Metabolic Reprogramming of Chemoresistant Cancer Cells and the Potential Significance of Metabolic Regulation in the Reversal of Cancer Chemoresistance



Abstract
1. Introduction
2. Metabolic Reprogramming Contributes to Chemoresistance
2.1. Oxidative Stress Adaptation
2.2. Adaptation of Lipid Metabolism
2.3. Bioenergetic Adaptation
2.4. Dependence on Glucose and Glycolysis
2.5. Synthesis of Polyamine
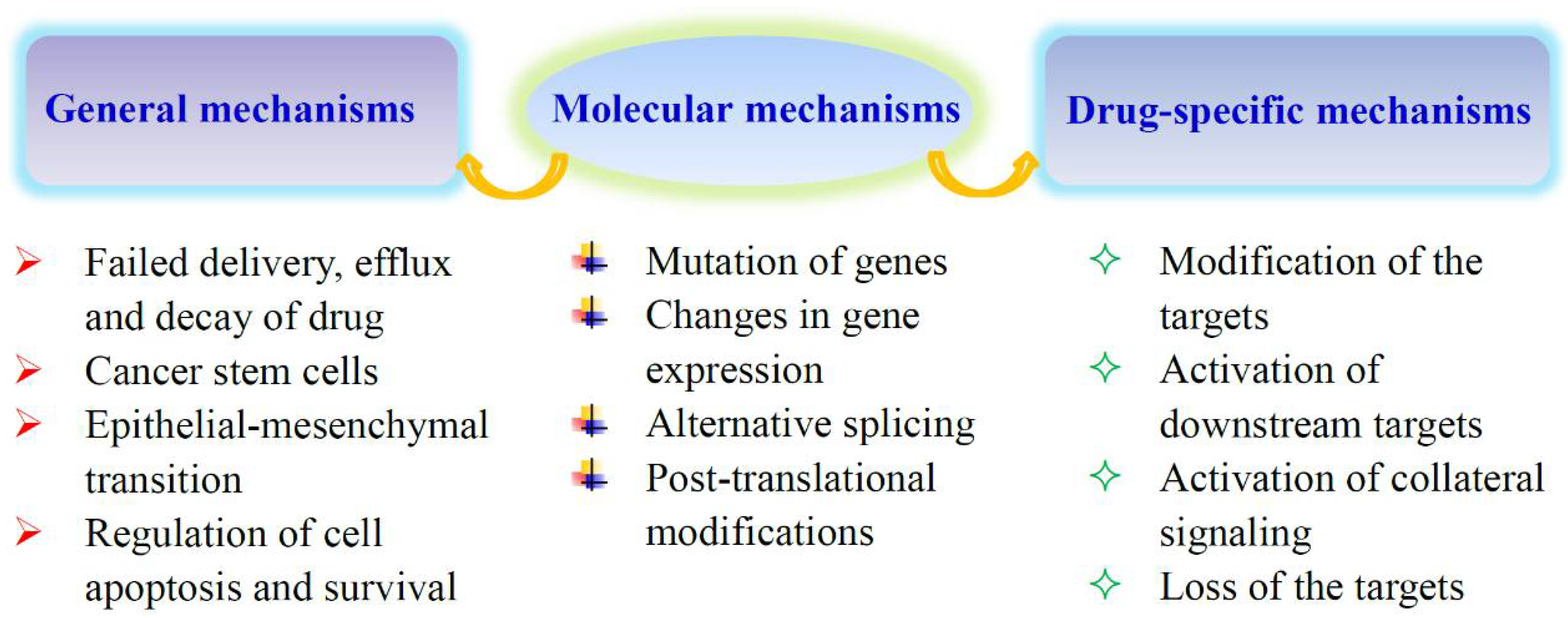
3. The Potential Mechanisms of Metabolic Alterations in Resistant Cells
4. Potential Reversal Effect of Metabolic Regulation on Chemoresistant Tumor Cells
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Aleksakhina, S.N.; Kashyap, A.; Imyanitov, E.N. Mechanisms of acquired tumor drug resistance. Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 188310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhuang, X.; Zong, L.; Liu, S.; Liu, Z.; Song, F. Investigations on the cell metabolomics basis of multidrug resistance from tumor cells by ultra-performance liquid chromatography-mass spectrometry. Anal. Bioanal. Chem. 2016, 408, 5843–5854. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Cao, H.; Qi, X.; Li, H.; Ye, P.; Wang, Z.; Wang, D.; Sun, M. Research Progress in Reversal of Tumor Multi-Drug Resistance via Natural Products. Anticancer Agents Med. Chem. 2017, 17, 1466–1476. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Qiao, S.; Koh, S.B.; Vivekanandan, V.; Salunke, D.; Patra, K.C.; Zaganjor, E.; Ross, K.; Mizukami, Y.; Jeanfavre, S.; Chen, A.; et al. REDD1 loss reprograms lipid metabolism to drive progression of RAS mutant tumors. Genes Dev. 2020, 34, 751–756. [Google Scholar] [CrossRef]
- Lee, N.C.W.; Carella, M.A.; Papa, S.; Bubici, C. High Expression of Glycolytic Genes in Cirrhosis Correlates With the Risk of Developing Liver Cancer. Front. Cell Dev. Biol. 2018, 6, 138. [Google Scholar] [CrossRef] [PubMed]
- Vernucci, E.; Abrego, J.; Gunda, V.; Shukla, S.K.; Dasgupta, A.; Rai, V.; Chaika, N.; Buettner, K.; Illies, A.; Yu, F.; et al. Metabolic Alterations in Pancreatic Cancer Progression. Cancers 2019, 12, 2. [Google Scholar] [CrossRef]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef]
- Cardoso, M.R.; Santos, J.C.; Ribeiro, M.L.; Talarico, M.C.R.; Viana, L.R.; Derchain, S.F.M. A Metabolomic Approach to Predict Breast Cancer Behavior and Chemotherapy Response. Int. J. Mol. Sci. 2018, 19, 617. [Google Scholar] [CrossRef]
- Maria, R.M.; Altei, W.F.; Selistre-de-Araujo, H.S.; Colnago, L.A. Impact of chemotherapy on metabolic reprogramming: Characterization of the metabolic profile of breast cancer MDA-MB-231 cells using H-1 HR-MAS NMR spectroscopy. J. Pharm. Biomed. 2017, 146, 324–328. [Google Scholar] [CrossRef]
- Rahman, M.; Hasan, M.R. Cancer Metabolism and Drug Resistance. Metabolites 2015, 5, 571–600. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Cao, M.; Liu, J.; Yang, Q.; Miao, X.; Go, V.L.W.; Lee, P.W.N.; Xiao, G.G. Metabolic Regulation in Mitochondria and Drug Resistance. Adv. Exp. Med. Biol. 2017, 1038, 149–171. [Google Scholar] [CrossRef]
