
Experience with the Urinary Tetrasaccharide Metabolite for Pompe Disease in the Diagnostic Laboratory



Abstract
:1. Introduction
2. Results
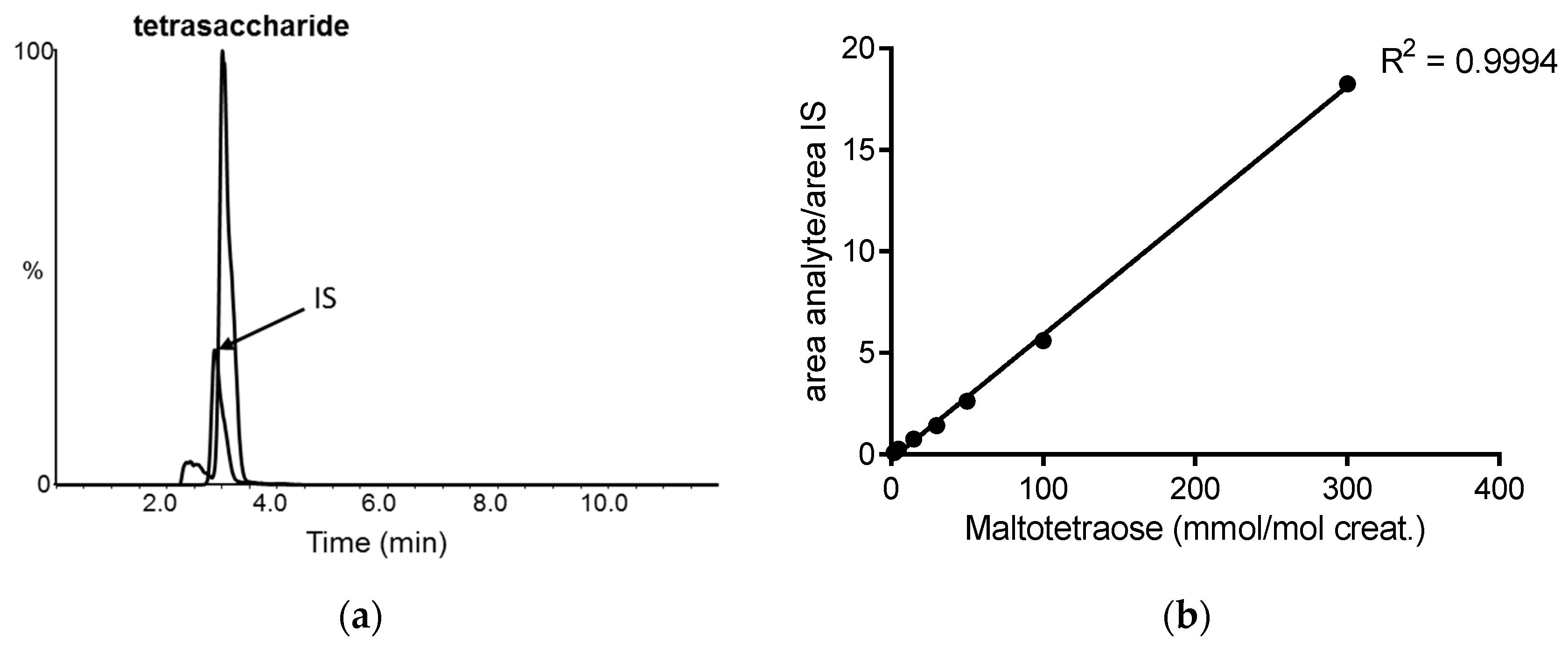
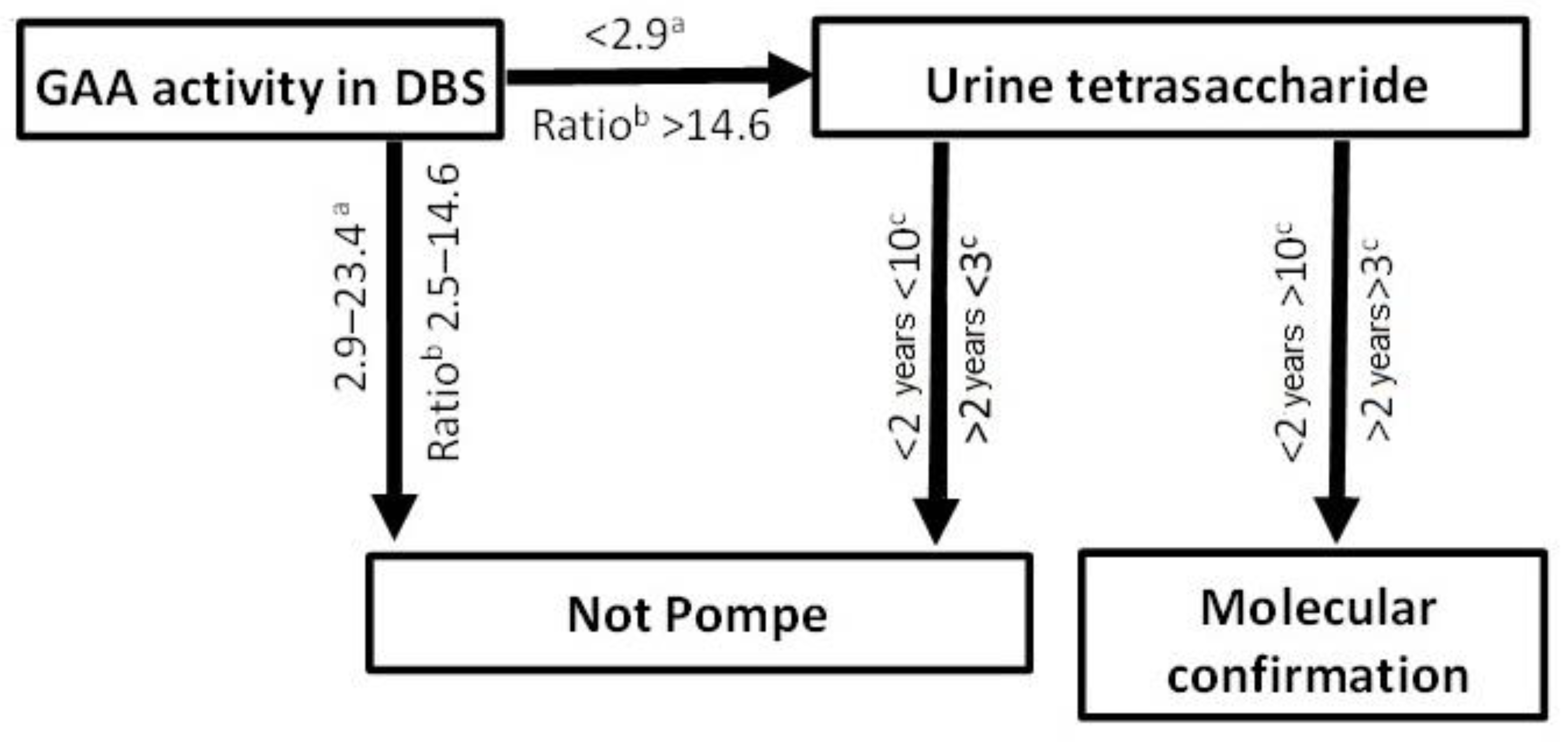
2.1. Urine Tetrasaccharide Method Validation
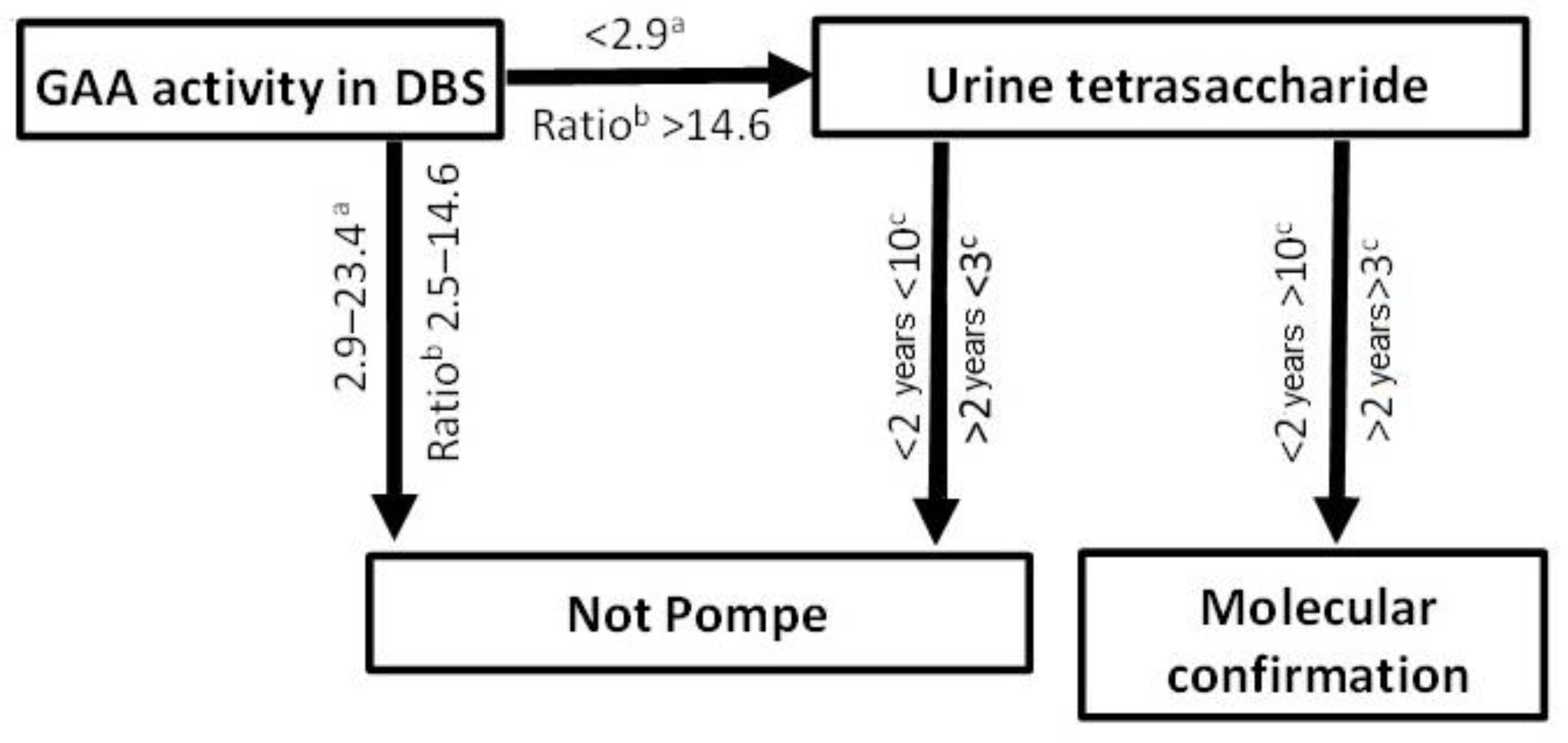
2.2. Diagnosis of PD
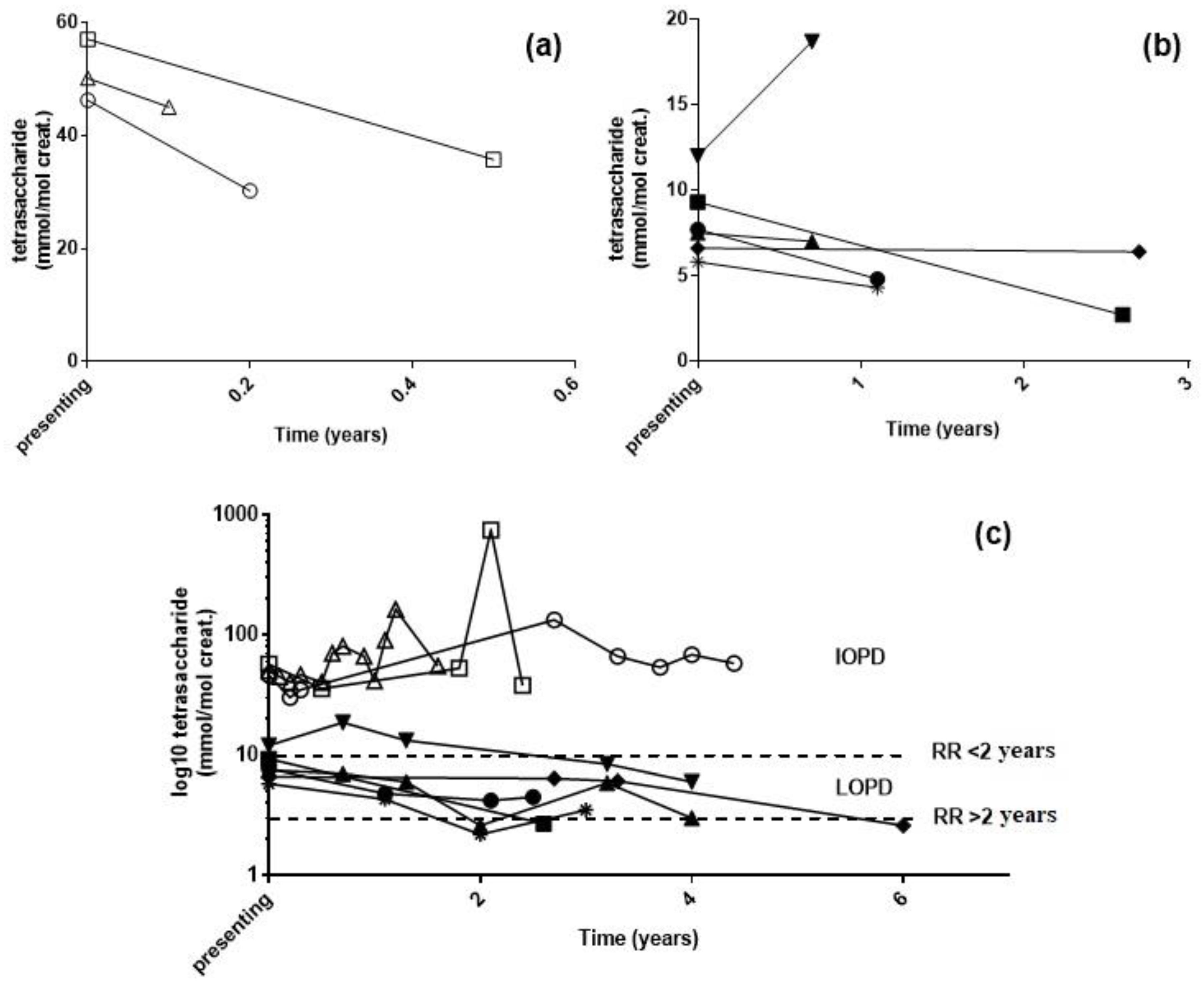
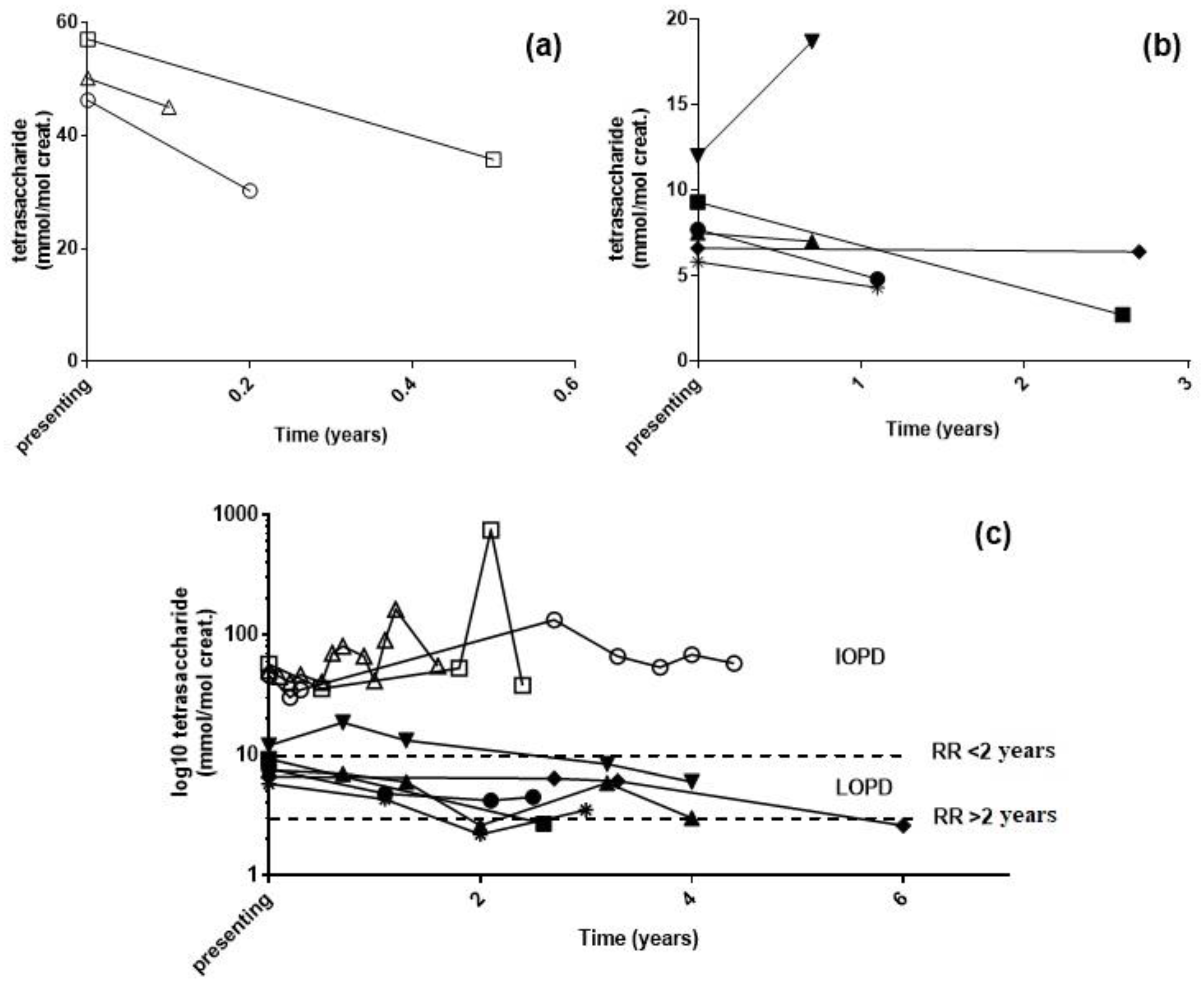
2.3. Longitudinal Monitoring of PD Patients
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Patient Samples
4.3. Urinary Tetrasaccharide Determination
4.4. GAA Activity in Dried Blood Spots
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van den Hout, H.M.; Hop, W.; van Diggelen, O.P.; Smeitink, J.A.M.; Smit, G.P.A.; Poll-The, B.T.T.; Bakker, H.D.; Loonen, M.C.B.; de Klerk, J.B.C.; Reuser, A.J.J.; et al. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from literature. Pediatrics 2003, 112, 332–340. [Google Scholar] [CrossRef] [Green Version]
- Kishnani, P.S.; Hwu, W.L.; Mandel, H.; Nicolino, M.; Yong, F.; Corzo, D. Infantile-Onset Pompe Disease Natural History Study Group. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J. Pediatr. 2006, 148, 671–676. [Google Scholar] [CrossRef]
- Wokke, J.H.J.; Escolar, D.M.; Pestronk, A.; Jaffe, K.M.; Carter, G.T.; van den Berg, L.H.; Florence, J.M.; Mayhew, J.; Skrinar, A.; Corzo, D.; et al. Clinical features of late-onset Pompe disease: A prospective cohort study. Muscle Nerve 2008, 38, 1236–1245. