.; Escrivá, C.; Dromant, M.; Borrás, C.; Viña, J. Lipid peroxidation as measured by chromatographic determination of malondialdehyde. Human plasma reference values in health and disease. Arch. Biochem. Biophys. 2021, 709, 108941. [Google Scholar] [CrossRef]
- Boenzi, S.; Diodato, D. Biomarkers for mitochondrial energy metabolism diseases. Essays Biochem. 2018, 62, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Weaver, L.K. Carbon monoxide poisoning. In Hyperbaric Oxygen Therapy Indications, 14th ed.; Moon, R.E., Ed.; Best Publishing Company: North Palm Beach, FL, USA, 2019; pp. 81–104. [Google Scholar]
- Reisberg, B.; Ferris, S.H.; de Leon, M.J.; Crook, T. The global deterioration scale for assessment of primary degenerative dementia. Am. J. Psychiatry 1982, 139, 1136–1139. [Google Scholar] [CrossRef] [Green Version]
- Cho, D.H.; Ko, S.M.; Son, J.W.; Park, E.J.; Cha, Y.S. Myocardial injury and fibrosis from acute carbon monoxide poisoning: A prospective observational study. JACC Cardiovasc. Imaging 2021, 14, 1758–1770. [Google Scholar] [CrossRef]
- Lee, Y.; Cha, Y.S.; Kim, S.H.; Kim, H. Effect of hyperbaric oxygen therapy initiation time in acute carbon monoxide poisoning. Crit. Care Med. 2021, 49, e910–e919. [Google Scholar] [CrossRef]
- Kim, S.J.; Thom, S.R.; Kim, H.; Hwang, S.O.; Lee, Y.; Park, E.J.; Lee, S.J.; Cha, Y.S. Effects of adjunctive therapeutic hypothermia combined with hyperbaric oxygen therapy in acute severe carbon monoxide poisoning. Crit. Care Med. 20210, 48, e706–e714. [Google Scholar] [CrossRef]
- Paul, R.H.; Cohen, R.A.; Moser, D.J.; Zawacki, T.; Ott, B.R.; Gordon, N.; Stone, W. The global deterioration scale: Relationships to neuropsychological performance and activities of daily living in patients with vascular dementia. J. Geriatr. Psychiatry Neurol. 2002, 15, 50–54. [Google Scholar] [CrossRef]
- Eisdorfer, C.; Cohen, D.; Paveza, G.J.; Ashford, J.W.; Luchins, D.J.; Gorelick, P.B.; Hirschman, R.S.; Freels, S.A.; Levy, P.S.; Semla, T.P.; et al. An empirical evaluation of the global deterioration scale for staging alzheimer’s disease. Am. J. Psychiatry 1992, 149, 190–194. [Google Scholar] [CrossRef]
- Ozge, C.; Ozge, A.; Unal, O. Cognitive and functional deterioration in patients with severe copd. Behav. Neurol. 2006, 17, 121–130. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Total | Favorable Outcome | Poor Outcome | p-Value | |
|---|---|---|---|---|
| N | 51 (100) | 38 (74.5) | 13 (25.5) | |
| Age (years) | 47 (32, 60) | 41 (24, 49) | 60 (52, 71) | <0.001 |
| Sex (male) | 41 (80.3) | 30 (78.9) | 11 (84.6) | 0.999 |
| Intention of self-harm | 25 (49) | 21 (55.3) | 4 (30.8) | 0.199 |
| Source of CO | ||||
| No fire | 48 (94.1) | 35 (92.1) | 13 (100) | 0.561 |