- Zhang, C.; Ma, Q.; Shi, Y.; Li, X.; Wang, M.; Wang, J.; Ge, J.; Chen, Z.; Wang, Z.; Jiang, H. A novel 5-fluorouracil-resistant human esophageal squamous cell carcinoma cell line Eca-109/5-FU with significant drug resistance-related characteristics. Oncol. Rep. 2017, 37, 2942–2954. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, Y.; Hao, L.; Hou, A.; Chen, X.; Li, Y.; Wang, R.; Luo, P.; Ruan, Z.; Ou, J.; et al. Macrophages induce resistance to 5-fluorouracil chemotherapy in colorectal cancer through the release of putrescine. Cancer Lett. 2016, 381, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Jang, W.J.; Choi, B.; Joo, S.H.; Jeong, C.H. Comparative metabolomic analysis of HPAC cells following the acquisition of erlotinib resistance. Oncol. Lett. 2017, 13, 3437–3444. [Google Scholar] [CrossRef]
- You, X.; Jiang, W.; Lu, W.; Zhang, H.; Yu, T.; Tian, J.; Wen, S.; Garcia-Manero, G.; Huang, P.; Hu, Y. Metabolic reprogramming and redox adaptation in sorafenib-resistant leukemia cells: Detected by untargeted metabolomics and stable isotope tracing analysis. Cancer Commun. 2019, 39, 17. [Google Scholar] [CrossRef] [PubMed]
- Cruz, I.N.; Coley, H.M.; Kramer, H.B.; Madhuri, T.K.; Safuwan, N.A.; Angelino, A.R.; Yang, M. Proteomics Analysis of Ovarian Cancer Cell Lines and Tissues Reveals Drug Resistance-Associated Proteins. Cancer Genom. Proteom. 2017, 14, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wu, D.; Bao, L.; Yin, T.; Lei, D.; Yu, J.; Tong, X. 6PGD inhibition sensitizes hepatocellular carcinoma to chemotherapy via AMPK activation and metabolic reprogramming. Biomed. Pharmacother. 2019, 111, 1353–1358. [Google Scholar] [CrossRef]
- Ghosh, J.C.; Siegelin, M.D.; Vaira, V.; Faversani, A.; Tavecchio, M.; Chae, Y.C.; Lisanti, S.; Rampini, P.; Giroda, M.; Caino, M.C.; et al. Adaptive mitochondrial reprogramming and resistance to PI3K therapy. J. Natl. Cancer Inst. 2015, 107, dju502. [Google Scholar] [CrossRef] [PubMed]
- Montopoli, M.; Bellanda, M.; Lonardoni, F.; Ragazzi, E.; Dorigo, P.; Froldi, G.; Mammi, S.; Caparrotta, L. “Metabolic Reprogramming” in Ovarian Cancer Cells Resistant to Cisplatin. Curr. Cancer Drug Targets 2011, 11, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Catanzaro, D.; Gaude, E.; Orso, G.; Giordano, C.; Guzzo, G.; Rasola, A.; Ragazzi, E.; Caparrotta, L.; Frezza, C.; Montopoli, M. Inhibition of glucose-6-phosphate dehydrogenase sensitizes cisplatin-resistant cells to death. Oncotarget 2015, 6, 30102–30114. [Google Scholar] [CrossRef] [PubMed]
- Masamha, C.P.; LaFontaine, P. Molecular targeting of glutaminase sensitizes ovarian cancer cells to chemotherapy. J. Cell. Biochem. 2018, 119, 6136–6145. [Google Scholar] [CrossRef] [PubMed]
- Galicia-Vazquez, G.; Aloyz, R. Ibrutinib Resistance Is Reduced by an Inhibitor of Fatty Acid Oxidation in Primary CLL Lymphocytes. Front. Oncol. 2018, 8, 411. [Google Scholar] [CrossRef]
