
Metabolism, HDACs, and HDAC Inhibitors: A Systems Biology Perspective


Abstract
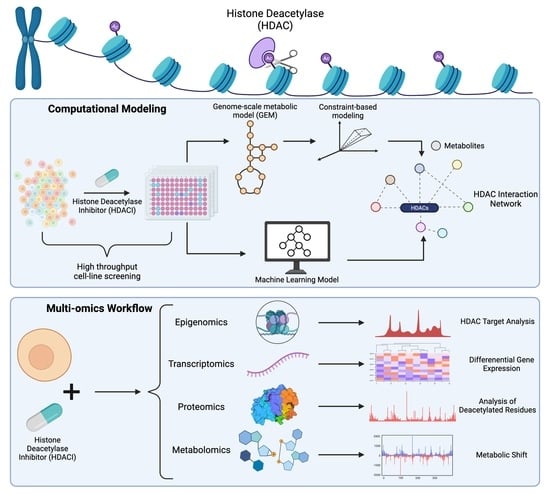
1. Introduction
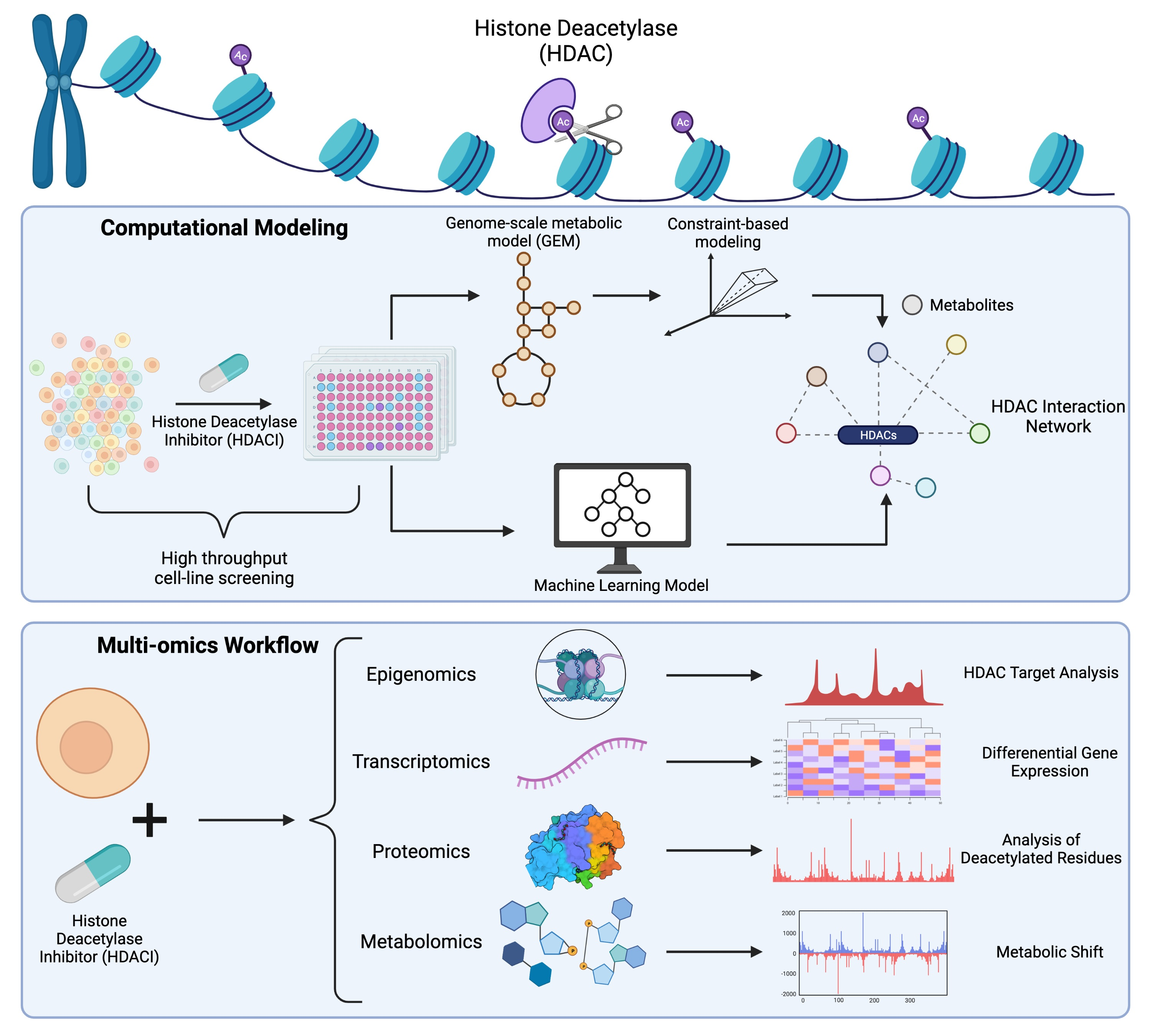
2. Relationship between HDACs, HDACIs, and Metabolism
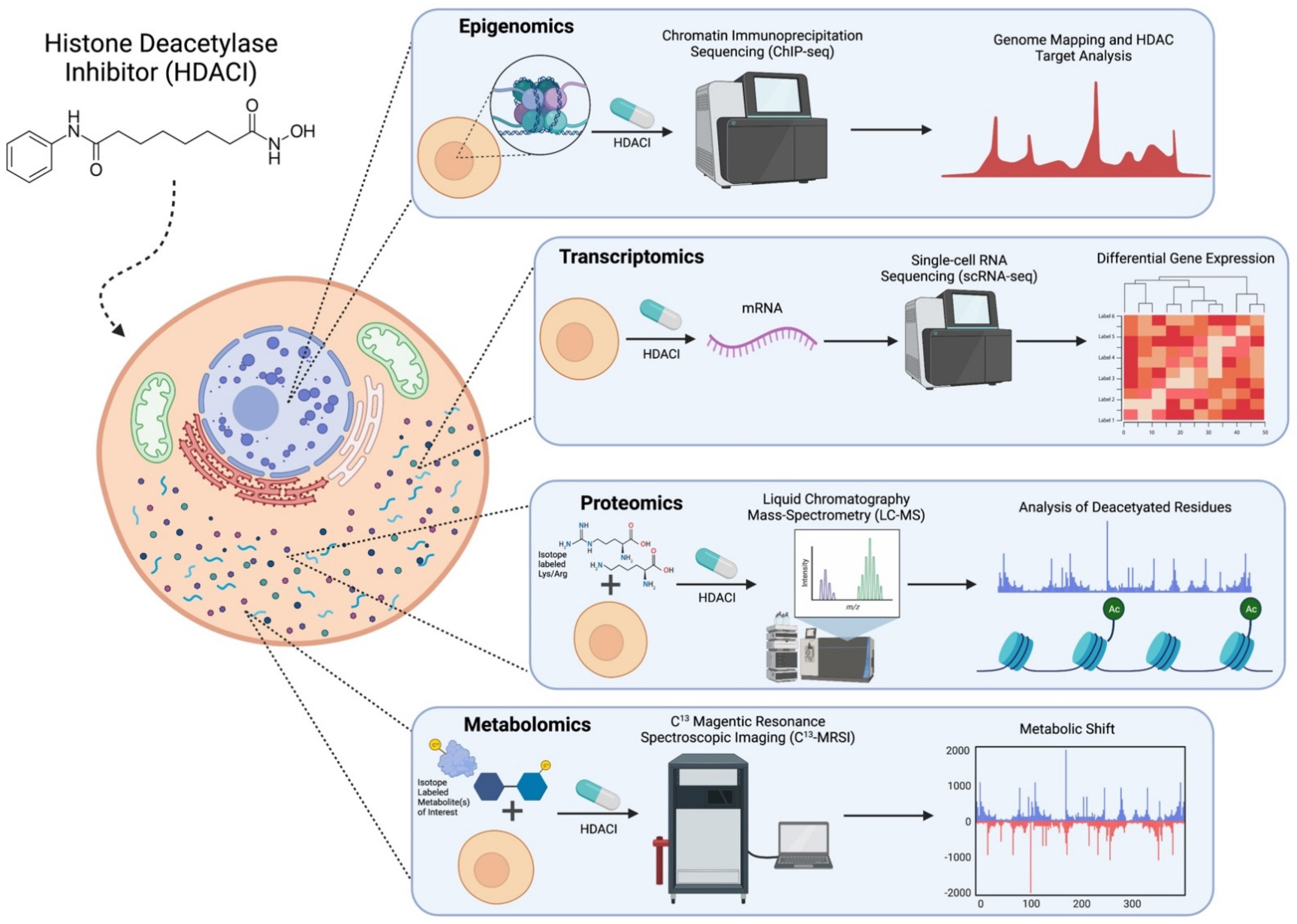
3. Emerging Technologies to Study Interactions between HDACs and Metabolism
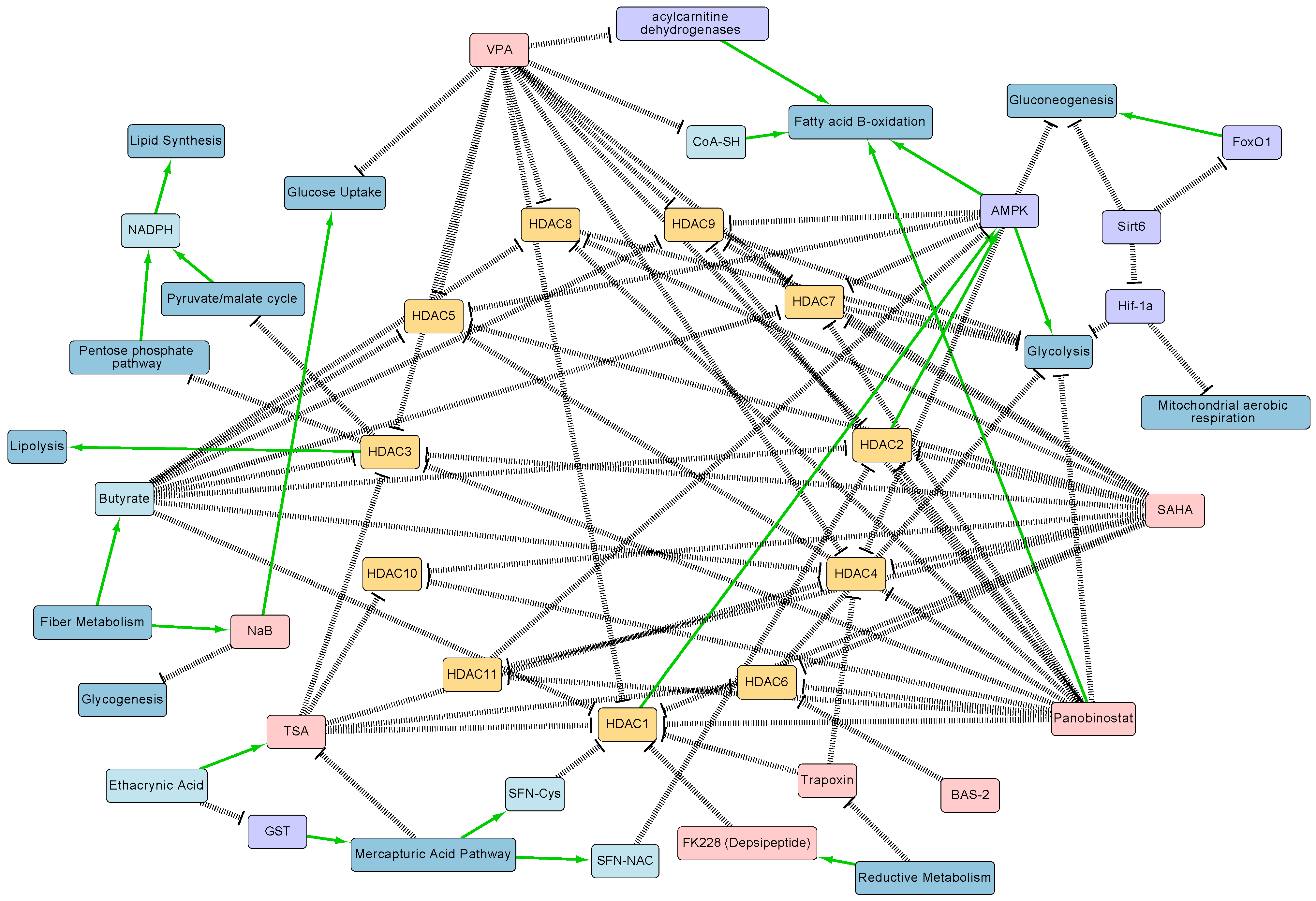
3.1. Epigenomics
3.2. Transcriptomics
3.3. Proteomics
3.4. Metabolomics
3.5. High Throughput Cell Line Screening
3.6. Genome-Scale Metabolic Modeling
3.7. Microbiome Profiling
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BS-seq | Bisulfite Sequencing |
| CAROM | Comparative Analysis of Regulators of Metabolism |
| CBM | Constraint-Based Modeling |
| CCLE | Cancer Cell Line Encyclopedia |
| CE-MS | Capillary Electrophoresis Mass Spectrometry |
| ChIP-seq | Chromatin Immunoprecipitation Sequencing |
| CTRP | Cancer Therapeutics Response Portal |
| ESC | Embryonic Stem Cell |
| FACS | Fluorescence-Activated Cell Sorting |
| FBP1 | Fructose-1,6-bisphosphate |
| FK228 | Depsipeptide (active form is Romidepsin) |
| GC-MS | Gas Chromatography Mass Spectrometry |
| GCP | Global Chromatin Profiling |
| GEM | Genome-Scale Metabolic Model |
| GLUT1 | Glucose Transporter Type 1 |
| HAT | Histone Acetyltransferase |
| HCC | Hepatocellular Carcinoma |
| HDAC | Histone Deacetylase |
| HXK1 | Hexokinase 1 |
| LC-MS | Liquid Chromatography Mass Spectrometry |
| MNase | Micrococcal Nuclease |
| MRSI | Magnetic Resonance Spectroscopic Imaging |
| MS | Mass Spectrometry |
| NaB | Sodium Butyrate |
| NMR | Nuclear Magnetic Resonance |
| NPC | Neuronal Precursor Cell |
| PBAT | Post-Bisulfite Adapter-Tagging |
| PPP | Pentose Phosphate Pathway |
| RNA-seq | RNA Sequencing |
| SAHA | Suberoylanilide Hydroxamic Acid (Vorinostat) |
| SCFA | Short-Chain Fatty Acids |
| scRNA-seq | Single-Cell RNA Sequencing |
| scRRBS | Single-Cell Reduced-Representation Bisulfite Sequencing |
| SFC-MS | Supercritical Fluid Chromatography Mass Spectrometry |
| SILAC | Stable Isotope Labeling by Amino Acids in Cell Culture |
| TPX | Trapoxin |
| TSA | Trichostatin A |
| VPA | Valproate |
References
- Berni Canani, R.; Di Costanzo, M.; Leone, L. The epigenetic effects of butyrate: Potential therapeutic implications for clinical practice. Clin. Epigenetics 2012, 4, 4. [Google Scholar] [CrossRef]
