
Synthesis and Biological Evaluation of Some Coumarin–Triazole Conjugates as Potential Anticancer Agents

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Instrumentations
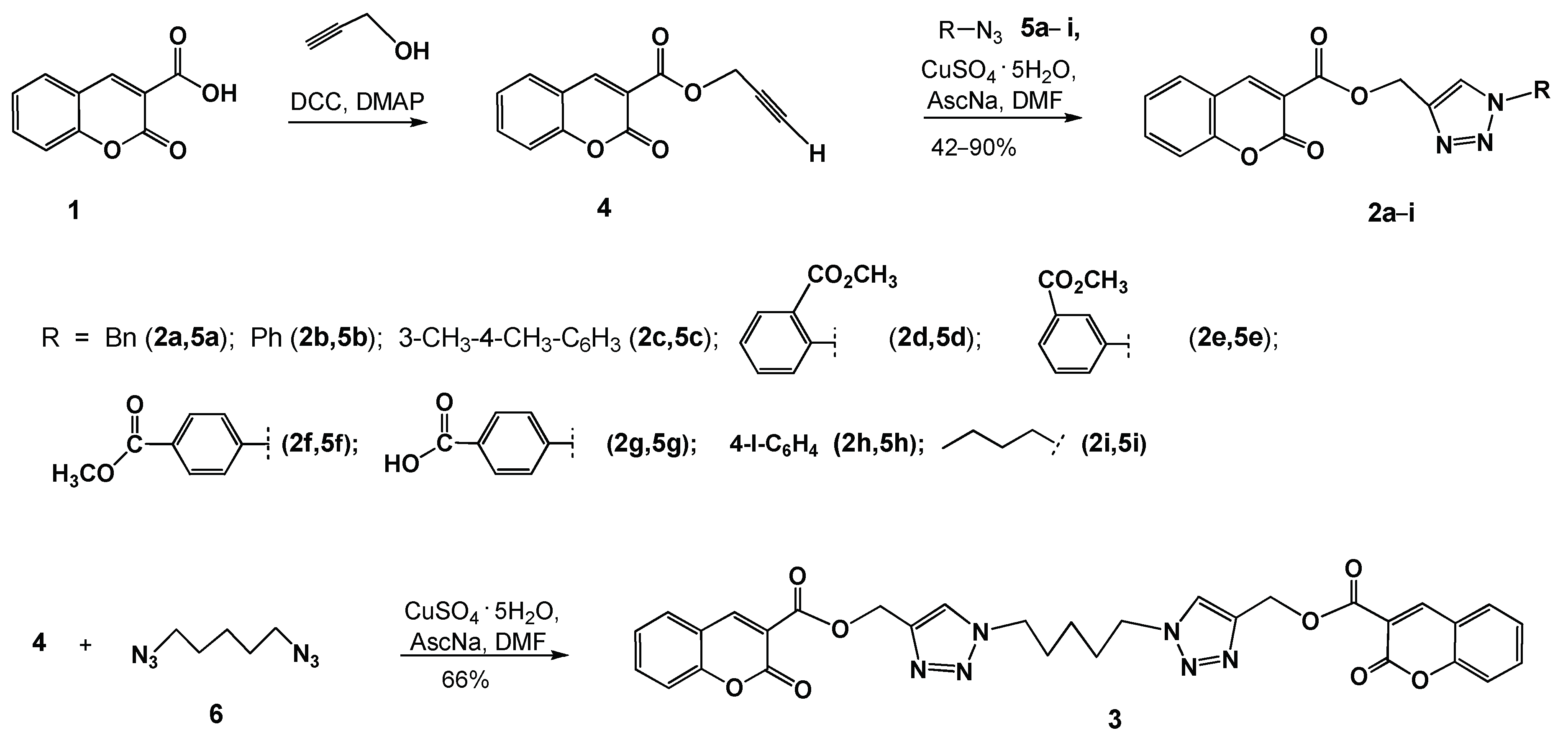
2.2. Synthesis
General Procedure for the Synthesis of Coumarino-1,2,3-triazolyl derivatives (2a–i)
2.3. Characterization of Compounds 2a–i
Synthesis of (1,1′-(Pentane-1,5-diyl)bis(1H-1,2,3-triazole-4,1-diyl))bis(methylene) bis-(2-oxo-2H-chromene-3-carboxylate) (3)
2.4. Biological Studies
2.4.1. Cell Culture and Cytotoxicity Assay
2.4.2. Cell Cycle Analysis
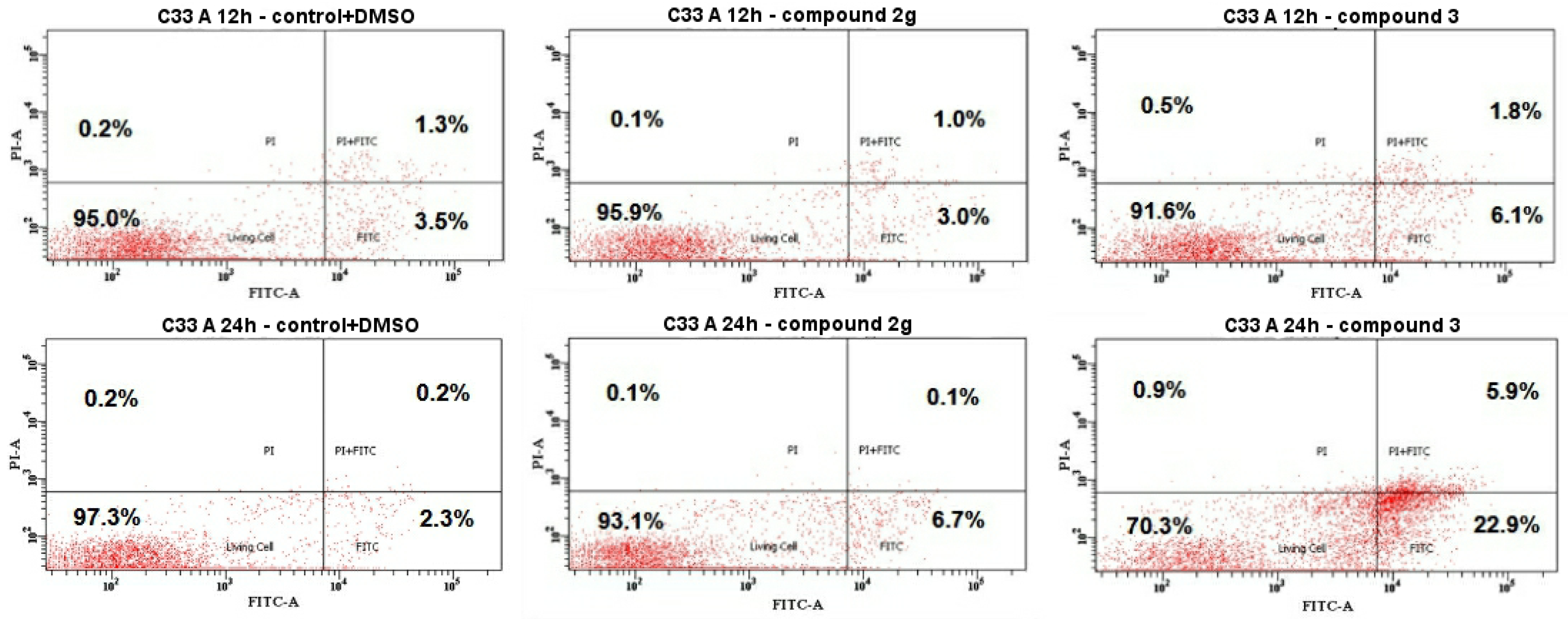
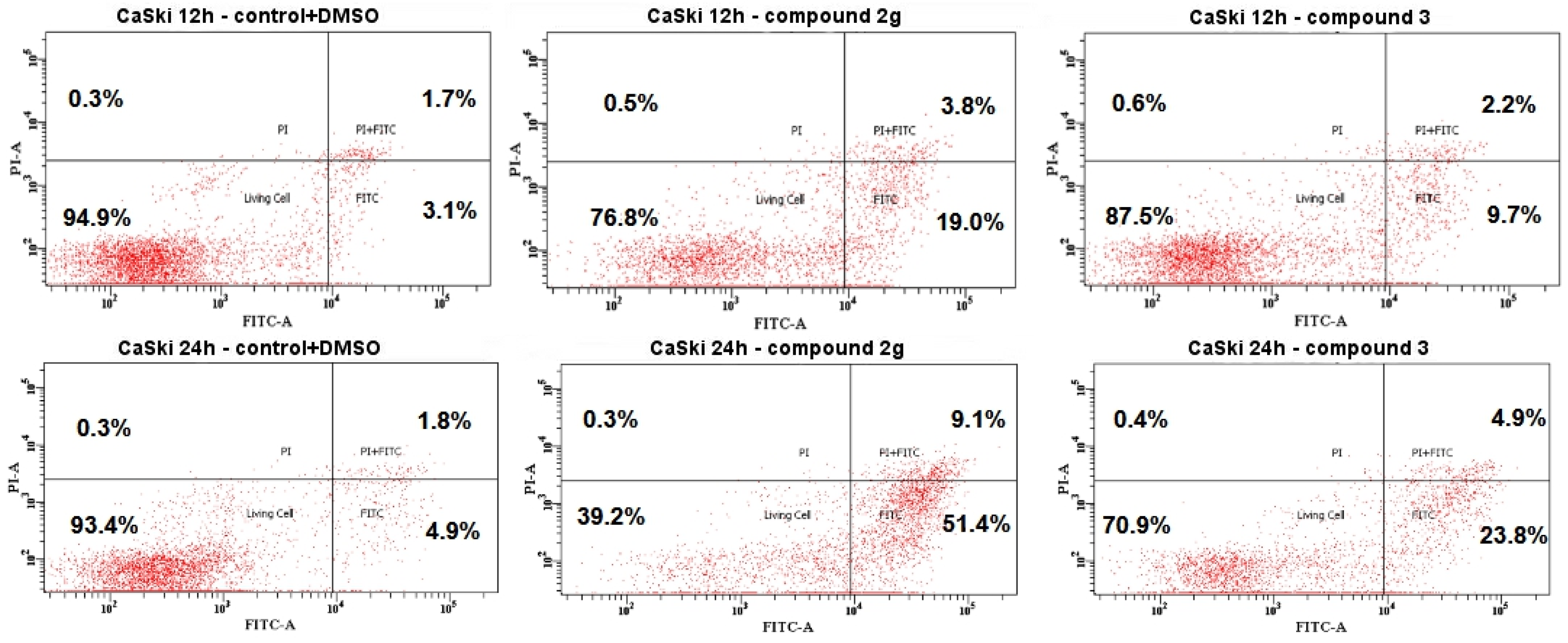
2.4.3. Cell Death by Annexin V-FITC/PI Staining Analyzed by Flow Cytometry
3. Results
3.1. Chemistry
3.2. Biological Study
3.2.1. Cytotoxicity Assay
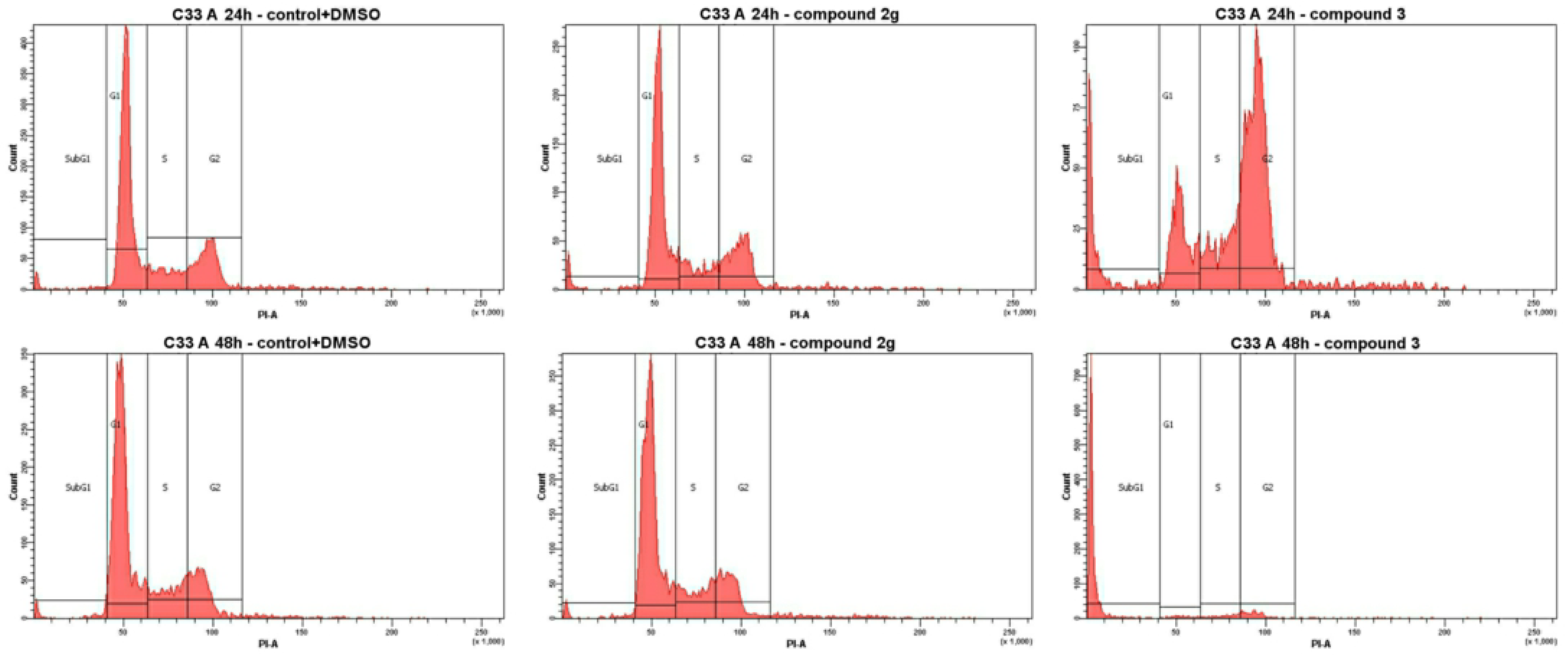
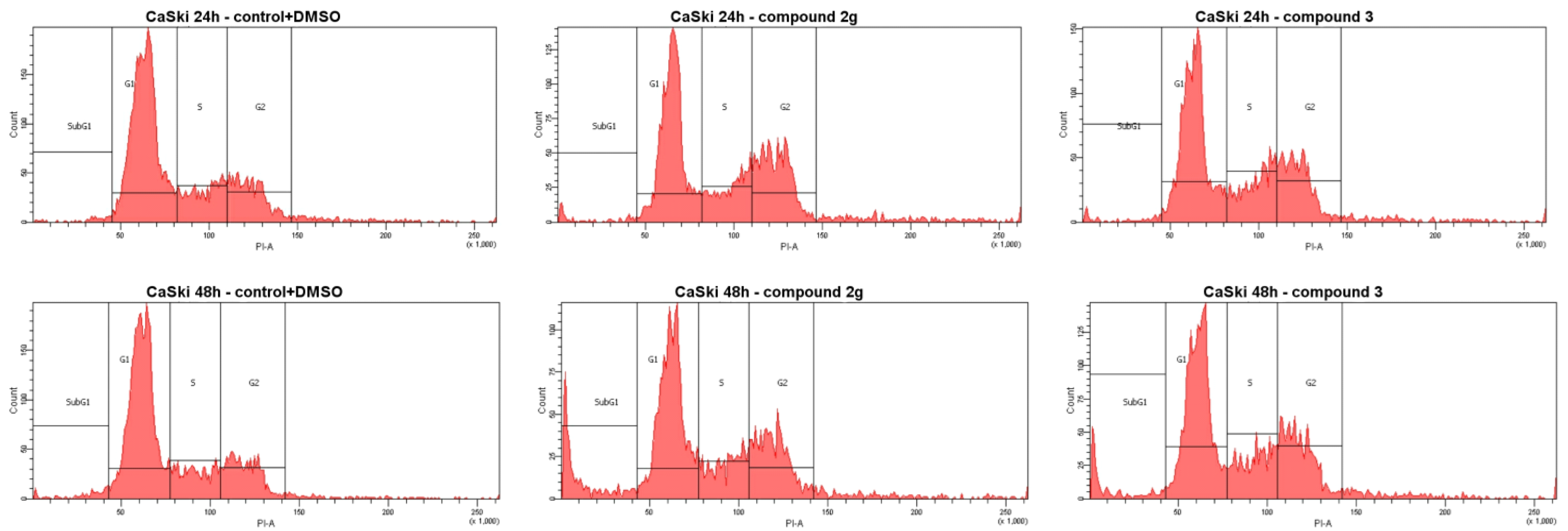
