Lysophospholipids as Predictive Markers of ST-Elevation Myocardial Infarction (STEMI) and Non-ST-Elevation Myocardial Infarction (NSTEMI)


Abstract
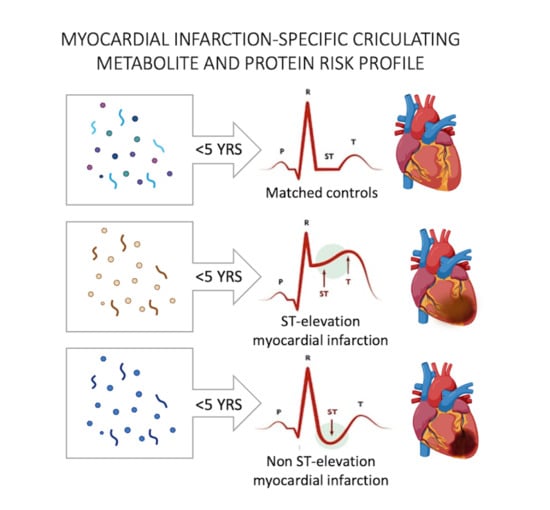
1. Introduction
2. Results
2.1. Cohort Characteristics
2.2. Metabolomics and Protein Panel Analysis
2.3. Infarction Risk Metabolite Profiles
2.4. Infarction Risk Protein Profiles
3. Discussion
4. Materials and Methods
4.1. Baseline Measurements
4.2. Metabolomics Analysis
4.3. Protein Analysis
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Global Burden of Diseases Nutrition and Chronic Diseases Expert Group (NutriCoDE); Castetbon, K. Cardiovascular disease, chronic kidney disease, and diabetes mortality burden of cardiometabolic risk factors from 1980 to 2010: A comparative risk assessment. Lancet Diabetes Endocrinol. 2014, 2, 634–647. [Google Scholar] [CrossRef]
- Benziger, C.P.; Roth, G.A.; Moran, A.E. The Global Burden of Disease Study and the Preventable Burden of NCD. Glob. Heart 2016, 11, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Hamm, C.W.; Bassand, J.-P.; Agewall, S.; Bax, J.J.; Boersma, E.; Bueno, H.; Caso, P.; Dudek, D.; Gielen, S.; Huber, K.; et al. ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: The Task Force for the management of acute coronary syndromes (ACS) in patients presenting without persistent ST-segment elevation of the European Society of Cardiology (ESC). Eur. Heart J. 2011, 32, 2999–3054. [Google Scholar] [CrossRef] [PubMed]
- Pasterkamp, G.; Den Ruijter, H.M.; Libby, P. Temporal shifts in clinical presentation and underlying mechanisms of atherosclerotic disease. Nat. Rev. Cardiol. 2017, 14, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Zaliaduonyte-Peksiene, D.; Lesauskaite, V.; Liutkevičienė, R.; Tamakauskas, V.; Kviesulaitis, V.; Sinkunaite-Marsalkiene, G.; Simonyte, S.; Maciulskyte, S.; Tamuleviciute-Prasciene, E.; Gustiene, O.; et al. Association of the genetic and traditional risk factors of ischaemic heart disease with STEMI and NSTEMI development. J. Renin-Angiotensin-Aldosterone Syst. 2017, 18, 1470320317739987. [Google Scholar] [CrossRef]
- Anderson, K.M.; Odell, P.M.; Wilson, P.W.; Kannel, W.B. Cardiovascular disease risk profiles. Am. Heart J. 1991, 121, 293–298. [Google Scholar] [CrossRef]
- Jneid, H.; Addison, D.; Bhatt, D.L.; Fonarow, G.C.; Gokak, S.; Grady, K.L.; Green, L.A.; A Heidenreich, P.; Ho, P.M.; Jurgens, C.Y.; et al. 2017 AHA/ACC Clinical Performance and Quality Measures for Adults With ST-Elevation and Non–ST-Elevation Myocardial Infarction: A Report of the American College of Cardiology/American Heart Association Task Force on Performance Measures. Circ. Cardiovasc. Qual. Outcomes 2017, 10, e000032. [Google Scholar] [CrossRef]
- Law, S.-H.; Chan, M.-L.; Marathe, G.K.; Parveen, F.; Chen, C.-H.; Ke, L.-Y. An Updated Review of Lysophosphatidylcholine Metabolism in Human Diseases. Int. J. Mol. Sci. 2019, 20, 1149. [Google Scholar] [CrossRef]
- Schober, A.; Siess, W. Lysophosphatidic acid in atherosclerotic diseases. Br. J. Pharmacol. 2012, 167, 465–482. [Google Scholar] [CrossRef]
- Fourcade, O.; Simon, M.-F.; Viodé, C.; Rugani, N.; Leballe, F.; Ragab, A.; Fournié, B.; Sarda, L.; Chap, H. Secretory phospholipase A2 generates the novel lipid mediator lysophosphatidic acid in membrane microvesicles shed from activated cells. Cell 1995, 80, 919–927. [Google Scholar] [CrossRef]