- Rajendran, P.; Williams, D.E.; Ho, E.; Dashwood, R.H. Metabolism as a key to histone deacetylase inhibition. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 181–199. [Google Scholar] [CrossRef]
- Wardell, S.E.; Ilkayeva, O.R.; Wieman, H.L.; Frigo, D.E.; Rathmell, J.C.; Newgard, C.B.; McDonnell, D.P. Glucose metabolism as a target of histone deacetylase inhibitors. Mol. Endocrinol. 2009, 23, 388–401. [Google Scholar] [CrossRef]
- Chiaradonna, F.; Cirulli, C.; Palorini, R.; Votta, G.; Alberghina, L. New Insights into the Connection Between Histone Deacetylases, Cell Metabolism, and Cancer. Antioxid. Redox Signal. 2015, 23, 30–50. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Zhao, S. Metabolic changes in cancer: Beyond the Warburg effect. Acta Biochim. Biophys. Sin. 2013, 45, 18–26. [Google Scholar] [CrossRef]
- Cho, M.; Choi, E.; Kim, J.H.; Kim, H.; Kim, H.M.; Lee, J.I.; Hwang, K.-C.; Kim, H.-J.; Han, G. Lactam-based HDAC inhibitors for anticancer chemotherapy: Restoration of RUNX3 by posttranslational modification and epigenetic control. ChemMedChem 2014, 9, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Bae, S.-C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011, 3, 166–179. [Google Scholar] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Wright, L.H.; Menick, D.R. A class of their own: Exploring the nondeacetylase roles of class IIa HDACs in cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H199–H206. [Google Scholar] [CrossRef]
- Mrakovcic, M.; Kleinheinz, J.; Fröhlich, L.F. p53 at the Crossroads between Different Types of HDAC Inhibitor-Mediated Cancer Cell Death. Int. J. Mol. Sci. 2019, 20, 2415. [Google Scholar] [CrossRef]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhang, M.; Zhou, Y.; Guo, W.; Yi, M.; Zhang, Z.; Ding, Y.; Wang, Y. The application of histone deacetylases inhibitors in glioblastoma. J. Exp. Clin. Cancer Res. 2020, 39, 138. [Google Scholar] [CrossRef] [PubMed]
- Knutson, S.K.; Chyla, B.J.; Amann, J.M.; Bhaskara, S.; Huppert, S.S.; Hiebert, S.W. Liver-specific deletion of histone deacetylase 3 disrupts metabolic transcriptional networks. EMBO J. 2008, 27, 1017–1028. [Google Scholar] [CrossRef]
- Feng, D.; Liu, T.; Sun, Z.; Bugge, A.; Mullican, S.E.; Alenghat, T.; Liu, X.S.; Lazar, M.A. A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science 2011, 331, 1315–1319. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Miller, R.A.; Patel, R.T.; Chen, J.; Dhir, R.; Wang, H.; Zhang, D.; Graham, M.J.; Unterman, T.G.; Shulman, G.I.; et al. Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat. Med. 2012, 18, 934–942. [Google Scholar] [CrossRef]