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Corzo, D.; Nicolino, M.; Byrne, B.; Mandel, H.; Hwu, W.L.; Leslie, N.; Levine, J.; Spencer, C.; McDonald, M.; et al. Recombinant human acid α-glucosidase: Major clinical benefits in infantile-onset Pompe disease. Neurology 2007, 68, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Schoser, B.; Stewart, A.; Kanters, S.; Hamed, A.; Jansen, J.; Chan, K.; Karamouzian, M.; Toscano, A. Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: A systematic review and meta-analysis. J. Neurol. 2017, 264, 621–630. [Google Scholar] [CrossRef]
- Burton, B.K.; Kronn, D.F.; Hwu, W.L.; Kishnani, P.S.; Pompe Disease Newborn Screening Working Group. The initial valuation of patients after positive newborn screening: Recommended algorithms leading to a confirmed diagnosis of Pompe disease. Pediatrics 2017, 140, S14–S23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Kallwass, H.; Young, S.P.; Carr, C.; Dai, J.; Kishnani, P.S.; Millington, D.S.; Keutzer, J.; Chen, Y.-T.; Bali, D. Comparison of maltose and acarbose as inhibitors of maltase-glucoamylase activity in assaying acid alpha-glucosidase activity in dried blood spots for the diagnosis of infantile Pompe disease. Genet. Med. 2006, 8, 302–306. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Scott, C.R.; Chamoles, N.A.; Ghavami, A.; Pinto, B.M.; Turecek, F.; Gelb, M.H. Direct multiplex assay of lysosomal enzymes in dried blood spots for newborn screening. Clin. Chem. 2004, 50, 1785–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dajnoki, A.; Mühl, A.; Fekete, G.; Keutzer, J.; Orsini, J.; Dejesus, V.; Zhang, X.K.; Bodamer, O.A. Newborn screening for Pompe disease by measuring acid alpha-glucosidase activity using tandem mass spectrometry. Clin. Chem. 2008, 54, 1624–1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kallwass, H.; Carr, C.; Gerrein, J.; Titlow, M.; Pomponio, R.; Bali, D.; Dai, J.; Kishnani, P.; Skrinar, A.; Corzo, D.; et al. Rapid diagnosis of late-onset Pompe disease by fluorometric assay of α-glucosidase activities in dried blood spots. Mol. Genet. Metab. 