
An Extensive Metabolomics Workflow to Discover Cardiotoxin-Induced Molecular Perturbations in Microtissues

 , , ,
, , ,
Abstract
1. Introduction
2. Results and Discussion
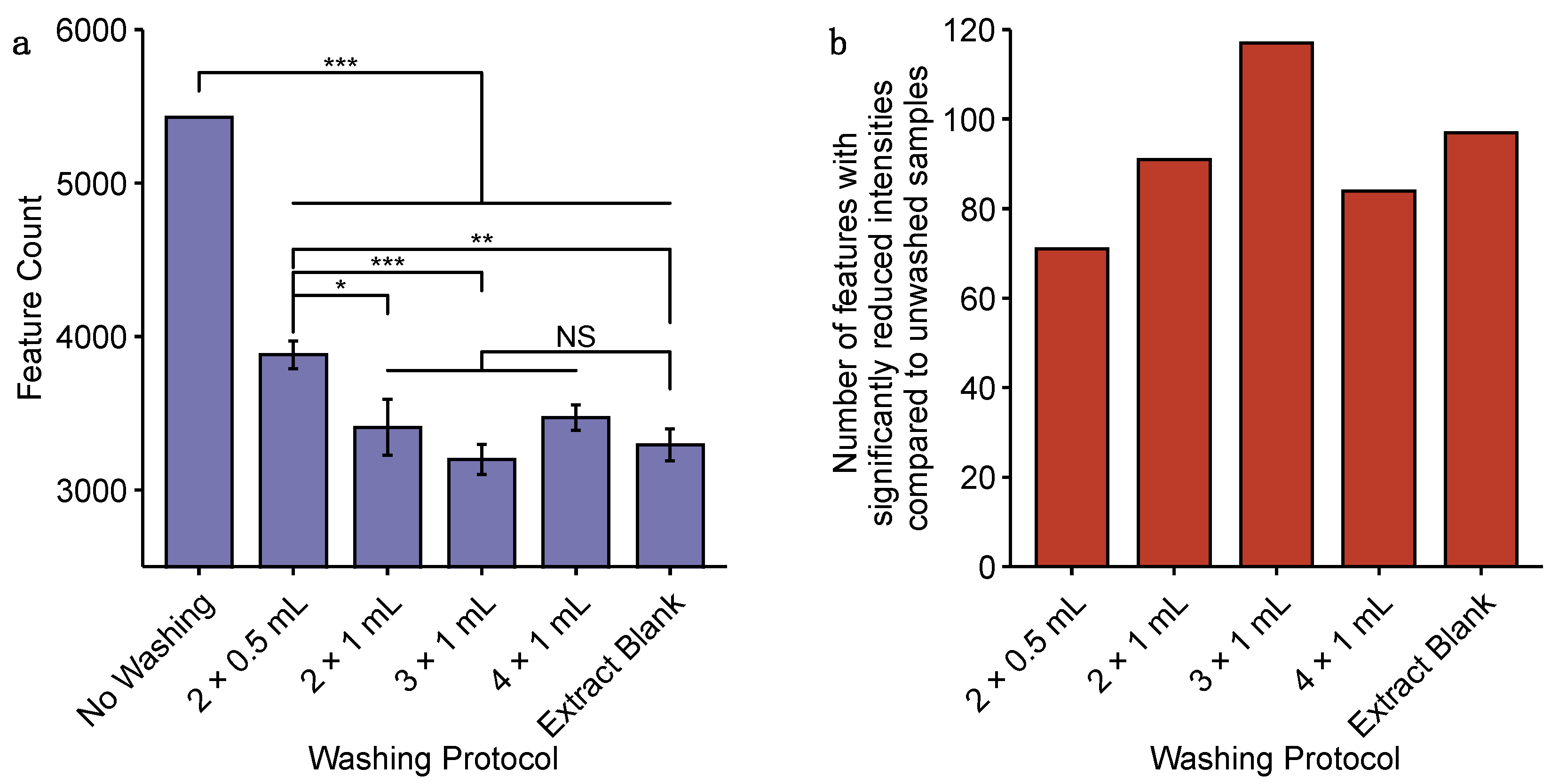
2.1. Comparison of Approaches for the Sampling of Cardiac Microtissues for Untargeted Metabolomics
2.2. Sensitivity and Reproducibility of nESI-DIMS Untargeted Metabolomics Measurements Using Very Low Biomass Samples
2.3. Probing Molecular Responses of Cardiac Microtissues to Sunitinib Exposure
2.3.1. Phenotypic Measurements Demonstrate Adversity at 72 h
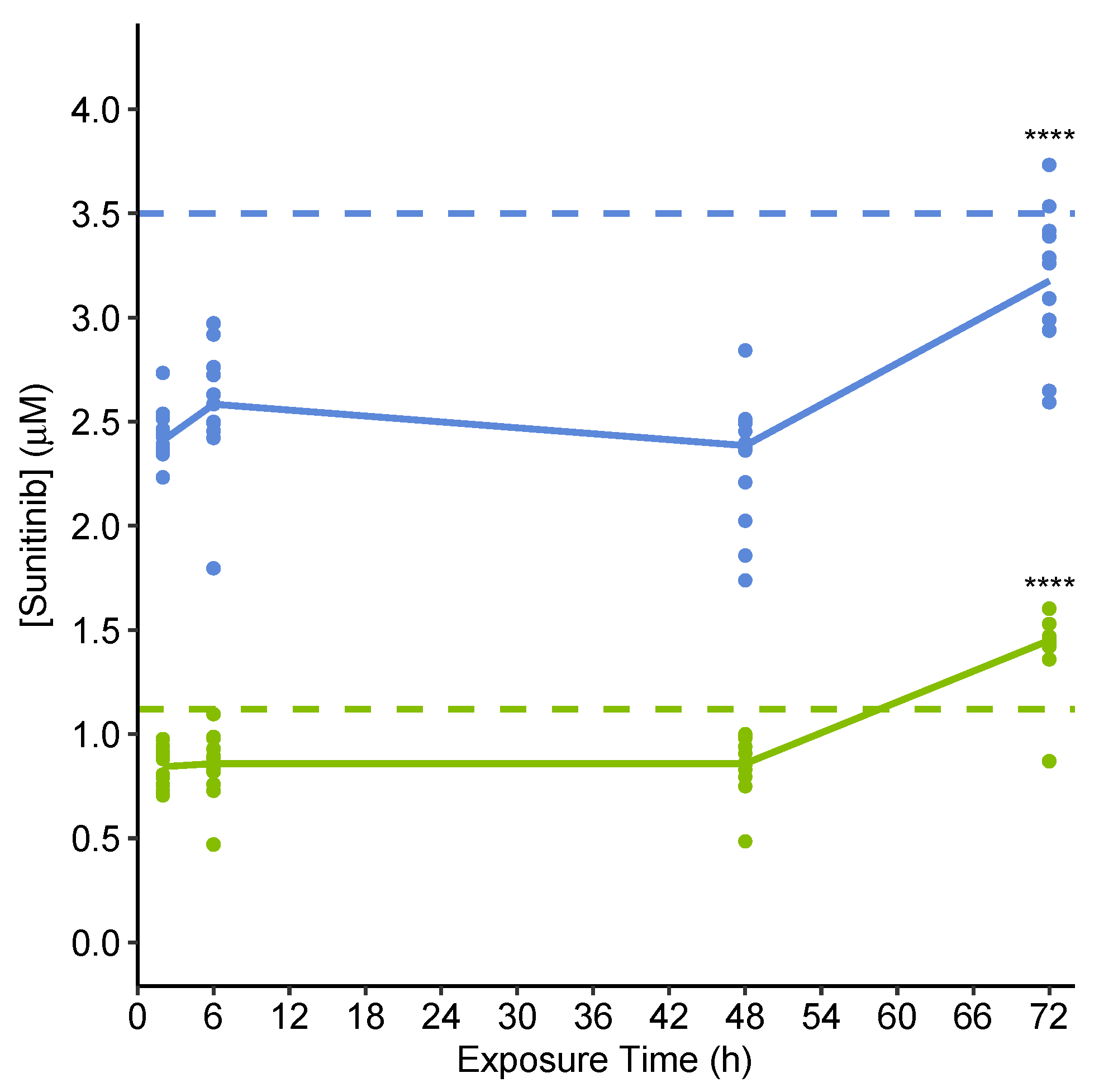
2.3.2. Quantification of Extracellular Sunitinib in Spent Culture Medium Indicates Uptake into Microtissues
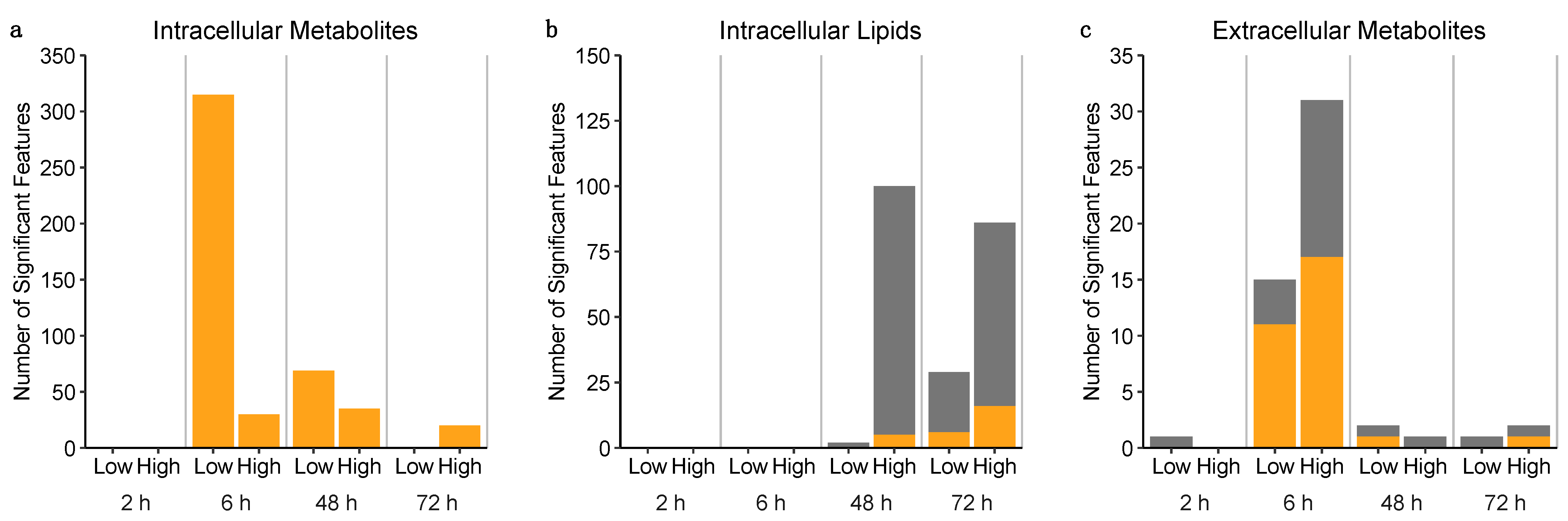
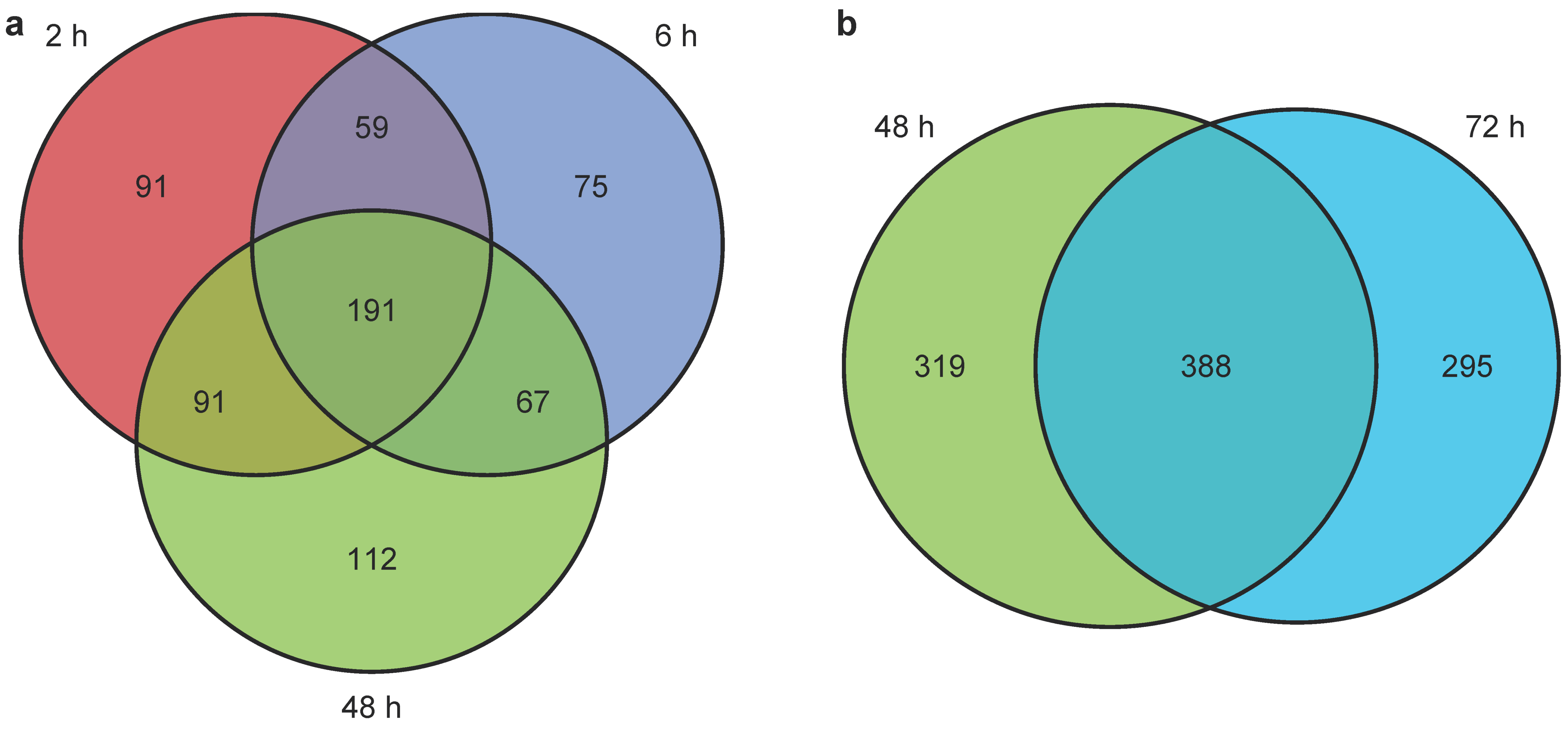
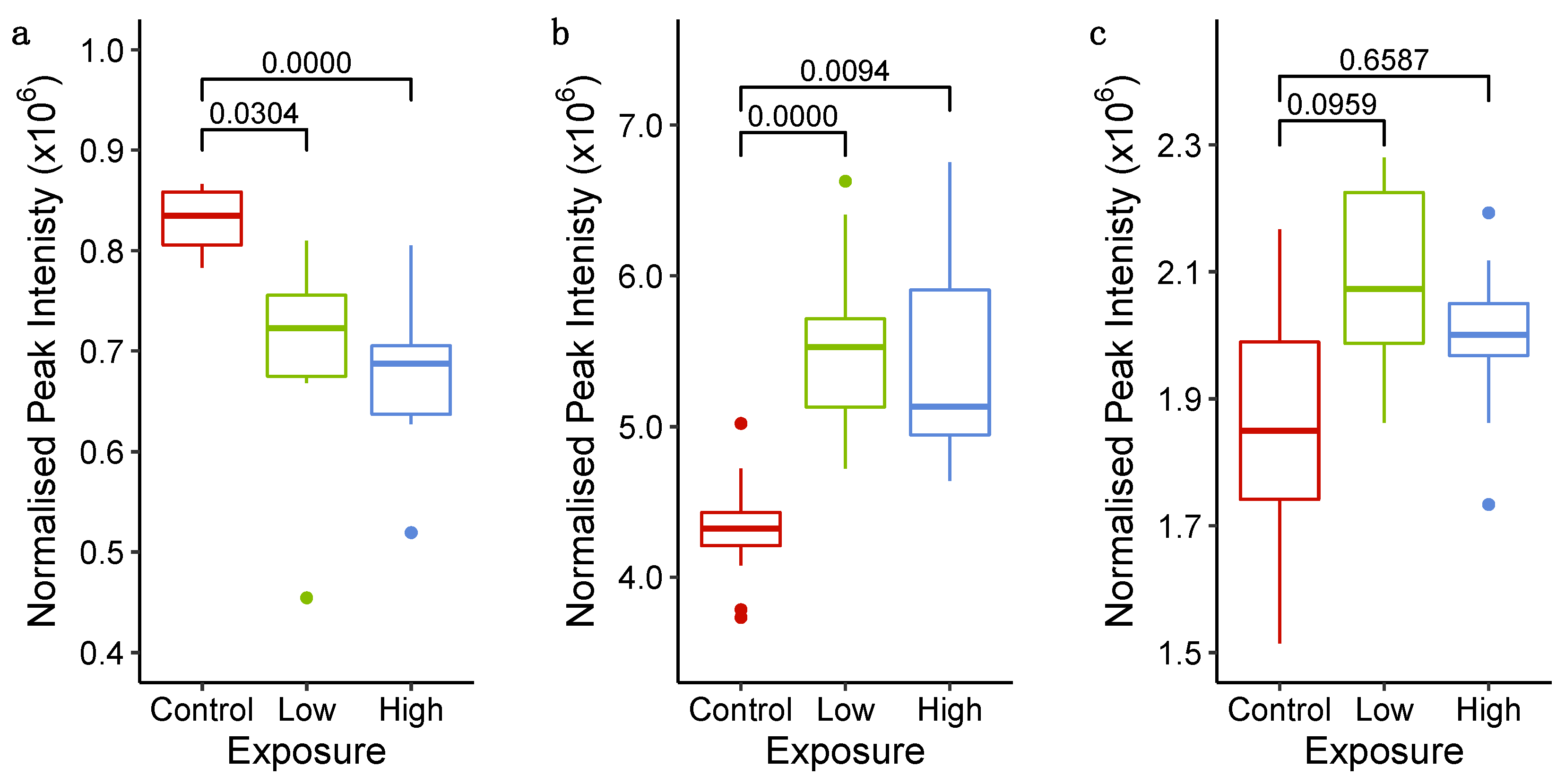
2.3.3. Untargeted Metabolomics and Lipidomics Reveal Time-Dependent Sunitinib-Induced Intracellular and Extracellular Perturbations in Cardiac Microtissues
3. Materials and Methods
3.1. Cell Culturing and Sunitinib Exposure
3.2. High-Content Biology Assay
3.3. Microtissue Sample Collection
3.3.1. Microtissue Sampling by Centrifugation
3.3.2. Microtissue Sampling by Filtration
3.4. Intracellular nESI-DIMS Untargeted Metabolomics and Lipidomics
3.4.1. Extraction of Intracellular Metabolites
3.4.2. Extraction of Intracellular Lipids
3.4.3. Resuspension of Dried Extracts
3.4.4. Data Acquisition by nESI-DIMS
3.4.5. Processing and Annotation of nESI-DIMS Data
3.5. UHPLC-MS(/MS) Untargeted Metabolomics and Quantification of Sunitinib
3.5.1. Extraction of Polar Metabolites from Culture Medium
3.5.2. Preparation of Sunitinib Calibration Standards, Toxicokinetic QCs and Blanks
3.5.3. Data Acquisition by UHPLC-MS(/MS)
3.5.4. Processing and Annotation of UHPLC-MS(/MS) Untargeted Metabolomics Data
3.5.5. Quantification of Sunitinib
3.6. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cook, D.; Brown, D.; Alexander, R.; March, R.; Morgan, P.; Satterthwaite, G.; Pangalos, M.N. Lessons learned from the fate of AstraZeneca′s drug pipeline: A five-dimensional framework. Nat. Rev. Drug Discov. 2014, 13, 419–431. [Google Scholar] [CrossRef]