- Diehl, P.; Nienaber, F.; Zaldivia, M.T.K.; Stamm, J.; Siegel, P.M.; Mellett, N.A.; Wessinger, M.; Wang, X.; McFadyen, J.D.; Bassler, N.; et al. Lysophosphatidylcholine is a Major Component of Platelet Microvesicles Promoting Platelet Activation and Reporting Atherosclerotic Plaque Instability. Thromb. Haemost. 2019, 119, 1295–1310. [Google Scholar] [CrossRef] [PubMed]
- Wennberg, P.; Wensley, F.; Di Angelantonio, E.; Johansson, L.; Boman, K.; Rumley, A.; Lowe, G.; Hallmans, G.; Danesh, J.; Jansson, J.-H. Haemostatic and inflammatory markers are independently associated with myocardial infarction in men and women. Thromb. Res. 2012, 129, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Evangelou, A. Platelet-activating factor (PAF): Implications for coronary heart and vascular diseases. Prostaglandins Leukot. Essent. Fat. Acids 1994, 50, 1–28. [Google Scholar] [CrossRef]
- Kurano, M.; Kano, K.; Dohi, T.; Matsumoto, H.; Igarashi, K.; Nishikawa, M.; Ohkawa, R.; Ikeda, H.; Miyauchi, K.; Daida, H.; et al. Different origins of lysophospholipid mediators between coronary and peripheral arteries in acute coronary syndrome. J. Lipid Res. 2017, 58, 433–442. [Google Scholar] [CrossRef]
- Morisawa, T.; Nakagomi, A.; Kohashi, K.; Kusama, Y.; Shimizu, W. Serum Tartrate-resistant Acid Phosphatase-5b Levels are Associated with the Severity and Extent of Coronary Atherosclerosis in Patients with Coronary Artery Disease. J. Atheroscler. Thromb. 2017, 24, 1058–1068. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Winn, M.E.; Zehmer, J.K.; Gillette, W.K.; Lubkowski, J.T.; Pilon, A.L.; Kimura, S. Preclinical evaluation of human secretoglobin 3A2 in mouse models of lung development and fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2014, 306, L10–L22. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Blumenthal, D.K.; Terry, C.M.; He, Y.; Carlson, M.L.; Cheung, A.K. PDGF-induced proliferation in human arterial and venous smooth muscle cells: Molecular basis for differential effects of PDGF isoforms. J. Cell. Biochem. 2010, 112, 289–298. [Google Scholar] [CrossRef] [PubMed]
- De Leon, M.P.; Carubbi, F.; Di Donato, P.; Carulli, N.; De Leon, M.P. Cholesterol esterase activity of human intestinal mucosa. Dig. Dis. Sci. 1985, 30, 1053–1064. [Google Scholar] [CrossRef]
- Rodrigues, C.M.; Sola, S.; Nan, Z.; Castro, R.E.; Ribeiro, P.S.; Low, W.C.; Steer, C.J. Tauroursodeoxycholic acid reduces apoptosis and protects against neurological injury after acute hemorrhagic stroke in rats. Proc. Natl. Acad. Sci. USA 2003, 100, 6087–6092. [Google Scholar] [CrossRef]
- Graf, D.; Kurz, A.K.; Reinehr, R.; Fischer, R.; Kircheis, G.; Häussinger, D. Prevention of bile acid-induced apoptosis by betaine in rat liver. Hepatology 2002, 36, 829–839. [Google Scholar] [CrossRef]
- Ellulu, M.S.; Patimah, I.; Khaza’Ai, H.; Rahmat, A.; Abed, Y. Obesity and inflammation: The linking mechanism and the complications. Arch. Med. Sci. 2017, 4, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Meikle, P.J.; Summers, S.A. Sphingolipids and phospholipids in insulin resistance and related metabolic disorders. Nat. Rev. Endocrinol. 2017, 13, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Lee, S.-H.; Shin, M.-J.; Hwang, G.-S. Alteration in Metabolic Signature and Lipid Metabolism in Patients with Angina Pectoris and Myocardial Infarction. PLoS ONE 2015, 10, e0135228. [Google Scholar] [CrossRef] [PubMed]