2007, 90, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Tajima, Y.; Matsuzawa, F.; Aikawa, S.I.; Okumiya, T.; Yoshimizu, M.; Tsukimura, T.; Ikekita, M.; Tsujino, S.; Tsuji, A.; Edmunds, T.; et al. Structural and biochemical studies on Pompe disease and a “pseudodeficiency of acid α-glucosidase”. J. Hum. Genet. 2007, 52, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Niño, M.Y.; In’t Groen, S.L.; Bergsma, A.J.; van der Beek, N.A.M.E.; Kroos, M.; Hoogeveen-Westerveld, M.; van der Ploeg, A.T.; Pijnappel, W.W.M.P. Extension of the Pompe mutation database by linking disease-associated variants to clinical severity. Hum. Mutat. 2019, 40, 1954–1967. [Google Scholar] [CrossRef]
- Ficicioglu, C.; Ahrens-Nicklas, R.C.; Barch, J.; Cuddapah, S.R.; DiBoscio, B.S.; DiPerna, J.C.; Gordon, P.L.; Henderson, N.; Menello, C.; Luongo, N.; et al. Newborn screening for Pompe disease: Pennsylvania experience. Int. J. Neonatal Screen. 2020, 13, 89. [Google Scholar] [CrossRef] [PubMed]
- Fuller, M.; Duplock, S.; Turner, C.; Davey, P.; Brooks, D.A.; Hopwood, J.J.; Meikle, P.J. Mass spectrometric quantification of glycogen to assess primary substrate accumulation in the Pompe mouse. Anal. Biochem. 2012, 421, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Hallgren, P.; Hansson, G.; Henriksson, K.G.; Häger, A.; Lundblad, A.; Svensson, S. Increased excretion of a glucose-containing tetrasaccharide in the urine of a patient with glycogen storage disease type II (Pompe’s disease). Eur. J. Clin. Investig. 1974, 4, 429–433. [Google Scholar] [CrossRef]