- Laverty, H.; Benson, C.; Cartwright, E.; Cross, M.; Garland, C.; Hammond, T.; Holloway, C.; McMahon, N.; Milligan, J.; Park, B.; et al. How can we improve our understanding of cardiovascular safety liabilities to develop safer medicines? Br. J. Pharmacol. 2011, 163, 675–693. [Google Scholar] [CrossRef]
- Weaver, R.J.; Valentin, J.-P. Today′s Challenges to De-Risk and Predict Drug Safety in Human “Mind-the-Gap”. Toxicol. Sci. 2019, 167, 307–321. [Google Scholar] [CrossRef]
- Onakpoya, I.J.; Heneghan, C.J.; Aronson, J.K. Post-marketing withdrawal of 462 medicinal products because of adverse drug reactions: A systematic review of the world literature. BMC Med. 2016, 14, 10. [Google Scholar] [CrossRef]
- Gintant, G.; Sager, P.T.; Stockbridge, N. Evolution of strategies to improve preclinical cardiac safety testing. Nat. Rev. Drug Discov. 2016, 15, 457–471. [Google Scholar] [CrossRef]
- Li, X.; Zhang, R.; Zhao, B.; Lossin, C.; Cao, Z. Cardiotoxicity screening: A review of rapid-throughput in vitro approaches. Arch. Toxicol. 2016, 90, 1803–1816. [Google Scholar] [CrossRef]
- Pollard, C.E.; Abi Gerges, N.; Bridgland-Taylor, M.H.; Easter, A.; Hammond, T.G.; Valentin, J.P. An introduction to QT interval prolongation and non-clinical approaches to assessing and reducing risk. Br. J. Pharmacol. 2010, 159, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Pointon, A.; Abi-Gerges, N.; Cross, M.J.; Sidaway, J.E. Phenotypic profiling of structural cardiotoxins in vitro reveals dependency on multiple mechanisms of toxicity. Toxicol. Sci. Off. J. Soc. Toxicol 2013, 132, 317–326. [Google Scholar] [CrossRef][Green Version]
- Pointon, A.; Harmer, A.R.; Dale, I.L.; Abi-Gerges, N.; Bowes, J.; Pollard, C.; Garside, H. Assessment of cardiomyocyte contraction in human-induced pluripotent stem cell-derived cardiomyocytes. Toxicol. Sci. Off. J. Soc. Toxicol. 2015, 144, 227–237. [Google Scholar] [CrossRef]