- Lopes-Coelho, F.; Gouveia-Fernandes, S.; Goncalves, L.G.; Nunes, C.; Faustino, I.; Silva, F.; Felix, A.; Pereira, S.A.; Serpa, J. HNF1beta drives glutathione (GSH) synthesis underlying intrinsic carboplatin resistance of ovarian clear cell carcinoma (OCCC). Tumour Biol. 2016, 37, 4813–4829. [Google Scholar] [CrossRef] [PubMed]
- Nencioni, A.; Caffa, I.; Cortellino, S.; Longo, V.D. Fasting and cancer: Molecular mechanisms and clinical application. Nat. Rev. Cancer 2018, 18, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Sanderson, S.M.; Dai, Z.; Reid, M.A.; Cooper, D.E.; Lu, M.; Richie, J.P., Jr.; Ciccarella, A.; Calcagnotto, A.; Mikhael, P.G.; et al. Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature 2019, 572, 397–401. [Google Scholar] [CrossRef]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013, 4, e532. [Google Scholar] [CrossRef]
- Long, Y.; Tsai, W.B.; Wang, D.J.; Hawke, D.H.; Savaraj, N.; Feun, L.G.; Hung, M.C.; Chen, H.H.W.; Kuo, M.T. Argininosuccinate synthetase 1 (ASS1) is a common metabolic marker of chemosensitivity for targeted arginine- and glutamine-starvation therapy. Cancer Lett. 2017, 388, 54–63. [Google Scholar] [CrossRef]
- Zhou, M.; Zhao, Y.H.; Ding, Y.; Liu, H.; Liu, Z.X.; Fodstad, O.; Riker, A.I.; Kamarajugadda, S.; Lu, J.R.; Owen, L.B.; et al. Warburg effect in chemosensitivity: Targeting lactate dehydrogenase-A re-sensitizes Taxol-Resistant cancer cells to Taxol. Mol. Cancer 2010, 9, 1–12. [Google Scholar] [CrossRef]
- Jang, W.J.; Choi, B.; Song, S.H.; Lee, N.; Kim, D.J.; Lee, S.; Jeong, C.H. Multi-Omics analysis reveals that ornithine decarboxylase contributes to erlotinib resistance in pancreatic cancer cells. Oncotarget 2017, 8, 92727–92742. [Google Scholar] [CrossRef] [PubMed]
- Poisson, L.M.; Munkarah, A.; Madi, H.; Datta, I.; Hensley-Alford, S.; Tebbe, C.; Buekers, T.; Giri, S.; Rattan, R. A metabolomic approach to identifying platinum resistance in ovarian cancer. J. Ovarian Res. 2015, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, H.S.; Seo, Y.R. Understanding of ROS-Inducing Strategy in Anticancer Therapy. Oxidative Med. Cell Longev. 2019, 2019, 5381692. [Google Scholar] [CrossRef] [PubMed]
- Wangpaichitr, M.; Wu, C.; Li, Y.Y.; Nguyen, D.J.M.; Kandemir, H.; Shah, S.; Chen, S.; Feun, L.G.; Prince, J.S.; Kuo, M.T.; et al. Exploiting ROS and metabolic differences to kill cisplatin resistant lung cancer. Oncotarget 2017, 8, 49275–49292. [Google Scholar] [CrossRef] [PubMed]
- Wangpaichitr, M.; Kandemir, H.; Li, Y.Y.; Wu, C.; Nguyen, D.; Feun, L.G.; Kuo, M.T.; Savaraj, N. Relationship of Metabolic Alterations and PD-L1 Expression in Cisplatin Resistant Lung Cancer. Cell Dev. Biol. 2017, 6, 183. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Bermudez, A.; Laza-Briviesca, R.; Vicente-Blanco, R.J.; Garcia-Grande, A.; Coronado, M.J.; Laine-Menendez, S.; Palacios-Zambrano, S.; Moreno-Villa, M.R.; Ruiz-Valdepenas, A.M.; Lendinez, C.; et al. Cisplatin resistance involves a metabolic reprogramming through ROS and PGC-1alpha in NSCLC which can be overcome by OXPHOS inhibition. Free Radic. Biol. Med. 2019, 135, 167–181. [Google Scholar] [CrossRef]