3.2.2. Cell Cycle Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salem, M.A.; Marzouk, M.I.; El-Kazak, A.M. Synthesis and characterization of some new coumarins with in vitro antitumor and antioxidant activity and high protective effects against DNA damage. Molecules 2016, 21, 249–269. [Google Scholar] [CrossRef] [PubMed]
- Majhi, A.; Venkateswarlu, K.; Sasikumar, P. Coumarin based fluorescent probe for detecting heavy metal ions. J. Fluoresc. 2024, 34, 1453–1483. [Google Scholar] [CrossRef]
- Sokol, I.; Toma, M.; Krnić, M.; Macan, A.M.; Drenjančević, D.; Liekens, S.; Raić-Malić, S.; Gazivoda Kraljević, T. Transition metal-catalyzed synthesis of new 3-substituted coumarin derivatives as antibacterial and cytostatic agents. Future Med. Chem. 2021, 13, 1865–1884. [Google Scholar] [CrossRef]
- Ghouse, S.M.; Bahatam, K.; Angeli, A.; Pawar, G.; Chinchilli, K.K.; Yaddanapudi, V.M.; Mohammed, A.; Supuran, C.T.; Nanduri, S. Synthesis and biological evaluation of new 3-substituted coumarin derivatives as selective inhibitors of human carbonic anhydrase IX and XII. J. Enzym. Inhib. Med. Chem. 2023, 38, 2185760. [Google Scholar] [CrossRef]