- Doherty, K.R.; Talbert, D.R.; Trusk, P.B.; Moran, D.M.; Shell, S.A.; Bacus, S. Structural and functional screening in human induced-pluripotent stem cell-derived cardiomyocytes accurately identifies cardiotoxicity of multiple drug types. Toxicol. Appl. Pharmacol. 2015, 285, 51–60. [Google Scholar] [CrossRef]
- Doherty, K.R.; Wappel, R.L.; Talbert, D.R.; Trusk, P.B.; Moran, D.M.; Kramer, J.W.; Brown, A.M.; Shell, S.A.; Bacus, S. Multi-parameter in vitro toxicity testing of crizotinib, sunitinib, erlotinib, and nilotinib in human cardiomyocytes. Toxicol. Appl. Pharmacol. 2013, 272, 245–255. [Google Scholar] [CrossRef]
- Archer, C.R.; Sargeant, R.; Basak, J.; Pilling, J.; Barnes, J.R.; Pointon, A. Characterization and Validation of a Human 3D Cardiac Microtissue for the Assessment of Changes in Cardiac Pathology. Sci. Rep. 2018, 8, 10160. [Google Scholar] [CrossRef]
- Giacomelli, E.; Bellin, M.; Sala, L.; van Meer, B.J.; Tertoolen, L.G.; Orlova, V.V.; Mummery, C.L. Three-dimensional cardiac microtissues composed of cardiomyocytes and endothelial cells co-differentiated from human pluripotent stem cells. Development 2017, 144, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Soares, C.P.; Midlej, V.; Oliveira, M.E.W.D.; Benchimol, M.; Costa, M.L.; Mermelstein, C. 2D and 3D-Organized Cardiac Cells Shows Differences in Cellular Morphology, Adhesion Junctions, Presence of Myofibrils and Protein Expression. PLoS ONE 2012, 7, e38147. [Google Scholar] [CrossRef]
- Russell, S.; Wojtkowiak, J.; Neilson, A.; Gillies, R.J. Metabolic Profiling of healthy and cancerous tissues in 2D and 3D. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Hinson, J.T.; Chopra, A.; Lowe, A.; Sheng, C.C.; Gupta, R.M.; Kuppusamy, R.; O’Sullivan, J.; Rowe, G.; Wakimoto, H.; Gorham, J.; et al. Integrative Analysis of PRKAG2 Cardiomyopathy iPS and Microtissue Models Identifies AMPK as a Regulator of Metabolism, Survival, and Fibrosis. Cell Rep. 2016, 17, 3292–3304. [Google Scholar] [CrossRef]