- Kang, Y.P.; Torrente, L.; Falzone, A.; Elkins, C.M.; Liu, M.; Asara, J.M.; Dibble, C.C.; DeNicola, G.M. Cysteine dioxygenase 1 is a metabolic liability for non-small cell lung cancer. Elife 2019, 8, e45572. [Google Scholar] [CrossRef]
- Nunes, S.C.; Lopes-Coelho, F.; Gouveia-Fernandes, S.; Ramos, C.; Pereira, S.A.; Serpa, J. Cysteine boosters the evolutionary adaptation to CoCl2 mimicked hypoxia conditions, favouring carboplatin resistance in ovarian cancer. BMC Evol. Biol. 2018, 18, 97. [Google Scholar] [CrossRef]
- Nunes, S.C.; Ramos, C.; Lopes-Coelho, F.; Sequeira, C.O.; Silva, F.; Gouveia-Fernandes, S.; Rodrigues, A.; Guimaraes, A.; Silveira, M.; Abreu, S.; et al. Cysteine allows ovarian cancer cells to adapt to hypoxia and to escape from carboplatin cytotoxicity. Sci. Rep. 2018, 8, 1–17. [Google Scholar] [CrossRef]
- Yu, L.; Teoh, S.T.; Ensink, E.; Ogrodzinski, M.P.; Yang, C.; Vazquez, A.I.; Lunt, S.Y. Cysteine catabolism and the serine biosynthesis pathway support pyruvate production during pyruvate kinase knockdown in pancreatic cancer cells. Cancer Metab. 2019, 7, 1–13. [Google Scholar] [CrossRef]
- Combs, J.A.; DeNicola, G.M. The Non-Essential Amino Acid Cysteine Becomes Essential for Tumor Proliferation and Survival. Cancers 2019, 11, 678. [Google Scholar] [CrossRef]
- Stewart, D.A.; Winnike, J.H.; McRitchie, S.L.; Clark, R.F.; Pathmasiri, W.W.; Sumner, S.J. Metabolomics Analysis of Hormone-Responsive and Triple-Negative Breast Cancer Cell Responses to Paclitaxel Identify Key Metabolic Differences. J. Proteome Res. 2016, 15, 3225–3240. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Stokes, W.; Chater, E.; Roy, R.; de Bruin, E.; Hu, Y.; Liu, Z.; Smit, E.F.; Heynen, G.J.; Downward, J.; et al. Decreased glutathione biosynthesis contributes to EGFR T790M-driven erlotinib resistance in non-small cell lung cancer. Cell Discov. 2016, 2, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kominsky, D.J.; Klawitter, J.; Brown, J.L.; Boros, L.G.; Melo, J.V.; Eckhardt, S.G.; Serkova, N.J. Abnormalities in glucose uptake and metabolism in imatinib-resistant human BCR-ABL-positive cells. Clin. Cancer Res. 2009, 15, 3442–3450. [Google Scholar] [CrossRef] [PubMed]
- Dewar, B.J.; Keshari, K.; Jeffries, R.; Dzeja, P.; Graves, L.M.; Macdonald, J.M. Metabolic assessment of a novel chronic myelogenous leukemic cell line and an imatinib resistant subline by H-1 NMR spectroscopy. Metabolomics 2010, 6, 439–450. [Google Scholar] [CrossRef]
- van Asten, J.J.A.; Vettukattil, R.; Buckle, T.; Rottenberg, S.; van Leeuwen, F.; Bathen, T.F.; Heerschap, A. Increased levels of choline metabolites are an early marker of docetaxel treatment response in BRCA1-mutated mouse mammary tumors: An assessment by ex vivo proton magnetic resonance spectroscopy. J. Transl. Med. 2015, 13, 114. [Google Scholar] [CrossRef]