- Yang, J.; Jin, X.; Yan, Y.; Shao, Y.; Pan, Y.; Roberts, L.R.; Zhang, J.; Huang, H.; Jiang, J. Inhibiting histone deacetylases suppresses glucose metabolism and hepatocellular carcinoma growth by restoring FBP1 expression. Sci. Rep. 2017, 7, 43864. [Google Scholar] [CrossRef]
- Fadaka, A.; Ajiboye, B.; Ojo, O.; Adewale, O.; Olayide, I.; Emuowhochere, R. Biology of glucose metabolization in cancer cells. J. Oncol. Sci. 2017, 3, 45–51. [Google Scholar] [CrossRef]
- Amoêdo, N.D.; Rodrigues, M.F.; Pezzuto, P.; Galina, A.; da Costa, R.M.; de Almeida, F.C.L.; El-Bacha, T.; Rumjanek, F.D. Energy metabolism in H460 lung cancer cells: Effects of histone deacetylase inhibitors. PLoS ONE 2011, 6, e22264. [Google Scholar] [CrossRef]
- Deshmukh, A.; Deshpande, K.; Arfuso, F.; Newsholme, P.; Dharmarajan, A. Cancer stem cell metabolism: A potential target for cancer therapy. Mol. Cancer 2016, 15, 69. [Google Scholar] [CrossRef]
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [CrossRef]
- Fan, J.; Krautkramer, K.A.; Feldman, J.L.; Denu, J.M. Metabolic regulation of histone post-translational modifications. ACS Chem. Biol. 2015, 10, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Vogelauer, M.; Krall, A.S.; McBrian, M.A.; Li, J.-Y.; Kurdistani, S.K. Stimulation of histone deacetylase activity by metabolites of intermediary metabolism. J. Biol. Chem. 2012, 287, 32006–32016. [Google Scholar] [CrossRef]
- Wong, C.C.; Qian, Y.; Yu, J. Interplay between epigenetics and metabolism in oncogenesis: Mechanisms and therapeutic approaches. Oncogene 2017, 36, 3359–3374. [Google Scholar] [CrossRef]
- Cuperlovic-Culf, M.; Culf, A. Protein Acetylation as an Integral Part of Metabolism in Cancer Development and Progression. Am. J. Cancer Rev. 2014, 2, 6–28. [Google Scholar]
- Furumai, R.; Komatsu, Y.; Nishino, N.; Khochbin, S.; Yoshida, M.; Horinouchi, S. Potent histone deacetylase inhibitors built from trichostatin A and cyclic tetrapeptide antibiotics including trapoxin. Proc. Natl. Acad. Sci. USA 2001, 98, 87–92. [Google Scholar] [CrossRef]
- Kijima, M.; Yoshida, M.; Sugita, K.; Horinouchi, S.; Beppu, T. Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase. J. Biol. Chem. 1993, 268, 22429–22435. [Google Scholar] [CrossRef]