- Pointon, A.; Pilling, J.; Dorval, T.; Wang, Y.; Archer, C.; Pollard, C. From the Cover: High-Throughput Imaging of Cardiac Microtissues for the Assessment of Cardiac Contraction during Drug Discovery. Toxicol. Sci. Off. J. Soc. Toxicol. 2017, 155, 444–457. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.A.; Smith, A.M.; Gryshkova, V.; Donley, E.L.R.; Valentin, J.-P.; Burrier, R.E. A Targeted Metabolomics-Based Assay Using Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes Identifies Structural and Functional Cardiotoxicity Potential. Toxicol. Sci. 2020, 174, 218–240. [Google Scholar] [CrossRef]
- Ramirez, T.; Daneshian, M.; Kamp, H.; Bois, F.Y.; Clench, M.R.; Coen, M.; Donley, B.; Fischer, S.M.; Ekman, D.R.; Fabian, E.; et al. Metabolomics in toxicology and preclinical research. Altex 2013, 30, 209–225. [Google Scholar] [CrossRef]
- Viant, M.R.; Ebbels, T.M.D.; Beger, R.D.; Ekman, D.R.; Epps, D.J.T.; Kamp, H.; Leonards, P.E.G.; Loizou, G.D.; MacRae, J.I.; van Ravenzwaay, B.; et al. Use cases, best practice and reporting standards for metabolomics in regulatory toxicology. Nat. Commun. 2019, 10, 3041. [Google Scholar] [CrossRef]
- Brockmeier, E.K.; Hodges, G.; Hutchinson, T.H.; Butler, E.; Hecker, M.; Tollefsen, K.E.; Garcia-Reyero, N.; Kille, P.; Becker, D.; Chipman, K.; et al. The Role of Omics in the Application of Adverse Outcome Pathways for Chemical Risk Assessment. Toxicol. Sci. Off. J. Soc. Toxicol. 2017, 158, 252–262. [Google Scholar] [CrossRef]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef]
- Bouhifd, M.; Hartung, T.; Hogberg, H.T.; Kleensang, A.; Zhao, L. Review: Toxicometabolomics. J. Appl. Toxicol. 2013, 33, 1365–1383. [Google Scholar] [CrossRef]
- Li, Y.; Ju, L.; Hou, Z.; Deng, H.; Zhang, Z.; Wang, L.; Yang, Z.; Yin, J.; Zhang, Y. Screening, verification, and optimization of biomarkers for early prediction of cardiotoxicity based on metabolomics. J. Proteome. Res. 2015, 14, 2437–2445. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Reijmers, T.; Chen, L.; Van Der Heijden, R.; Wang, M.; Peng, S.; Hankemeier, T.; Xu, G.; Van Der Greef, J. Systems toxicology study of doxorubicin on rats using ultra performance liquid chromatography coupled with mass spectrometry based metabolomics. Metabolomics 2009, 5, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, T.; Strigun, A.; Verlohner, A.; Huener, H.A.; Peter, E.; Herold, M.; Bordag, N.; Mellert, W.; Walk, T.; Spitzer, M.; et al. Prediction of liver toxicity and mode of action using metabolomics in vitro in HepG2 cells. Arch. Toxicol. 2018, 92, 893–906. [Google Scholar] [CrossRef]