- Lennartsson, G.; Lundblad, A.; Sjöblad, S.; Svensson, S.; Öckerman, P.-A. Quantitation of a urinary tetrassacharide by gas chromatography and mass spectrometry. Biomed. Mass Spectrom. 1976, 3, 51–54. [Google Scholar] [CrossRef]
- Sluiter, W.; van den Bosch, J.C.; Goudriaan, D.A.; van Gelder, C.M.; de Vries, J.M.; Huijmans, J.G.M.; Reuser, A.J.J.; van der Ploeg, A.T.; Ruijter, G.J.G. Rapid ultraperformance liquid chromatography-tandem mass spectrometry assay for a characteristic glycogen-derived tetrasaccharide in Pompe disease and other glycogen storage diseases. Clin. Chem. 2012, 58, 1139–1147. [Google Scholar] [CrossRef] [Green Version]
- Manwaring, V.; Prunty, H.; Bainbridge, K.; Burke, D.; Finnegan, N.; Franses, R.; Lam, A.; Vellodi, A.; Heales, S. Urine analysis of glucose tetrasaccharide by HPLC; a useful marker for the investigation of patients with Pompe and other glycogen storage diseases. J. Inherit. Metab. Dis. 2012, 35, 311–316. [Google Scholar] [CrossRef]
- Piraud, M.; Pettazzoni, M.; de Antonio, M.; Vianey-Saban, C.; Froissart, R.; Chabrol, B.; Young, S.; Laforêt, P.; French Pompe Study Group. Urine glucose tetrasaccharide: A good biomarker for glycogenoses type III and III? A study of the French cohort. Mol. Genet. Metab. Rep. 2020, 23, 100583. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Young, S.P.; Hillman, S.L.; Van Hove, J.L.; Chen, Y.T.; Millington, D.S. Liquid chromatography assay for a glucose tetrasaccharide, a putative biomarker for the diagnosis of Pompe disease. Anal. Biochem. 2000, 287, 136–143. [Google Scholar] [CrossRef]