- Desai, D.; Salli, U.; Vrana, K.E.; Amin, S. SelSA, selenium analogs of SAHA as potent histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 2044–2047. [Google Scholar] [CrossRef]
- Lea, M.A.; Randolph, V.M.; Hodge, S.K. Induction of histone acetylation and growth regulation in eryrthroleukemia cells by 4-phenylbutyrate and structural analogs. Anticancer Res. 1999, 19, 1971–1976. [Google Scholar] [PubMed]
- Porter, N.J.; Christianson, D.W. Binding of the microbial cyclic tetrapeptide trapoxin A to the class I histone deacetylase HDAC8. ACS Chem. Biol. 2017, 12, 2281–2286. [Google Scholar] [CrossRef]
- Nguyen, T.T.T.; Zhang, Y.; Shang, E.; Shu, C.; Torrini, C.; Zhao, J.; Bianchetti, E.; Mela, A.; Humala, N.; Mahajan, A.; et al. HDAC inhibitors elicit metabolic reprogramming by targeting super-enhancers in glioblastoma models. J. Clin. Investig. 2020, 130, 3699–3716. [Google Scholar] [CrossRef] [PubMed]
- Clarke, K.; Young, C.; Liberante, F.; McMullin, M.-F.; Thompson, A.; Mills, K. The histone deacetylase inhibitor Romidepsin induces as a cascade of differential gene expression and altered histone H3K9 marks in myeloid leukaemia cells. Oncotarget 2019, 10, 3462–3471. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yadav, R.; Mishra, P.; Yadav, D. Histone deacetylase inhibitors: A prospect in drug discovery. Turk. J. Pharm. Sci. 2019, 16, 101–114. [Google Scholar] [CrossRef]
- Singh, A.; Patel, V.K.; Jain, D.K.; Patel, P.; Rajak, H. Panobinostat as pan-deacetylase inhibitor for the treatment of pancreatic cancer: Recent progress and future prospects. Oncol. Ther. 2016, 4, 73–89. [Google Scholar] [CrossRef]
- Clark, S.J.; Lee, H.J.; Smallwood, S.A.; Kelsey, G.; Reik, W. Single-cell epigenomics: Powerful new methods for understanding gene regulation and cell identity. Genome Biol. 2016, 17, 72. [Google Scholar] [CrossRef]
- Lo, P.-K.; Zhou, Q. Emerging techniques in single-cell epigenomics and their applications to cancer research. J. Clin. Genom. 2018, 1. [Google Scholar] [CrossRef]
- Shen, F.; Boccuto, L.; Pauly, R.; Srikanth, S.; Chandrasekaran, S. Genome-scale network model of metabolism and histone acetylation reveals metabolic dependencies of histone deacetylase inhibitors. Genome Biol. 2019, 20, 49. [Google Scholar] [CrossRef] [PubMed]
- Rafehi, H.; Kaspi, A.; Ziemann, M.; Okabe, J.; Karagiannis, T.C.; El-Osta, A. Systems approach to the pharmacological actions of HDAC inhibitors reveals EP300 activities and convergent mechanisms of regulation in diabetes. Epigenetics 2017, 12, 991–1003. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Kim, J.K.; Svensson, V.; Marioni, J.C.; Teichmann, S.A. The technology and biology of single-cell RNA sequencing. Mol. Cell 2015, 58, 610–620. [Google Scholar] [CrossRef]