- St-Coeur, P.D.; Poitras, J.J.; Cuperlovic-Culf, M.; Touaibia, M.; Morin, P. Investigating a signature of temozolomide resistance in GBM cell lines using metabolomics. J. Neuro-Oncol. 2015, 125, 91–102. [Google Scholar] [CrossRef]
- Zaal, E.A.; Wu, W.; Jansen, G.; Zweegman, S.; Cloos, J.; Berkers, C.R. Bortezomib resistance in multiple myeloma is associated with increased serine synthesis. Cancer Metab. 2017, 5, 7. [Google Scholar] [CrossRef]
- Wu, X.; Xia, J.; Zhang, J.; Zhu, Y.; Wu, Y.; Guo, J.; Chen, S.; Lei, Q.; Meng, B.; Kuang, C.; et al. Phosphoglycerate dehydrogenase promotes proliferation and bortezomib resistance through increasing reduced glutathione synthesis in multiple myeloma. Br. J. Haematol. 2020, 190, 52–66. [Google Scholar] [CrossRef]
- Cao, Y. Adipocyte and lipid metabolism in cancer drug resistance. J. Clin. Invest. 2019, 129, 3006–3017. [Google Scholar] [CrossRef] [PubMed]
- Musacchio, T.; Toniutti, M.; Kautz, R.; Torchilin, V.P. 1H NMR detection of mobile lipids as a marker for apoptosis: The case of anticancer drug-loaded liposomes and polymeric micelles. Mol. Pharm. 2009, 6, 1876–1882. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Luciani, A.M.; Grande, S.; Palma, A.; Rosi, A.; Giovannini, C.; Sapora, O.; Viti, V.; Guidoni, L. Characterization of 1H NMR detectable mobile lipids in cells from human adenocarcinomas. FEBS J. 2009, 276, 1333–1346. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Yeon, A.; Shahid, M.; Cho, E.; Sairam, V.; Figlin, R.; Kim, K.H.; Kim, J. Reprogrammed lipid metabolism in bladder cancer with cisplatin resistance. Oncotarget 2018, 9, 13231–13243. [Google Scholar] [CrossRef]
- Hong, W.; Zhao, Y.; Cao, L.; Cao, D.; Zhao, Z.; Jin, J. Metabolomics Study on the Differences of Endogenous Small Molecule between A549/DDP and A549 Cells Based on High Solution UPLC-TOF-MS. Zhongguo Fei Ai Za Zhi 2018, 21, 571–577. [Google Scholar] [CrossRef]
- Dar, S.; Chhina, J.; Mert, I.; Chitale, D.; Buekers, T.; Kaur, H.; Giri, S.; Munkarah, A.; Rattan, R. Bioenergetic Adaptations in Chemoresistant Ovarian Cancer Cells. Sci. Rep. 2017, 7, 1–17. [Google Scholar] [CrossRef]
- Puchades-Carrasco, L.; Pineda-Lucena, A. Metabolomics Applications in Precision Medicine: An Oncological Perspective. Curr. Top. Med. Chem. 2017, 17, 2740–2751. [Google Scholar] [CrossRef]
- Li, X.R.; Lu, J.; Kan, Q.C.; Li, X.L.; Fan, Q.; Li, Y.Q.; Huang, R.X.; Slipicevic, A.; Dong, H.P.; Eide, L.; et al. Metabolic reprogramming is associated with flavopiridol resistance in prostate cancer DU145 cells. Sci. Rep. 2017, 7, 1–20. [Google Scholar] [CrossRef]
- Staubert, C.; Bhuiyan, H.; Lindahl, A.; Broom, O.J.; Zhu, Y.; Islam, S.; Linnarsson, S.; Lehtio, J.; Nordstrom, A. Rewired metabolism in drug-resistant leukemia cells: A metabolic switch hallmarked by reduced dependence on exogenous glutamine. J. Biol. Chem. 2015, 290, 8348–8359. [Google Scholar] [CrossRef] [PubMed]
- Nowotarski, S.L.; Feith, D.J.; Shantz, L.M. Skin Carcinogenesis Studies Using Mouse Models with Altered Polyamines. Cancer Growth Metastasis 2015, 8, 17–27. [Google Scholar] [CrossRef]