- Canbay, E.; Vural, M.; Uçar, S.K.; Sezer, E.D.; Karasoy, H.; Vüceyar, A.N.; Çoker, M.; Sözmen, E.Y. The decision-making levels of urine tetrasaccharide for the diagnosis of Pompe disease in the Turkish population. J. Pediatr. Endocrinol. Metab. 2020, 33, 391–395. [Google Scholar] [CrossRef]
- Chien, Y.H.; Goldstein, J.L.; Hwu, W.L.; Smith, P.B.; Lee, N.C.; Chiang, S.C.; Tolun, A.A.; Zhang, H.; Vaisnins, A.E.; Millington, D.S.; et al. Baseline urinary tetrasaccharide concentrations in patients with infantile- and late-onset Pompe disease identified by newborn screening. JIMD Rep. 2015, 19, 67–73. [Google Scholar]
- An, Y.; Young, S.P.; Kishnani, P.S.; Millington, D.S.; Amalfitano, A.; Corz, D.; Chen, Y.T. Glucose tetrasaccharide as a biomarker for monitoring the therapeutic response to enzyme replacement therapy for Pompe disease. Mol. Genet. Metab. 2005, 85, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Young, S.P.; Zhang, H.; Corzo, D.; Thurberg, B.L.; Bali, D.; Kishnani, P.S.; Millington, D.S. Long-term monitoring of patients with infantile-onset Pompe disease on enzyme replacement therapy using a urinary glucose tetrasaccharide biomarker. Genet. Med. 2009, 11, 536–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.A.; Case, L.E.; Herburt, M.; DeArmey, S.; Jones, H.; Crisp, K.; Zimmermand, K.; ElMallah, M.; Young, S.P.; Kishnani, P.S. Higher dosing of alglucosidase alfa improves outcomes in children with Pompe disease: A clinical study and review of the literature. Genet. Med. 2020, 22, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Lundblad, A.; Svensson, S.; Yamashina, I.; Ohta, M. Increased urinary excretion of a glucose-containing tetrasaccharide in patients with Duchenne muscular dystrophy. FEBS Lett. 1979, 97, 249–252. [Google Scholar] [PubMed] [Green Version]