| Fire | 3 (5.9) | 3 (7.9) | 0 (0) | |
| Maximal CO exposure time (h) | 3.5 (1.48, 8) | 2.71 (1, 5) | 8 (4.92, 12) | 0.002 |
| Time from rescue to ED (h) | 3.07 (1.52, 4.85) | 2.98 (1.73, 4.6) | 3.35 (1.48, 8.3) | 0.482 |
| GCS at the ED | 12 (8, 15) | 12 (8, 15) | 8 (8, 12) | 0.006 |
| Current smoker | 22 (43.1) | 19 (50) | 3 (23.1) | 0.114 |
| Current drinker * | 22 (43.1) | 17 (44.7) | 5 (38.5) | 0.693 |
| Comorbidities | ||||
| Diabetes mellitus | 7 (13.7) | 4 (10.5) | 3 (23.1) | 0.352 |
| Hypertension | 8 (15.7) | 4 (10.5) | 4 (30.8) | 0.179 |
| Dyslipidaemia | 7 (13.7) | 5 (13.2) | 2 (15.4) | 0.999 |
| Lung diseases | 1 (2) | 1 (2.6) | 0 (0) | 0.999 |
| Cardiovascular diseases | 3 (5.9) | 3 (7.9) | 0 (0) | 0.561 |
| Liver diseases | 1 (2) | 1 (2.6) | 0 (0) | 0.999 |
| Psychiatric diseases | 7 (13.7) | 7 (18.4) | 0 (0) | 0.169 |
| Symptoms and signs at the ED | ||||
| Loss of consciousness | 43 (84.3) | 30 (78.9) | 13 (100) | 0.096 |
| Shock | 4 (7.8) | 1 (2.6) | 3 (23.1) | 0.046 |
| Seizure | 1 (2) | 1 (2.6) | 0 (0) | 0.999 |
| Time elapsed from rescue to HBO2 therapy (h) | 4.97 (3.37, 9.08) | 4.63 (3.3, 7.09) | 6.58 (4.17, 16.1) | 0.098 |
| Biochemical markers | ||||
| CO-Hb (%) # | 24.1 ± 15.2 | 23.1 ± 15.8 | 27.2 ± 13.5 | 0.294 |
| Bicarbonate (mmol/L) # | 22 ± 4.1 | 23 ± 3 | 18.8 ± 5.2 | 0.005 |
| Lactate (mmol/L) | 2.3 (1.4, 3.3) | 2.2 (1.2, 3.1) | 2.7 (2.1, 8.5) | 0.042 |
| Creatine kinase (U/L) | 143 (103, 370) | 129 (95, 212) | 1379 (299, 7531) | <0.001 |
| Troponin I (pg/mL) | 38.8 (4.2, 541.5) | 12.1 (2.5, 229.6) | 541.5 (282, 2884) | <0.001 |
| Complications | ||||
| Acute kidney injury | 4 (7.8) | 0 (0) | 4 (30.8) | 0.003 |
| Rhabdomyolysis | 12 (23.5) | 2 (5.3) | 10 (76.9) | <0.001 |
| Myocardial injury | 25 (49.0) | 13 (34.2) | 12 (92.3) | <0.001 |
| Pneumonia | 10 (19.6) | 2 (5.3) | 8 (61.5) | <0.001 |
| Total | Favorable Outcome | Poor Outcome | p-Value | |
|---|---|---|---|---|
| GDF15initial (ng/mL) | 0.92 (0.4, 2.72) | 0.65 (0.33, 1.25) | 2.72 (1.65, 4.07) | <0.001 |
| FGF21initial (ng/mL) | 0.8 (0.44, 1.85) | 0.71 (0.4, 1.87) | 1.01 (0.56, 3.63) | 0.374 |
| 8OHdGinitial (pg/mL) | 22.1 (12.9, 38.3) | 20.3 (12.0, 33.0) | 38.3 (20.1, 65.5) | 0.026 |
| MDAinitial (nmol/mL) | 1.94 (0.9, 2.61) | 1.95 (0.89, 2.58) | 1.52 (0.97, 2.66) | 0.721 |
| GDF151d-HBO2 (ng/mL) | 0.66 (0.37, 1.13) | 0.47 (0.33, 0.83) | 1.13 (0.89, 1.37) | <0.001 |
| FGF211d-HBO2 (ng/mL) | 0.31 (0.17, 0.54) | 0.32 (0.19, 0.62) | 0.22 (0.14, 0.41) | 0.260 |
| 8-OHdG1d-HBO2 (pg/mL) | 17.3 (11.7, 25.5) | 17.5 (12.2, 26.0) | 13.8 (8.7, 26.7) | 0.425 |
| MDA1d-HBO2 (nmol/mL) | 1.56 (0.76, 2.36) | 1.75 (0.82, 2.56) | 1.33 (0.68, 1.84) | 0.154 |
| Δ% of GDF15 | −33.4 (−55.9, −11.7) | −26.2 (−46.1, −1.4) | −47.6 (−76.3, −30.7) | 0.005 |