- Cuykx, M.; Rodrigues, R.M.; Laukens, K.; Vanhaecke, T.; Covaci, A. In vitro assessment of hepatotoxicity by metabolomics: A review. Arch. Toxicol. 2018, 92, 3007–3029. [Google Scholar] [CrossRef]
- Southam, A.D.; Weber, R.J.; Engel, J.; Jones, M.R.; Viant, M.R. A complete workflow for high-resolution spectral-stitching nanoelectrospray direct-infusion mass-spectrometry-based metabolomics and lipidomics. Nat. Protoc. 2017, 12, 310–328. [Google Scholar] [CrossRef]
- Malinowska, J.M.; Palosaari, T.; Sund, J.; Carpi, D.; Bouhifd, M.; Weber, R.J.M.; Whelan, M.; Viant, M.R. Integrating in vitro metabolomics with a 96-well high-throughput screening platform. Metabolomics 2021, in press. [Google Scholar]
- Bordag, N.; Janakiraman, V.; Nachtigall, J.; Gonzalez Maldonado, S.; Bethan, B.; Laine, J.P.; Fux, E. Fast Filtration of Bacterial or Mammalian Suspension Cell Cultures for Optimal Metabolomics Results. PLoS ONE 2016, 11, e0159389. [Google Scholar] [CrossRef]
- ICH. S3A: Toxicokinetics: A Guidance for Assessing Systemic Exposure in Toxicology Studies; Springer: New York, NY, USA, 1995. [Google Scholar]
- Nyman, A.-M.; Schirmer, K.; Ashauer, R. Importance of Toxicokinetics for Interspecies Variation in Sensitivity to Chemicals. Environ. Sci. Techol. 2014, 48, 5946–5954. [Google Scholar] [CrossRef]
- Bowen, T.J.; Southam, A.D.; Hall, A.R.; Lloyd, G.R.; Macdonald, R.; Wilson, A.; Pointon, A.; Weber, R.J.M.; Viant, M.R. Simultaneously discovering the fate and biochemical effects of a xenobiotic through untargeted metabolomics. Nat. Commun. 2021. (under review). [Google Scholar]