- Cao, B.; Li, M.J.; Zha, W.B.; Zhao, Q.J.; Gu, R.R.; Liu, L.S.; Shi, J.; Zhou, J.; Zhou, F.; Wu, X.L.; et al. Metabolomic approach to evaluating adriamycin pharmacodynamics and resistance in breast cancer cells. Metabolomics 2013, 9, 960–973. [Google Scholar] [CrossRef] [PubMed]
- Klawitter, J.; Anderson, N.; Klawitter, J.; Christians, U.; Leibfritz, D.; Eckhardt, S.G.; Serkova, N.J. Time-Dependent effects of imatinib in human leukaemia cells: A kinetic NMR-profiling study. Br. J. Cancer 2009, 100, 923–931. [Google Scholar] [CrossRef]
- Sasada, S.; Miyata, Y.; Tsutani, Y.; Tsuyama, N.; Masujima, T.; Hihara, J.; Okada, M. Metabolomic analysis of dynamic response and drug resistance of gastric cancer cells to 5-fluorouracil. Oncol. Rep. 2013, 29, 925–931. [Google Scholar] [CrossRef]
- Wu, W.; Wang, Q.; Yin, F.; Yang, Z.; Zhang, W.; Gabra, H.; Li, L. Identification of proteomic and metabolic signatures associated with chemoresistance of human epithelial ovarian cancer. Int. J. Oncol. 2016, 49, 1651–1665. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, X.; Wang, L.; Chen, S. The sweet trap in tumors: Aerobic glycolysis and potential targets for therapy. Oncotarget 2016, 7, 38908–38926. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, X.; Sun, X.Q.; Wang, L.T.; Chen, S.W. The Glycolytic Switch in Tumors: How Many Players Are Involved? J. Cancer 2017, 8, 3430–3440. [Google Scholar] [CrossRef]
- Shimanishi, M.; Ogi, K.; Sogabe, Y.; Kaneko, T.; Dehari, H.; Miyazaki, A.; Hiratsuka, H. Silencing of GLUT-1 inhibits sensitization of oral cancer cells to cisplatin during hypoxia. J. Oral Pathol. Med. 2013, 42, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Goode, G.; Gunda, V.; Chaika, N.V.; Purohit, V.; Yu, F.; Singh, P.K. MUC1 facilitates metabolomic reprogramming in triple-negative breast cancer. PLoS ONE 2017, 12, e0179098. [Google Scholar] [CrossRef]
- Tripathi, S.C.; Fahrmann, J.F.; Celiktas, M.; Aguilar, M.; Marini, K.D.; Jolly, M.K.; Katayama, H.; Wang, H.; Murage, E.N.; Dennison, J.B.; et al. MCAM Mediates Chemoresistance in Small-Cell Lung Cancer via the PI3K/AKT/SOX2 Signaling Pathway. Cancer Res. 2017, 77, 4414–4425. [Google Scholar] [CrossRef]
- Qian, X.; Xu, W.; Xu, J.; Shi, Q.; Li, J.; Weng, Y.; Jiang, Z.; Feng, L.; Wang, X.; Zhou, J.; et al. Enolase 1 stimulates glycolysis to promote chemoresistance in gastric cancer. Oncotarget 2017, 8, 47691–47708. [Google Scholar] [CrossRef]
- Fujisawa, K.; Terai, S.; Takami, T.; Yamamoto, N.; Yamasaki, T.; Matsumoto, T.; Yamaguchi, K.; Owada, Y.; Nishina, H.; Noma, T.; et al. Modulation of anti-cancer drug sensitivity through the regulation of mitochondrial activity by adenylate kinase 4. J. Exp. Clin. Cancer Res. 2016, 35, 48. [Google Scholar] [CrossRef]
- Amoroso, M.R.; Matassa, D.S.; Agliarulo, I.; Avolio, R.; Maddalena, F.; Condelli, V.; Landriscina, M.; Esposito, F. Stress-Adaptive Response in Ovarian Cancer Drug Resistance: Role of TRAP1 in Oxidative Metabolism-Driven Inflammation. Adv. Protein Chem. Struct. Biol. 2017, 108, 163–198. [Google Scholar] [CrossRef] [PubMed]