- Heiner-Fokkema, M.R.; van der Krogt, J.; de Boer, F.; Fokkert-Wilts, M.J.; Maatman, R.G.; Hoogeveen, I.J.; Derks, T.G. The multiple faces of urinary glucose tetrasaccharide as biomarker for patients with hepatic glycogen storage diseases. Genet. Med. 2020, 22, 1915–1916. [Google Scholar] [CrossRef]
- Kumlien, J.; Chester, M.A.; Lindberg, B.S.; Pizzo, P.; Zopf, D.; Lundblad, A. Urinary excretion of a glucose-containing tetrasaccharide. A parameter for increased degradation of glycogen. Clin. Chim. Acta 1988, 176, 39–48. [Google Scholar] [CrossRef]
- Young, S.P.; Piraud, M.; Goldstein, J.L.; Zhang, H.; Rehder, C.; Laforet, P.; Kishnani, P.S.; Millington, D.S.; Bashir, M.R.; Bali, D.S. Assessing disease severity in Pompe disease: The roles of a urinary glucose tetrasaccharide biomarker and imaging techniques. Am. J. Med. Genet. 2012, 160, 50–58. [Google Scholar] [CrossRef]
- Chan, J.; Desai, A.K.; Kazi, Z.B.; Corey, K.; Austin, S.; Hobson-Webb, L.D.; Case, L.E.; Jones, H.N.; Kishnani, P.S. The emerging phenotype of late-onset Pompe disease: A systematic literature review. Mol. Genet. Metab. 2017, 120, 163–172. [Google Scholar] [CrossRef]
- van Capelle, C.I.; van der Meijden, J.C.; van den Hout, J.M.; Jaeken, J.; Baethmann, M.; Voit, T.; Kroos, M.A.; Derks, T.G.; Rubio-Gozalbo, M.E.; Willemsen, M.A.; et al. Childhood Pompe disease: Clinical spectrum and genotype in 31 patients. Orphanet J. Rare Dis. 2016, 11, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, Y.H.; Tsai, W.H.; Chang, C.L.; Chiu, P.C.; Chou, Y.Y.; Tsai, F.J.; Wong, S.L.; Lee, N.C.; Hwu, W.L. Earlier and higher dosing of alglucosidase alfa improve outcomes in patients with infantile-onset Pompe disease: Evidence from real-world experiences. Mol. Genet. Metab. Rep. 2020, 23, 100591. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
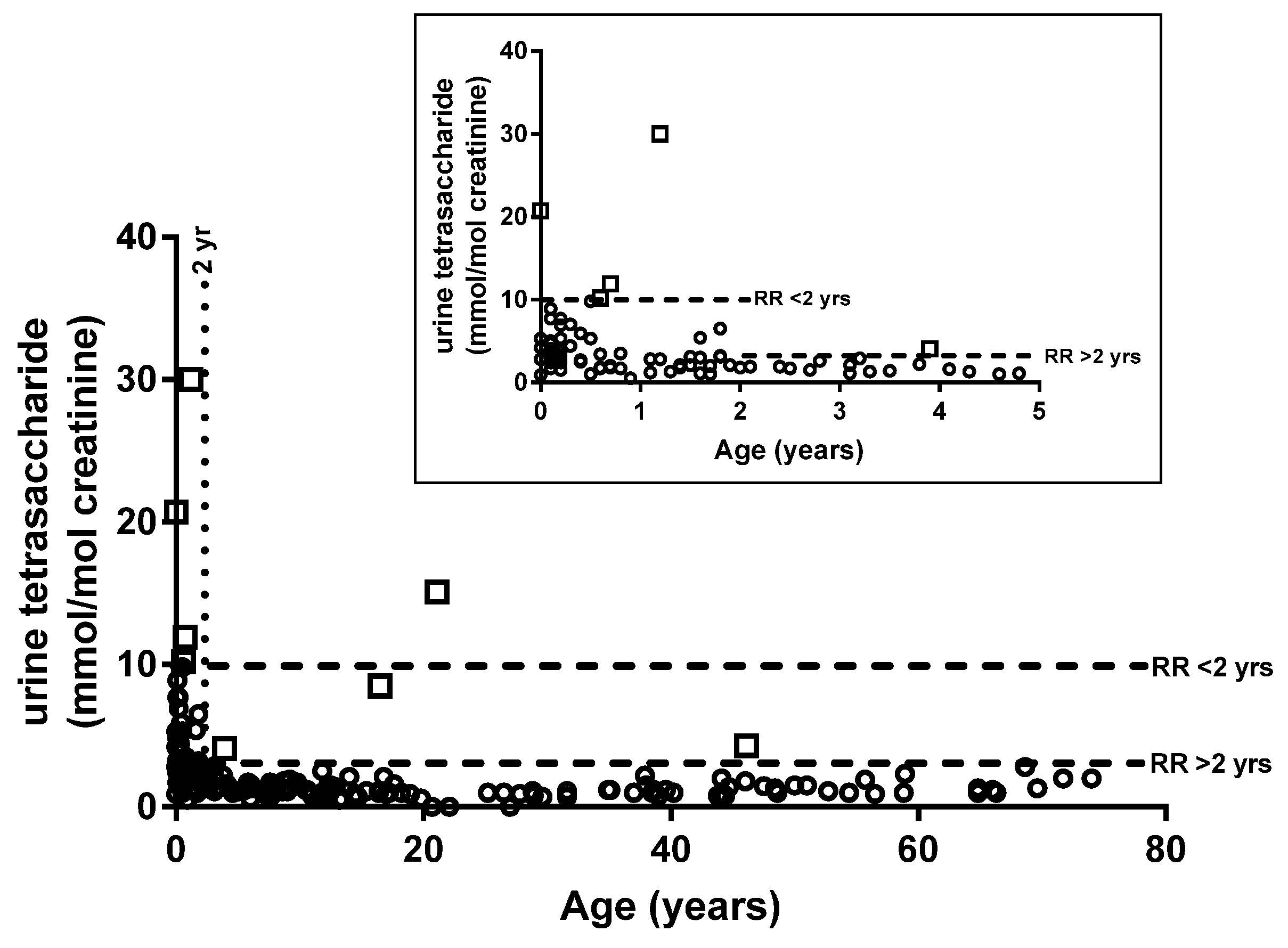{kind=link}
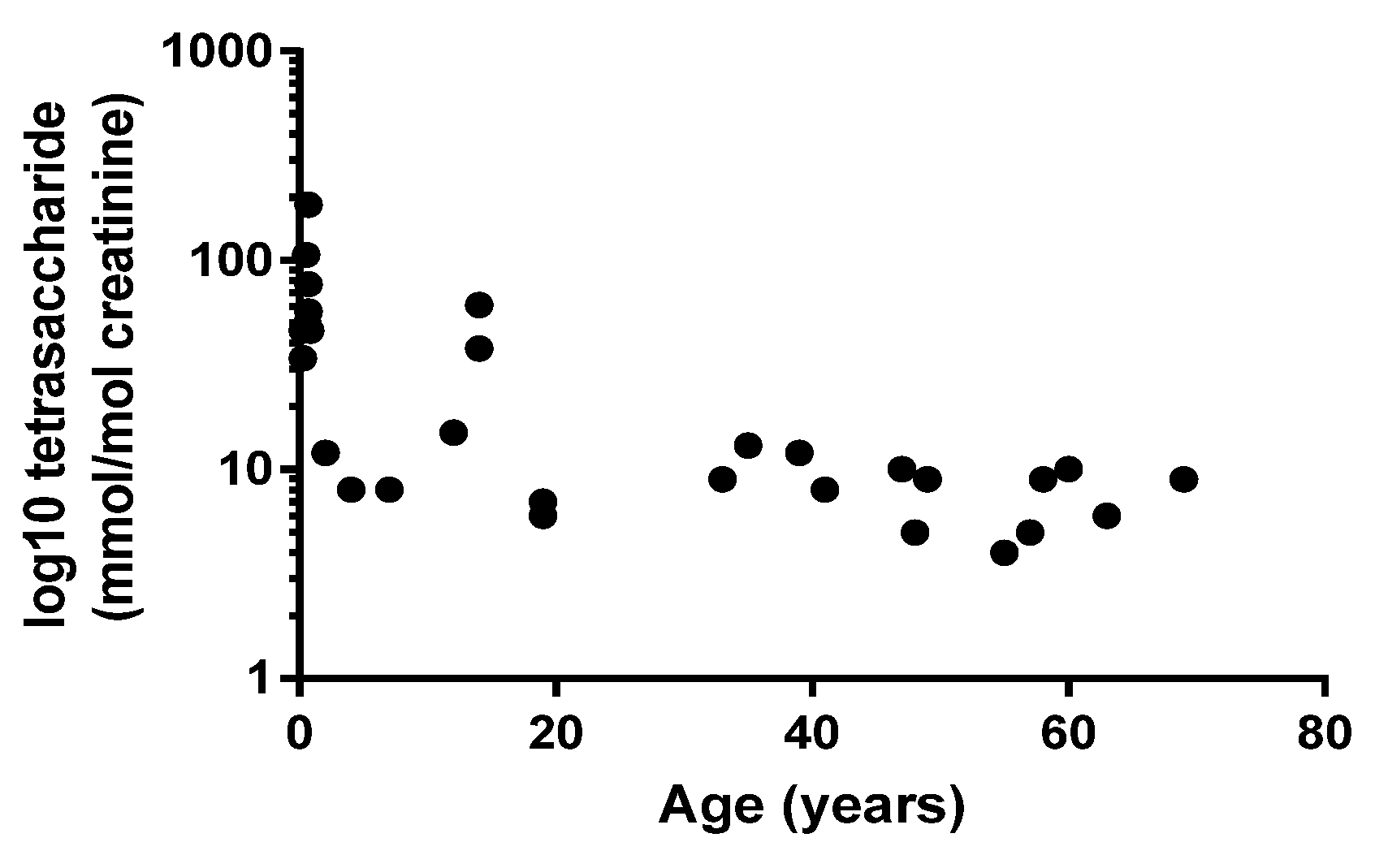{kind=link}
{kind=link}
{kind=link}
| Group | Age (years) | GAA Activity (pmol/spot/h) a | Ratio Total/GAA Activity b | Urine Tetrasaccharide (mmol/mol creatinine) c | Genotype | |
|---|---|---|---|---|---|---|
| Confirmed | IOPD | 0.6 | 0.0 | 482 | 50 | c.953T>C/c.953T>C |
| IOPD | 0.7 | 0.9 | 32 | 57 | c.266G>T/c.2815_2816del | |
| IOPD | 0.7 | 0.7 | 89 | 77 | Genotype not known † | |
| IOPD | 0.25 | 0.9 | 59 | 46 | Genotype not known † | |
| IOPD | 0.8 | 0.3 | 142 | 46 | c.323C>A homozygous | |
| IOPD | 0.7 | 0.8 | 62 | 185 | c.[2155G>C; 1726C>A; 2065G>A] homozygous | |
| IOPD | 0.5 | 0.7 | 65 | 106 | Genotype not known | |
| LOPD | 47 | 0.9 | 57 | 10 | c.-32-13T>G/c.1827del | |
| LOPD | 69 | 0.7 | 39 | 9 | c.-32-13T>G/c.953T>C | |
| LOPD | 7 | 0.0 | 4417 | 8 | c.-32-13T>G/c.1735G>A | |
| LOPD | 41 | 0.8 | 31 | 8 | c.-32-13T>G/c.307T>G | |
| LOPD | 4 | 0.2 | 260 | 8 | c.1827del/c.[-32-13T>G; 2275G>A] d | |
| LOPD | 14 | 0.3 | 101 | 61 | Genotype not known e | |
| LOPD | 14 | 0.6 | 95 | 38 | Genotype not known e | |
| LOPD | 2 | 0.5 | 88 | 12 | c.1827del/c.[-32-13T>G; 2275G>A] d | |
| LOPD | 19 | 0.5 | 116 | 7 | c.-32-13T>G/c.307T>G | |
| LOPD | 58 | 0.9 | 51 | 9 | c.-32-13T>G/c.1827del | |
| LOPD | 12 | 0.4 | 69 | 15 | Genotype not known | |
| LOPD | 19 | 0.4 | 132 | 6 | c.-32-13T>G/c.482_483del | |