- Van Der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta (BBA) Biomembr. 2017, 1859, 1558–1572. [Google Scholar] [CrossRef] [PubMed]
- Levkau, B. HDL-S1P: Cardiovascular functions, disease-associated alterations, and therapeutic applications. Front. Pharmacol. 2015, 6, 243. [Google Scholar] [CrossRef]
- Morris, T.G.; Borland, S.J.; Clarke, C.J.; Wilson, C.; Hannun, Y.A.; Ohanian, V.; Canfield, A.E.; Ohanian, J. Sphingosine 1-phosphate activation of ERM contributes to vascular calcification. J. Lipid Res. 2017, 59, 69–78. [Google Scholar] [CrossRef]
- Alewijnse, A.E.; Peters, S.L. Sphingolipid signalling in the cardiovascular system: Good, bad or both? Eur. J. Pharmacol. 2008, 585, 292–302. [Google Scholar] [CrossRef]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A Branched-Chain Amino Acid-Related Metabolic Signature that Differentiates Obese and Lean Humans and Contributes to Insulin Resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef]
- Wischmeyer, P.E.; Jayakar, D.; Williams, U.; Singleton, K.; Riehm, J.; Bacha, E.; Jeevanandam, V.; Christians, U.; Serkova, N. Single dose of glutamine enhances myocardial tissue metabolism, glutathione content, and improves myocardial function after ischemia-reperfusion injury. J. Parenter. Enter. Nutr. 2003, 27, 396–403. [Google Scholar] [CrossRef]
- Nickel, A.; Löffler, J.; Maack, C. Myocardial energetics in heart failure. Basic Res. Cardiol. 2013, 108, 1–20. [Google Scholar] [CrossRef]
- Makrecka-Kuka, M.; Sevostjanovs, E.; Vilks, K.; Volska, K.; Antone, U.; Kuka, J.; Makarova, E.; Pugovics, O.; Dambrova, M.; Liepinsh, E. Plasma acylcarnitine concentrations reflect the acylcarnitine profile in cardiac tissues. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- McCoin, C.S.; Knotts, T.A.; Adams, S.H. Acylcarnitines—Old actors auditioning for new roles in metabolic physiology. Nat. Rev. Endocrinol. 2015, 11, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Forslund, A.-S.; Jansson, J.-H.; Soederberg, S.; Wennberg, M.; Eliasson, M. Greater decreases in cholesterol levels among individuals with high cardiovascular risk than among the general population: The northern Sweden MONICA study 1994 to 2014. Eur. Heart J. 2016, 37, 1985–1992. [Google Scholar] [CrossRef] [PubMed]
- Norberg, M.; Wall, S.; Boman, K.; Weinehall, L. The Västerbotten Intervention Programme: Background, design and implications. Glob. Health Action 2010, 3, 34. [Google Scholar] [CrossRef] [PubMed]
- Stegmayr, B.; Lundberg, V.; Asplund, K. The events registration and survey procedures in the Northern Sweden MONICA Project. Scand. J. Public Health 2003, 31, 9–17. [Google Scholar] [CrossRef]
- Prineas, R.; Crow, R.; Blackburn, H. The Minnesota Code Manual of Electrocardiographic Findings; John Wright-PSG, Inc.: Littleton, MA, USA, 1982. [Google Scholar]
- Jonsson, P.; Wuolikainen, A.; Thysell, E.; Chorell, E.; Stattin, P.; Wikström, P.; Antti, H. Constrained randomization and multivariate effect projections improve information extraction and biomarker pattern discovery in metabolomics studies involving dependent samples. Metabolomics 2015, 11, 1667–1678. [Google Scholar] [CrossRef]