- Fekir, K.; Dubois-Pot-Schneider, H.; Desert, R.; Daniel, Y.; Glaise, D.; Rauch, C.; Morel, F.; Fromenty, B.; Musso, O.; Cabillic, F.; et al. Retrodifferentiation of Human Tumor Hepatocytes to Stem Cells Leads to Metabolic Reprogramming and Chemoresistance. Cancer Res. 2019, 79, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- Osborne, M.J.; de Oliveira, L.C.; Volpon, L.; Zahreddine, H.A.; Borden, K.L.B. Overcoming Drug Resistance through the Development of Selective Inhibitors of UDP-Glucuronosyltransferase Enzymes. J. Mol. Biol. 2019, 431, 258–272. [Google Scholar] [CrossRef]
- Zhou, J.X.; Wink, M. Reversal of Multidrug Resistance in Human Colon Cancer and Human Leukemia Cells by Three Plant Extracts and Their Major Secondary Metabolites. Medicines 2018, 5, 123. [Google Scholar] [CrossRef] [PubMed]
- Zong, L.; Cheng, G.; Liu, S.; Pi, Z.; Liu, Z.; Song, F. Reversal of multidrug resistance in breast cancer cells by a combination of ursolic acid with doxorubicin. J. Pharm. Biomed. Anal. 2019, 165, 268–275. [Google Scholar] [CrossRef]
- Alonezi, S.; Tusiimire, J.; Wallace, J.; Dufton, M.J.; Parkinson, J.A.; Young, L.C.; Clements, C.J.; Park, J.K.; Jeon, J.W.; Ferro, V.A.; et al. Metabolomic Profiling of the Synergistic Effects of Melittin in Combination with Cisplatin on Ovarian Cancer Cells. Metabolites 2017, 7, 14. [Google Scholar] [CrossRef]
- Lin, L.; Li, L.; Chen, X.; Zeng, B.; Lin, T. Preliminary evaluation of the potential role of beta-elemene in reversing erlotinib-resistant human NSCLC A549/ER cells. Oncol. Lett. 2018, 16, 3380–3388. [Google Scholar] [CrossRef]
- Capella Roca, B.; Lao, N.; Barron, N.; Doolan, P.; Clynes, M. An arginase-based system for selection of transfected CHO cells without the use of toxic chemicals. J. Biol. Chem. 2019, 294, 18756–18768. [Google Scholar] [CrossRef]
- Bayet-Robert, M.; Morvan, D. Metabolomics reveals metabolic targets and biphasic responses in breast cancer cells treated by curcumin alone and in association with docetaxel. PLoS ONE 2013, 8, e57971. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Samples | Analysis Approach | Drugs | Metabolic Changes | Reference |
|---|---|---|---|---|
| Ovarian cancer cells | NMR | Cisplatin | GSH, mobile lipids↑ | [21] |
| Lung cancer cells | Fluorimeter | Cisplatin | ROS↑ | [35] |
| ESCC cells | NMR | 5-Fluorouracil | Glutamate↓, lactate, glutamine↑ | [14] |
| TAMs | GC-TOF-MS | 5-Fluorouracil | Putrescine↑ | [15] |
| Pancreatic cancer cells | MS | Erlotinib | Short-chain acylcarnitines, lysophosphatidylcholines↑, acyl-alkyl-phosphatidylcholines, sphingolipid↓ | [16] |
| Lung cancer cells | NMR | Erlotinib | GSH↓ | [44] |
| Leukemia cells | UHPLC-ESI-MS | Sorafenib | GSH↑ | [17] |
| CML cells | NMR, GC-MS | Imatinib | Lactate ↑ | [45] |
| CML cells | NMR | Imatinib | Creatine, phosphocreatine↑, lactate↓ | [46] |
| Breast cancer cells | NMR | Paclitaxel | Glutamine, glutamate, GSH↑, lysine, proline, valine↓ | [43] |