- Srivatsan, S.R.; McFaline-Figueroa, J.L.; Ramani, V.; Saunders, L.; Cao, J.; Packer, J.; Pliner, H.A.; Jackson, D.L.; Daza, R.M.; Christiansen, L.; et al. Massively multiplex chemical transcriptomics at single-cell resolution. Science 2020, 367, 45–51. [Google Scholar] [CrossRef]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef]
- KhalKhal, E.; Rezaei-Tavirani, M.; Rostamii-Nejad, M. Pharmaceutical Advances and Proteomics Researches. Iran J. Pharm. Res. 2019, 18, 51–67. [Google Scholar] [CrossRef]
- Karpievitch, Y.V.; Polpitiya, A.D.; Anderson, G.A.; Smith, R.D.; Dabney, A.R. Liquid Chromatography Mass Spectrometry-Based Proteomics: Biological and Technological Aspects. Ann. Appl. Stat. 2010, 4, 1797–1823. [Google Scholar] [CrossRef]
- Schölz, C.; Weinert, B.T.; Wagner, S.A.; Beli, P.; Miyake, Y.; Qi, J.; Jensen, L.J.; Streicher, W.; McCarthy, A.R.; Westwood, N.J.; et al. Acetylation site specificities of lysine deacetylase inhibitors in human cells. Nat. Biotechnol. 2015, 33, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Bryson, B.D.; White, F.M. Quantitative Profiling of Lysine Acetylation Reveals Dynamic Crosstalk between Receptor Tyrosine Kinases and Lysine Acetylation. PLoS ONE 2015, 10, e0126242. [Google Scholar] [CrossRef] [PubMed]
- Bheda, P.; Swatkoski, S.; Fiedler, K.L.; Boeke, J.D.; Cotter, R.J.; Wolberger, C. Biotinylation of lysine method identifies acetylated histone H3 lysine 79 in Saccharomyces cerevisiae as a substrate for Sir2. Proc. Natl. Acad. Sci. USA 2012, 109, E916–E925. [Google Scholar] [CrossRef] [PubMed]
- Abelin, J.G.; Patel, J.; Lu, X.; Feeney, C.M.; Fagbami, L.; Creech, A.L.; Hu, R.; Lam, D.; Davison, D.; Pino, L.; et al. Reduced-representation Phosphosignatures Measured by Quantitative Targeted MS Capture Cellular States and Enable Large-scale Comparison of Drug-induced Phenotypes. Mol. Cell. Proteomics 2016, 15, 1622–1641. [Google Scholar] [CrossRef]
- Creech, A.L.; Taylor, J.E.; Maier, V.K.; Wu, X.; Feeney, C.M.; Udeshi, N.D.; Peach, S.E.; Boehm, J.S.; Lee, J.T.; Carr, S.A.; et al. Building the Connectivity Map of epigenetics: Chromatin profiling by quantitative targeted mass spectrometry. Methods 2015, 72, 57–64. [Google Scholar] [CrossRef]