- Parsons, H.M.; Ekman, D.R.; Collette, T.W.; Viant, M.R. Spectral relative standard deviation: A practical benchmark in metabolomics. Analyst 2009, 134, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Sarvin, B.; Lagziel, S.; Sarvin, N.; Mukha, D.; Kumar, P.; Aizenshtein, E.; Shlomi, T. Fast and sensitive flow-injection mass spectrometry metabolomics by analyzing sample-specific ion distributions. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Deng, J.; Zhang, G.; Neubert, T.A. Metabolomic Analysis of Glioma Cells Using Nanoflow Liquid Chromatography—Tandem Mass Spectrometry. In Glioblastoma: Methods and Protocols; Placantonakis, D.G., Ed.; Springer: New York, NY, USA, 2018; pp. 125–134. [Google Scholar] [CrossRef]
- Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, U.; Nemade, H.; Wagh, V.; Gaspar, J.A.; Ellis, J.K.; Srinivasan, S.P.; Spitkovski, D.; Nguemo, F.; Louisse, J.; Bremer, S.; et al. Identification of genomic biomarkers for anthracycline-induced cardiotoxicity in human iPSC-derived cardiomyocytes: An in vitro repeated exposure toxicity approach for safety assessment. Arch. Toxicol. 2016, 90, 2763–2777. [Google Scholar] [CrossRef]
- Hsu, H.-C.; Chen, C.-Y.; Chen, M.-F. N-3 polyunsaturated fatty acids decrease levels of doxorubicin-induced reactive oxygen species in cardiomyocytes—Involvement of uncoupling protein UCP2. J. Biomed. Sci. 2014, 21, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Q.; Chen, M.T.; Zhang, R.; Zhang, Y.; Li, W.; Li, Y.G. Docosahexaenoic Acid Attenuates Doxorubicin-induced Cytotoxicity and Inflammation by Suppressing NF-kappaB/iNOS/NO Signaling Pathway Activation in H9C2 Cardiac Cells. J. Cardiovasc. Pharmacol. 2016, 67, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Weber, R.J.M.; Zhou, J. DIMSpy: Python package for processing direct-infusion mass spectrometry-based metabolomics and lipidomics data. Zenodo 2020. [Google Scholar] [CrossRef]
- Lloyd, G.R.; Jankevics, A.; Weber, R.J.M. Struct: An R/Bioconductor-based framework for standardized metabolomics data analysis and beyond. Bioinformatics 2021, 36, 5551–5552. [Google Scholar] [CrossRef]
- Kirwan, J.A.; Broadhurst, D.I.; Davidson, R.L.; Viant, M.R. Characterising and correcting batch variation in an automated direct infusion mass spectrometry (DIMS) metabolomics workflow. Anal. Bioanal. Chem. 2013, 405, 5147–5157. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Sud, M.; Fahy, E.; Cotter, D.; Brown, A.; Dennis, E.A.; Glass, C.K.; Merrill, A.H.; Murphy, R.C.; Raetz, C.R.H.; Russell, D.W.; et al. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 2007, 35, D527–D532. [Google Scholar] [CrossRef]