- Gao, L.; Wang, F.; Chen, Y.; Li, F.; Han, B.; Liu, D. The antithrombotic activity of natural and synthetic coumarins. Fitoterapia 2021, 154, 104947. [Google Scholar] [CrossRef]
- Ramsis, T.M.; Ebrahim, M.A.; Fayed, E.A. Synthetic coumarin derivatives with anticoagulation and antiplatelet aggregation inhibitory effects. Med. Chem. Res. 2023, 32, 2269–2278. [Google Scholar] [CrossRef]
- Tan, X.; Soualmia, F.; Furio, L.; Renard, J.-F.; Kempen, I.; Qin, L.; Pagano, M.; Pirotte, B.; El Amri, C.; Hovnanian, A.; et al. Toward the first class of suicide inhibitors of kallikreins involved in skin diseases. J. Med. Chem. 2015, 58, 598–612. [Google Scholar] [CrossRef]
- Hanke, S.; Tindall, C.A.; Pippel, J.; Ulbricht, D.; Pirotte, B.; Reboud-Ravaux, M.; Heiker, J.T.; Sträter, N. Structural studies on the inhibitory binding mode of aromatic coumarinic esters to human kallikrein-related peptidase 7. J. Med. Chem. 2020, 63, 5723–5733. [Google Scholar] [CrossRef]