- Litichevskiy, L.; Peckner, R.; Abelin, J.G.; Asiedu, J.K.; Creech, A.L.; Davis, J.F.; Davison, D.; Dunning, C.M.; Egertson, J.D.; Egri, S.; et al. A Library of Phosphoproteomic and Chromatin Signatures for Characterizing Cellular Responses to Drug Perturbations. Cell Syst. 2018, 6, 424–443.e7. [Google Scholar] [CrossRef]
- Alcarraz-Vizán, G.; Boren, J.; Lee, W.-N.P.; Cascante, M. Histone deacetylase inhibition results in a common metabolic profile associated with HT29 differentiation. Metabolomics 2010, 6, 229–237. [Google Scholar] [CrossRef]
- Cuperlovic-Culf, M.; Touaibia, M.; St-Coeur, P.-D.; Poitras, J.; Morin, P., Jr.; Culf, A.S. Metabolic Effects of Known and Novel HDAC and SIRT Inhibitors in Glioblastomas Independently or Combined with Temozolomide. Metabolites 2014, 4, 807–830. [Google Scholar] [CrossRef]
- Kubala, E.; Muñoz-Álvarez, K.A.; Topping, G.; Hundshammer, C.; Feuerecker, B.; Gómez, P.A.; Pariani, G.; Schilling, F.; Glaser, S.J.; Schulte, R.F.; et al. Hyperpolarized 13C Metabolic Magnetic Resonance Spectroscopy and Imaging. J. Vis. Exp. 2016. [Google Scholar] [CrossRef]
- Radoul, M.; Najac, C.; Viswanath, P.; Mukherjee, J.; Kelly, M.; Gillespie, A.M.; Chaumeil, M.M.; Eriksson, P.; Santos, R.D.; Pieper, R.O.; et al. HDAC inhibition in glioblastoma monitored by hyperpolarized 13C MRSI. NMR Biomed. 2019, 32, e4044. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modeling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Mannan, A.M.; Yvone, G.M.; Ross, K.N.; Zhang, Y.-L.; Marton, M.A.; Taylor, B.R.; Crenshaw, A.; Gould, J.Z.; Tamayo, P.; et al. High-throughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat. Biotechnol. 2016, 34, 419–423. [Google Scholar] [CrossRef]
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A.; et al. Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 2020, 1, 235–248. [Google Scholar] [CrossRef]
- Gu, C.; Kim, G.B.; Kim, W.J.; Kim, H.U.; Lee, S.Y. Current status and applications of genome-scale metabolic models. Genome Biol. 2019, 20, 121. [Google Scholar] [CrossRef]
- Smith, K.; Shen, F.; Lee, H.J.; Chandrasekaran, S. Metabolic signatures of regulation by phosphorylation and acetylation. bioRxiv 2021, 838243. [Google Scholar] [CrossRef]
- Bultman, S.J. Interplay between diet, gut microbiota, epigenetic events, and colorectal cancer. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef]