- Southam, A.D.; Pursell, H.; Frigerio, G.; Jankevics, A.; Weber, R.J.M.; Dunn, W.B. Characterization of Monophasic Solvent-Based Tissue Extractions for the Detection of Polar Metabolites and Lipids Applying Ultrahigh-Performance Liquid Chromatography—Mass Spectrometry Clinical Metabolic Phenotyping Assays. J. Proteom. Res. 2020, 20, 831–840. [Google Scholar] [CrossRef]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- Smith, C.A.; Want, E.J.; O′Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Dodder, N. OrgMassSpecR: Organic Mass Spectrometry, R Package Version 0.5-3. Available online: https://CRAN.R-project.org/package=OrgMassSpecR (accessed on 31 August 2021).
- Sumner, L.W.; Urbanczyk-Wochniak, E.; Broeckling, C.D. Metabolomics data analysis, visualization, and integration. Methods Mol. Biol. 2007, 406, 409–436. [Google Scholar] [CrossRef]
- Pang, Z.; Chong, J.; Li, S.; Xia, J. MetaboAnalystR 3.0: Toward an Optimized Workflow for Global Metabolomics. Metabolites 2020, 10, 186. [Google Scholar] [CrossRef]
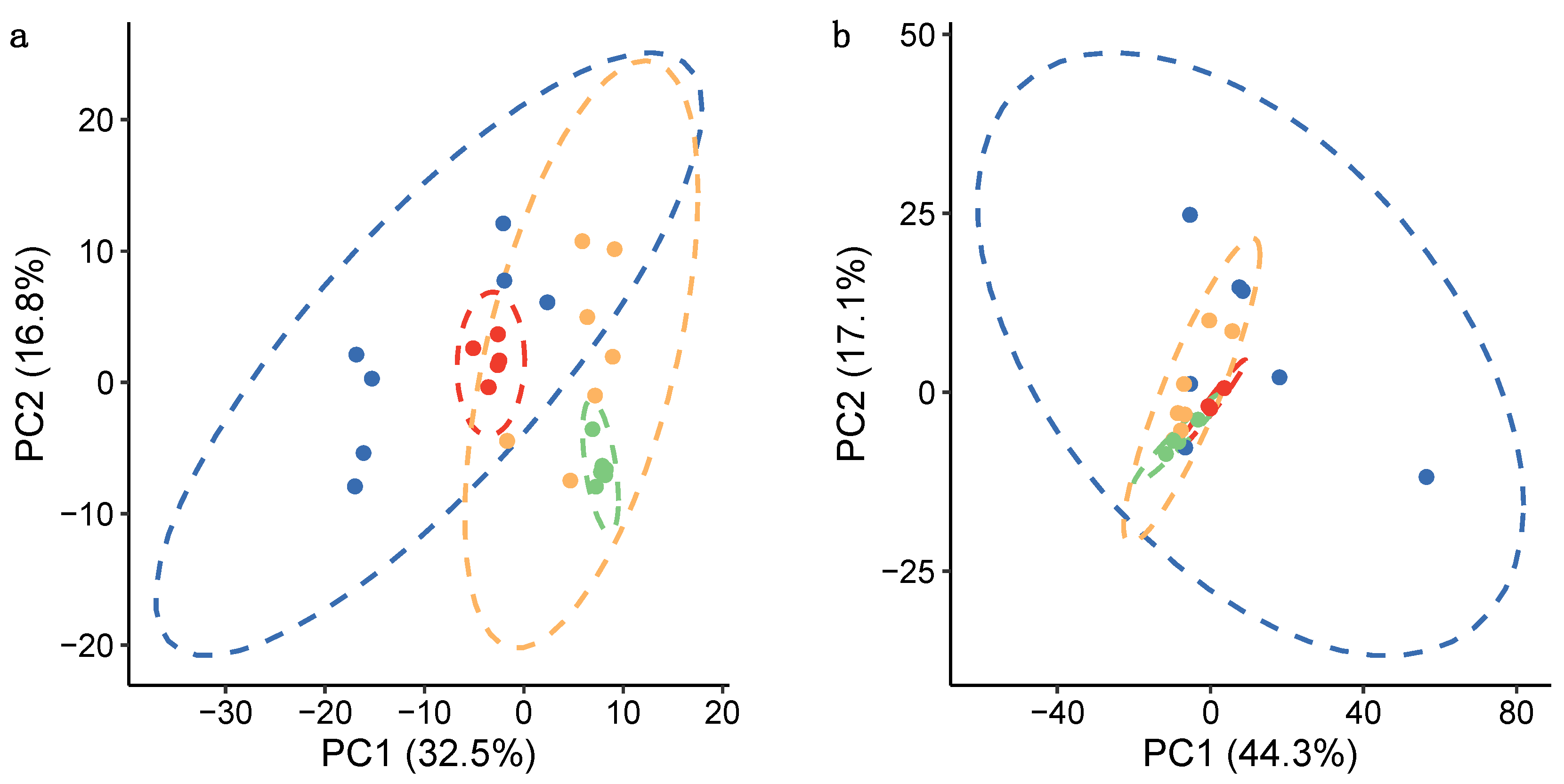
 centrifugation—biological replicates;
centrifugation—biological replicates;  centrifugation—technical replicates;
centrifugation—technical replicates;  filtration—biological replicates; and
filtration—biological replicates; and  filtration—technical replicates. Dashed lines show 95% confidence intervals for each group of samples.
centrifugation—biological replicates; centrifugation—technical replicates; filtration—biological replicates; and filtration—technical replicates. Dashed lines show 95% confidence intervals for each group of samples.
filtration—technical replicates. Dashed lines show 95% confidence intervals for each group of samples.
centrifugation—biological replicates; centrifugation—technical replicates; filtration—biological replicates; and filtration—technical replicates. Dashed lines show 95% confidence intervals for each group of samples.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay | Metabolomics | Lipidomics | ||