- Assarsson, E.; Lundberg, M.; Holmquist, G.; Björkesten, J.; Thorsen, S.B.; Ekman, D.; Eriksson, A.; Dickens, E.R.; Ohlsson, S.; Edfeldt, G.; et al. Homogenous 96-Plex PEA Immunoassay Exhibiting High Sensitivity, Specificity, and Excellent Scalability. PLoS ONE 2014, 9, e95192. [Google Scholar] [CrossRef]
- Lennart Eriksson, J.T. Svante Wold CV-ANOVA for significance testing of PLS and OPLS models. J. Chemo Metr. 2008, 22, 594–600. [Google Scholar] [CrossRef]
- Efron, B.; Gong, G. A Leisurely Look at the Bootstrap, the Jack-knife, and Cross-validation. Am. Stat. 1983, 37, 36–48. [Google Scholar]
- Bom, M.J.; Van Der Heijden, D.J.; Kedhi, E.; Van Der Heyden, J.; Meuwissen, M.; Knaapen, P.; Timmer, S.A.; Van Royen, N. Early Detection and Treatment of the Vulnerable Coronary Plaque. Circ. Cardiovasc. Imaging 2017, 10. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| STEMI | NSTEMI | |||||
|---|---|---|---|---|---|---|
| Cases (n = 50) | Referents (n = 50) | p-Value | Cases (n = 50) | Referents (n = 50) | p-Value | |
| Age at screening, years | 42.0 (5.2) | 42.1 (5.5) | 0.91 | 45.8 (5.1) | 45.8 (5.1) | 0.99 |
| Age at MI/SCD, years | 46.8 (5.5) | 50.6 (5.5) | ||||
| Sex (% male) | 96 | 96 | 1 | 86 | 86 | 1 |
| Smoking (%) | 48 | 10.4 | <0.001 | 42.9 | 15.2 | 0.003 |
| Diabetes (%) | 10 | 0 | 0.02 | 6 | 4 | 0.64 |
| Hypertension (%) | 54 | 36 | 0.07 | 46 | 26 | 0.04 |
| Low level of education (%) | 32 | 36 | 0.67 | 42.6 | 25.5 | 0.08 |
| BMI, kg/m2 a | 26.6 (4.0) | 25.5 (3.8) | 0.25 | 27.1 (4.5) | 25.3 (4.0) | 0.004 |
| SBT, mmHg | 135.8 (14.8) | 131.2 (14.9) | 0.12 | 132.7 (13.3) | 128.9 (12.2) | 0.15 |
| DBT, mmHg | 88.4 (11.2) | 83.1 (9.8) | 0.01 | 87.3 (9.0) | 82.5 (9.3) | 0.013 |
| Cholesterol, mmol/L | 6.27 (1.19) | 5.97 (1.24) | 0.21 | 6.68 (0.99) | 6.13 (1.19) | 0.015 |
| Glucose, mmol/L a | 5.10 (0.90) | 5.16 (0.63) | 0.87 | 5.10 (1.25) | 5.28 (0.63) | 0.69 |
| CRP, ng/L a | 1.15 (1.85) | 1.24 (2.34) | 0.81 | 2.29 (3.31) | 1.06 (1.11) | 0.004 |
| ApoB/apoA1 ratio | 0.99 (0.33) | 0.79 (0.21) | 0.001 | 1.00 (0.24) | 0.82 (0.26) | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chorell, E.; Olsson, T.; Jansson, J.-H.; Wennberg, P. Lysophospholipids as Predictive Markers of ST-Elevation Myocardial Infarction (STEMI) and Non-ST-Elevation Myocardial Infarction (NSTEMI). Metabolites 2021, 11, 25. https://doi.org/10.3390/metabo11010025
Chorell E, Olsson T, Jansson J-H, Wennberg P. Lysophospholipids as Predictive Markers of ST-Elevation Myocardial Infarction (STEMI) and Non-ST-Elevation Myocardial Infarction (NSTEMI). Metabolites. 2021; 11(1):25. https://doi.org/10.3390/metabo11010025
Chicago/Turabian StyleChorell, Elin, Tommy Olsson, Jan-Håkan Jansson, and Patrik Wennberg. 2021. "Lysophospholipids as Predictive Markers of ST-Elevation Myocardial Infarction (STEMI) and Non-ST-Elevation Myocardial Infarction (NSTEMI)" Metabolites 11, no. 1: 25. https://doi.org/10.3390/metabo11010025
APA StyleChorell, E., Olsson, T., Jansson, J.-H., & Wennberg, P. (2021). Lysophospholipids as Predictive Markers of ST-Elevation Myocardial Infarction (STEMI) and Non-ST-Elevation Myocardial Infarction (NSTEMI). Metabolites, 11(1), 25. https://doi.org/10.3390/metabo11010025