- Yuille, S.; Reichardt, N.; Panda, S.; Dunbar, H.; Mulder, I.E. Human gut bacteria as potent class I histone deacetylase inhibitors in vitro through production of butyric acid and valeric acid. PLoS ONE 2018, 13, e0201073. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H.; Lin, D.-W.; Eames, A.; Chandrasekaran, S. Next-Generation Genome-Scale Metabolic Modeling through Integration of Regulatory Mechanisms. Metabolites 2021, 11, 606. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Kou, J.; Qin, J.; Li, L.; Zhang, Z.; Pan, Y.; Xue, Y.; Du, W. NADPH levels affect cellular epigenetic state by inhibiting HDAC3-Ncor complex. Nat. Metab. 2021, 3, 75–89. [Google Scholar] [CrossRef]
- Dowling, C.M.; Hollinshead, K.E.R.; Di Grande, A.; Pritchard, J.; Zhang, H.; Dillon, E.T.; Haley, K.; Papadopoulos, E.; Mehta, A.K.; Bleach, R.; et al. Multiple screening approaches reveal HDAC6 as a novel regulator of glycolytic metabolism in triple-negative breast cancer. Sci. Adv. 2021, 7, eabc4897. [Google Scholar] [CrossRef]
- Wellen, K.E.; Snyder, N.W. Should we consider subcellular compartmentalization of metabolites, and if so, how do we measure them? Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Piazza, I.; Kochanowski, K.; Cappelletti, V.; Fuhrer, T.; Noor, E.; Sauer, U.; Picotti, P. A Map of Protein-Metabolite Interactions Reveals Principles of Chemical Communication. Cell 2018, 172, 358–372.e23. [Google Scholar] [CrossRef]
- Karpac, J.; Jasper, H. Metabolic homeostasis: HDACs take center stage. Cell 2011, 145, 497–499. [Google Scholar] [CrossRef] [PubMed]
- New, M.; Olzscha, H.; La Thangue, N.B. HDAC inhibitor-based therapies: Can we interpret the code? Mol. Oncol. 2012, 6, 637–656. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| HDACI | HDAC Isoform Selectivity | Notable Structural Characteristics | Metabolic Relationship | Sources |
|---|---|---|---|---|
| Trapoxin (TPX) | HDAC1,4 | Cyclic tetrapeptide Epoxyketone group | Activity decreased by reductive metabolism | [2,30] |
| Depsipeptide/FK228 (Romidepsin) | HDAC1,2 HDAC4, 6 (weaker) | Bicyclic peptide Activated by disulfide bond reduction | Activity increased by reductive metabolism Decreases glycolysis by suppressing c-Myc (glycolysis regulator) | [2,31,32,33] |
| Butyrate | HDAC1,2,3,4,5,7,8,9 (Class I, IIa) | Short-chain fatty acid anion (deprotonated carboxyl group) | Produced by gastrointestinal metabolism of fiber | [1] |