| Δ% of FGF21 | −63.0 (−84.9, −10.0) | −47.8 (−82.4, −6.7) | −82.1 (−88.9, −37.2) | 0.061 |
| Δ% of 8-OHdG # | −25.0 ± 33.6 | −14.4 ± 31.0 | −56.1 ± 18.0 | <0.001 |
| Δ% of MDA | −9.2 (−23.2, 5.3) | −5.3 (−16.6, 9.0) | −26.9 (−43.3, −17.3) | <0.001 |
| Variables | Univariate Analyses | Multivariate Analyses (Adjusted OR) | |||
|---|---|---|---|---|---|
| Unadjusted OR | CO Prognosis Index | New Biomarkers Alone | Clinical Information | Clinico-Lab Data | |
| Age (years) | 1.07 (1.02–1.12) | – | N/A | – | – |
| GCS at the ED | 0.76 (0.61–0.94) | 0.50 (0.27–0.94) | N/A | 0.76 (0.60–0.97) | 0.74 (0.56–0.99) |
| CO exposure time (h) | 1.22 (1.06–1.41) | 1.45 (1.01–2.06) | N/A | 1.21 (1.05–1.40) | 1.19 (1.01–1.40) |
| Time elapsed from rescue to HBO2 (h) | 1.15 (1.02–1.29) | – | N/A | – | – |
| Bicarbonate (mmol/L) | 0.76 (0.63–0.93) | – | N/A | N/A | – |
| Lactate (mmol/L) | 1.38 (1.04–1.83) | – | N/A | N/A | – |
| Creatine kinase (mU/L) | 2.22 (0.99–4.97) | – | N/A | N/A | 2.54 (1.07–6.05) |
| Troponin I (µg/mL) | 2.06 (1.03–4.12) | – | N/A | N/A | – |
| GDF15baseline (ng/mL) | 1.47 (1.07–2.01) | – | – | N/A | N/A |
| 8OHdGbaseline (pg/mL) | 1.04 (1.01–1.08) | – | – | N/A | N/A |
| GDF151d-HBO2 (ng/mL) | 6.17 (1.59–23.9) | – | – | N/A | N/A |
| MDA1d-HBO2 (nmol/mL) | 0.55 (0.25–1.22) | – | – | N/A | N/A |
| Δ% of GDF15 | 0.97 (0.94–0.99) | 0.92 (0.85–1.00) | 0.96 (0.92–0.99) | N/A | N/A |
| Δ% of 8OHdG | 0.94 (0.90–0.98) | – | – | N/A | N/A |
| Δ% of MDA | 0.93 (0.89–0.98) | 0.84 (0.74–0.96) | 0.92(0.88–0.97) | N/A | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cha, Y.S.; Chang, J.S.; Kim, H.; Park, K.-S. Application of Mitochondrial and Oxidative Stress Biomarkers in the Evaluation of Neurocognitive Prognosis Following Acute Carbon Monoxide Poisoning. Metabolites 2022, 12, 201. https://doi.org/10.3390/metabo12030201
Cha YS, Chang JS, Kim H, Park K-S. Application of Mitochondrial and Oxidative Stress Biomarkers in the Evaluation of Neurocognitive Prognosis Following Acute Carbon Monoxide Poisoning. Metabolites. 2022; 12(3):201. https://doi.org/10.3390/metabo12030201
Chicago/Turabian StyleCha, Yong Sung, Jae Seung Chang, Hyun Kim, and Kyu-Sang Park. 2022. "Application of Mitochondrial and Oxidative Stress Biomarkers in the Evaluation of Neurocognitive Prognosis Following Acute Carbon Monoxide Poisoning" Metabolites 12, no. 3: 201. https://doi.org/10.3390/metabo12030201
APA StyleCha, Y. S., Chang, J. S., Kim, H., & Park, K.-S. (2022). Application of Mitochondrial and Oxidative Stress Biomarkers in the Evaluation of Neurocognitive Prognosis Following Acute Carbon Monoxide Poisoning. Metabolites, 12(3), 201. https://doi.org/10.3390/metabo12030201