| Mouse mammary tumor tissues | NMR | Paclitaxel | Choline↑ | [47] |
| Glioblastoma multiforme cells | NMR | Temozolomide | Alanine, choline, creatine, and phosphorylcholine↓ | [48] |
| Events. | Examples | References |
|---|---|---|
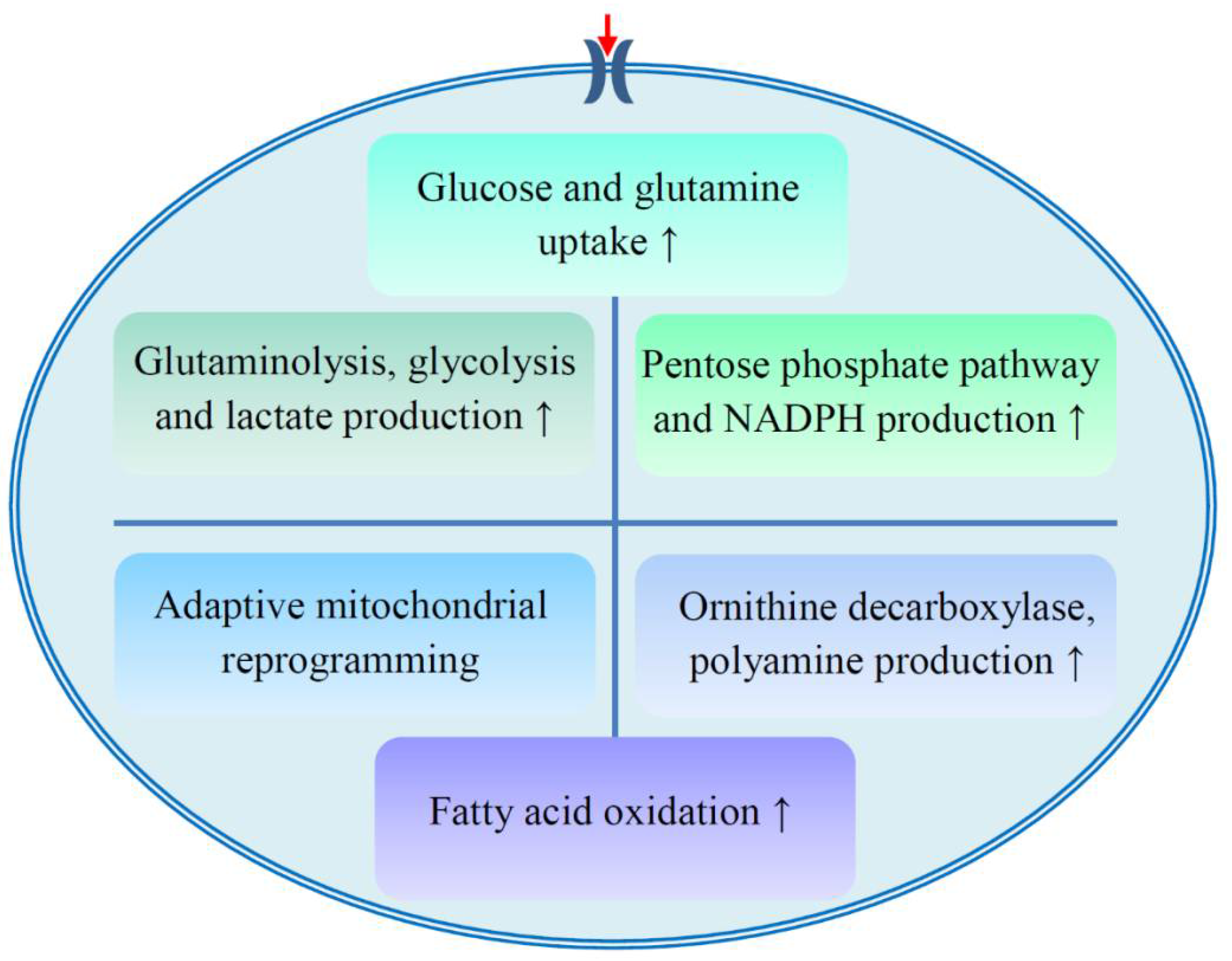
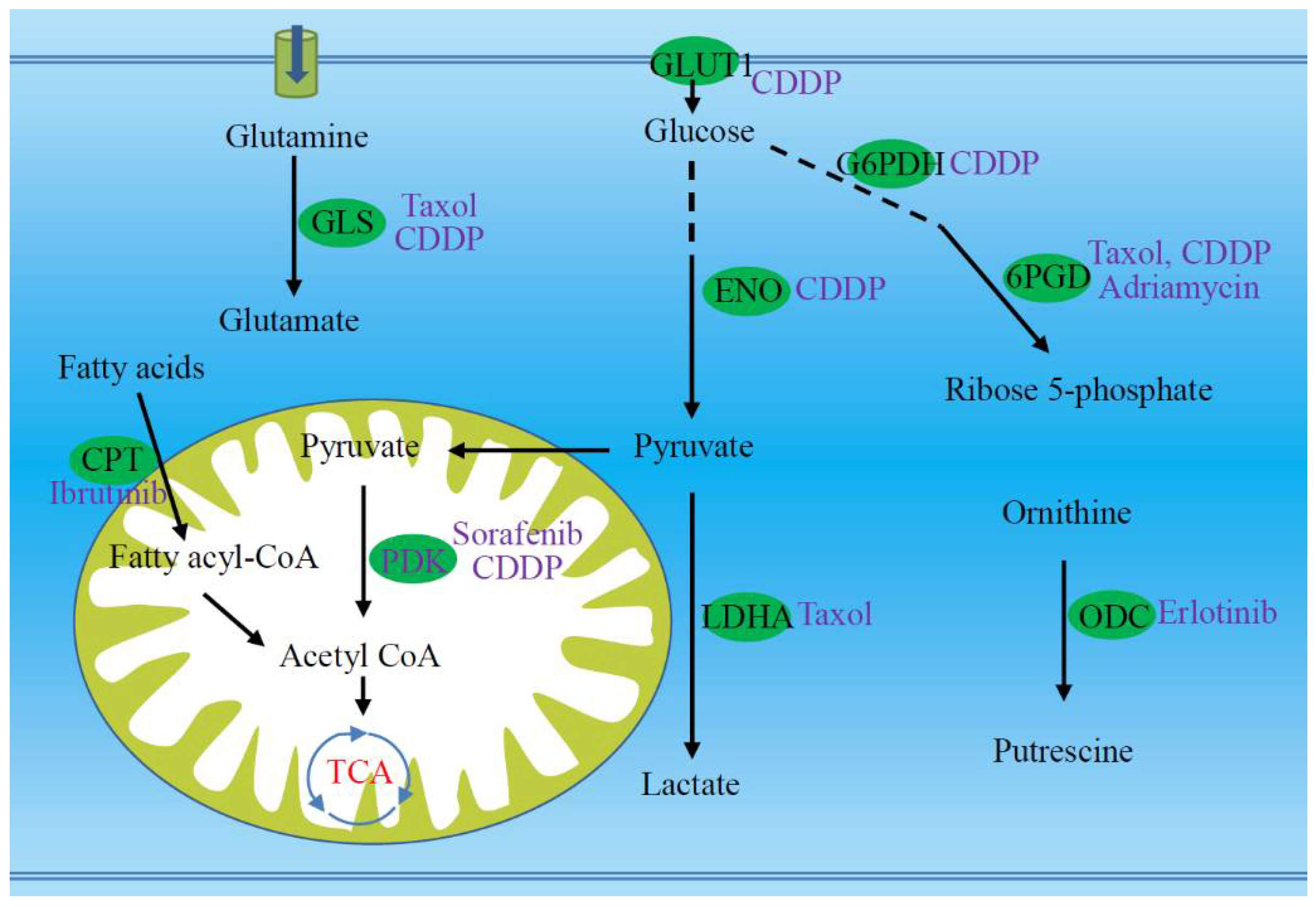
| Glucose and glutamine uptake | Mucin1 promotes glucose and glutamine uptake, which is associated with chemoresistance; the ibrutinib resistant cells absorb more glutamine; down-regulation of GLUT1 reduces the sensitivity of oral cancer cells to cisplatin. | [21,22,24,67,68] |
| Glutaminolysis, glycolysis and lactate production | MCAM regulates lactate production and chemosensitivity; enolase and GAPDH are carboplatin and paclitaxel resistance-related proteins; knockdown of enolase reverses cisplatin resistance; LDHA regulates the sensitivity of resistant cells to paclitaxel; glutaminase inhibitor increases the sensitivity of resistant cells to paclitaxel or cisplatin. | [18,23,30,69,70] |
| PPP and NADPH production | Expression and enzymatic activity of G6PDH are up-regulated in cisplatin resistant cells; 6PGD inhibition increases the sensitivity of HCC to paclitaxel, adriamycin, and cisplatin; NADPH accumulates in ibrutinib resistant cells. | [19,22,24] |
| Adaptive mitochondrial reprogramming | Resistance to PI3K therapy is related to adaptive mitochondrial reprogramming; AK4 knockdown increases the sensitivity to cisplatin; TRAP1 facilitates a metabolic shift toward OXPHOS, which is related to cisplatin resistance; PDK4 inhibition reverses the resistance of HCC stem cells to sorafenib or cisplatin. | [20,71,72,73] |
| Fatty acid oxidation | Inhibition of fatty acid oxidation re-sensitizes resistant cells to ibrutinib. | [24] |
| ODC and polyamine production | ODC-mediated production of putrescine contributes to the acquisition of erlotinib resistance. | [31] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Chen, S.; Yu, D. Metabolic Reprogramming of Chemoresistant Cancer Cells and the Potential Significance of Metabolic Regulation in the Reversal of Cancer Chemoresistance. Metabolites 2020, 10, 289. https://doi.org/10.3390/metabo10070289
Chen X, Chen S, Yu D. Metabolic Reprogramming of Chemoresistant Cancer Cells and the Potential Significance of Metabolic Regulation in the Reversal of Cancer Chemoresistance. Metabolites. 2020; 10(7):289. https://doi.org/10.3390/metabo10070289
Chicago/Turabian StyleChen, Xun, Shangwu Chen, and Dongsheng Yu. 2020. "Metabolic Reprogramming of Chemoresistant Cancer Cells and the Potential Significance of Metabolic Regulation in the Reversal of Cancer Chemoresistance" Metabolites 10, no. 7: 289. https://doi.org/10.3390/metabo10070289
APA StyleChen, X., Chen, S., & Yu, D. (2020). Metabolic Reprogramming of Chemoresistant Cancer Cells and the Potential Significance of Metabolic Regulation in the Reversal of Cancer Chemoresistance. Metabolites, 10(7), 289. https://doi.org/10.3390/metabo10070289