| LOPD | 35 | 0.5 | 61 | 13 | c.-32-13T>G/c.482_483del | |
| LOPD | 39 | 0.6 | 34 | 12 | Genotype not known | |
| LOPD | 19 | 0.8 | 37 | 6 | Genotype not known | |
| LOPD | 60 | 1.0 | 43 | 10 | c.-32-13T>G/c.1441T>C | |
| Het | 69 | 1.7 | 18 | 1 | c.-32-13T>G | |
| Diagnosed | IOPD | 0.25 | 0.2 | 200 | 34 | Genotype not known † |
| LOPD | 33 | 1.1 | 56 | 9 | Genotype not known | |
| LOPD | 48 | 0.2 | 149 | 5 | Genotype not known | |
| LOPD | 57 | 0.9 | 84 | 5 | c.-32-13T>G/c.1075+1G>T | |
| LOPD | 49 | 1.9 | 52 | 9 | c.-32-13T>G/c.2481+109_2646+38del | |
| LOPD | 63 | 0.2 | 246 | 6 | Genotype not known | |
| LOPD | 55 | 0.7 | 113 | 4 | Genotype not known | |
| Het | 1 | 2.7 | 14 | 1 | c.-32-13T>G | |
| Het | 27 | 2.4 | 13 | 1 | c.2481+109_2646+38del |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saville, J.T.; Fuller, M. Experience with the Urinary Tetrasaccharide Metabolite for Pompe Disease in the Diagnostic Laboratory. Metabolites 2021, 11, 446. https://doi.org/10.3390/metabo11070446
Saville JT, Fuller M. Experience with the Urinary Tetrasaccharide Metabolite for Pompe Disease in the Diagnostic Laboratory. Metabolites. 2021; 11(7):446. https://doi.org/10.3390/metabo11070446
Chicago/Turabian StyleSaville, Jennifer T., and Maria Fuller. 2021. "Experience with the Urinary Tetrasaccharide Metabolite for Pompe Disease in the Diagnostic Laboratory" Metabolites 11, no. 7: 446. https://doi.org/10.3390/metabo11070446
APA StyleSaville, J. T., & Fuller, M. (2021). Experience with the Urinary Tetrasaccharide Metabolite for Pompe Disease in the Diagnostic Laboratory. Metabolites, 11(7), 446. https://doi.org/10.3390/metabo11070446