|---|---|---|---|---|
| Sampling Method | Centrifugation | Filtration | Centrifugation | Filtration |
| Feature count | 1697 | 3169 | 3173 | 4919 |
| Technical variation: median RSD across technical replicates (%) | 17.0 | 12.9 | 13.0 | 20.4 |
| Total variation: median RSD across biological replicates (%) | 42.1 | 35.5 | 50.2 | 31.6 |
| Assay | Metabolomics | Lipidomics | ||||||
|---|---|---|---|---|---|---|---|---|
| Sample biomass (Number of pooled cardiac microtissues) | 7 | 14 | 21 | 28 | 7 | 14 | 21 | 28 |
| Feature count | 796 | 817 | 1030 | 1321 | 1427 | 1765 | 2336 | 2507 |
| Technical variation: median RSD across intra-study QCs (%) | 13.7 | 10.6 | 18.0 | 20.5 | 17.6 | 16.9 | 15.1 | 12.4 |
| Total variation: median RSD across biological replicates (%) | 37.1 | 39.0 | 40.5 | 31.9 | 33.4 | 37.9 | 34.7 | 27.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bowen, T.J.; Hall, A.R.; Lloyd, G.R.; Weber, R.J.M.; Wilson, A.; Pointon, A.; Viant, M.R. An Extensive Metabolomics Workflow to Discover Cardiotoxin-Induced Molecular Perturbations in Microtissues. Metabolites 2021, 11, 644. https://doi.org/10.3390/metabo11090644
Bowen TJ, Hall AR, Lloyd GR, Weber RJM, Wilson A, Pointon A, Viant MR. An Extensive Metabolomics Workflow to Discover Cardiotoxin-Induced Molecular Perturbations in Microtissues. Metabolites. 2021; 11(9):644. https://doi.org/10.3390/metabo11090644
Chicago/Turabian StyleBowen, Tara J., Andrew R. Hall, Gavin R. Lloyd, Ralf J. M. Weber, Amanda Wilson, Amy Pointon, and Mark R. Viant. 2021. "An Extensive Metabolomics Workflow to Discover Cardiotoxin-Induced Molecular Perturbations in Microtissues" Metabolites 11, no. 9: 644. https://doi.org/10.3390/metabo11090644
APA StyleBowen, T. J., Hall, A. R., Lloyd, G. R., Weber, R. J. M., Wilson, A., Pointon, A., & Viant, M. R. (2021). An Extensive Metabolomics Workflow to Discover Cardiotoxin-Induced Molecular Perturbations in Microtissues. Metabolites, 11(9), 644. https://doi.org/10.3390/metabo11090644