| Sodium Butyrate (NaB) | HDAC1,2,3,4,5,7,8,9 (Class I, IIa) | Short-chain fatty acid salt | Increases aerobic and mitochondrial metabolism | [1,19] |
| Trichostatin A (TSA) | HDAC1,3,4,6,10 | Hydroxamic acid | Increases aerobic and mitochondrial metabolism | [19,26] |
| Valproate (VPA) | HDAC1,2,3,4,5,7,8,9 (Class I, IIa) | Short-chain fatty acid | Decreases glycolysis and lipid metabolism | [3] |
| Vorinostat/Suberoylanilide Hydroxamic Acid (SAHA) | HDAC1,2,3,4,5,6,7,8,9,10,11 (Class I, II, IV) | Hydroxamic acid | Decreases glycolysis | [2] |
| Panobinostat (LBH-589) | HDAC1,2,3,4,5,6,7,8,9,10,11 (Class I, II, IV) | Hydroxamic acid | Decreases glycolysis by suppressing c-Myc (glycolysis regulator) | [31,34] |
| Technology | Primary Usage | Advantages | Disadvantages |
|---|---|---|---|
| Chromatin immunoprecipitation sequencing (ChIP-seq) | Epigenomics | Quantifies histone and other DNA-binding protein’s location on genome | High cost; reliance on highly sensitive and selective antibody |
| Single cell RNA sequencing (scRNA-seq) | Transcriptomics | Measures differentially expressed genes (e.g., response to HDACIs) in a variety of cell types | Poor cell quality control can lead to low-resolution results and inconsistent transcript coverage |
| Liquid chromatography-mass spectrometry (LC-MS) | Proteomics | Identifies residues of interest in post-translational modifications | High cost, sensitive to noise and it is not genome-scale unlike transcriptomics |
| 13C Magnetic Resonance Spectroscopic Imaging (13C-MRSI) | Metabolomics | Selectable, tracer metabolites of interest; minimally invasive in vivo | Expensive to achieve the resolution required for measuring metabolic shifts |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
King, J.; Patel, M.; Chandrasekaran, S. Metabolism, HDACs, and HDAC Inhibitors: A Systems Biology Perspective. Metabolites 2021, 11, 792. https://doi.org/10.3390/metabo11110792
King J, Patel M, Chandrasekaran S. Metabolism, HDACs, and HDAC Inhibitors: A Systems Biology Perspective. Metabolites. 2021; 11(11):792. https://doi.org/10.3390/metabo11110792
Chicago/Turabian StyleKing, Jacob, Maya Patel, and Sriram Chandrasekaran. 2021. "Metabolism, HDACs, and HDAC Inhibitors: A Systems Biology Perspective" Metabolites 11, no. 11: 792. https://doi.org/10.3390/metabo11110792
APA StyleKing, J., Patel, M., & Chandrasekaran, S. (2021). Metabolism, HDACs, and HDAC Inhibitors: A Systems Biology Perspective. Metabolites, 11(11), 792. https://doi.org/10.3390/metabo11110792