- Frédérick, R.; Robert, S.; Charlier, C.; Wouters, J.; Masereel, B.; Pochet, L. Mechanism-based thrombin inhibitors: Design, synthesis, and molecular docking of a new selective 2-oxo-2H-1-benzopyran derivative. J. Med. Chem. 2007, 50, 3645–3650. [Google Scholar] [CrossRef]
- Davoine, C.; Traina, A.; Evrard, J.; Lanners, S.; Fillet, M.; Pochet, L. Coumarins as factor XIIa inhibitors: Potency and selectivity improvements using a fragment-based strategy. Eur. J. Med. Chem. 2023, 259, 115636. [Google Scholar] [CrossRef]
- Liu, H.; Xia, D.G.; Chu, Z.W.; Hu, R.; Cheng, X.; Lv, X.H. Novel coumarin-thiazolyl ester derivatives as potential DNA gyrase Inhibitors: Design, synthesis, and antibacterial activity. Bioorg. Chem. 2020, 100, 103907. [Google Scholar] [CrossRef] [PubMed]
- Nelson, G.L.; Ronayne, C.T.; Solano, L.N.; Jonnalagadda, S.K.; Jonnalagadda, S.; Schumacher, T.J.; Gardner, Z.S.; Palle, H.; Mani, C.; Rumbley, J.; et al. Synthesis and biological evaluation of N, N-dialkylcarboxy coumarin-NO donor conjugates as potential anticancer agents. Bioorg. Med. Chem. Lett. 2021, 52, 128411. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Ding, L.; Tan, H.; Liu, C.-B.; He, L.-Q. Synthesis and antitumor activity evaluation of coumarin Mannich base derivatives. Chem. Biol. Drug Des. 2024, 103, e14389. [Google Scholar] [CrossRef]
- Jain, A.; Piplani, P. Exploring the chemistry and therapeutic potential of triazoles: A comprehensive literature. Mini Rev. Med. Chem. 2019, 19, 1298–1368. [Google Scholar]
- Guo, H.-Y.; Chen, Z.A.; Shen, Q.-K.; Quan, Z.-S. Application of triazoles in the structural modification of natural products. J. Enzyme Inhib. Med. Chem. 2021, 36, 1115–1144. [Google Scholar] [CrossRef]
- Alam, M.M. 1,2,3-Triazole hybrids as anticancer agents: A review. Arch. Pharm. 2022, 355, e2100158. [Google Scholar] [CrossRef]
- Rohman, N.; Ardiansah, B.; Wukirsari, T.; Judeh, Z. Recent trends in the synthesis and bioactivity of coumarin, coumarin-chalcone, and coumarin-triazole molecular hybrids. Molecules 2024, 29, 1026. [Google Scholar] [CrossRef]
- Pršir, K.; Horak, E.; Kralj, M.; Uzelac, L.; Liekens, S.; Steinberg, I.M.; Krištafor, S. Design, synthesis, spectroscopic characterisation and in vitro cytostatic evaluation of novel bis(coumarin-1,2,3-triazolyl)benzenes and hybrid coumarin-1,2,3-triazolyl-aryl derivatives. Molecules 2022, 27, 637. [Google Scholar] [CrossRef]
- Maiti, S.; Park, N.; Han, J.H.; Jeon, H.M.; Lee, J.H.; Bhuniya, S.; Kang, C.; Kim, J.S. Gemcitabine–coumarin–biotin conjugates: A target specific theranostic anticancer prodrug. J. Am. Chem. Soc. 2013, 135, 4567–4572. [Google Scholar] [CrossRef]
- Fan, Y.L.; Ke, X.; Liu, M. Coumarin–triazole hybrids and their biological activities. J. Heterocycl. Chem. 2018, 55, 791–802. [Google Scholar] [CrossRef]
- Slanova, K.; Todorov, L.; Belskaya, N.P.; Palafox, M.A.; Kostova, I.P. Developments in the application of 1,2,3-triazoles in cancer treatment. Recent Pat. Anti-Cancer Drug Discov. 2020, 15, 92–112. [Google Scholar] [CrossRef] [PubMed]
- López, S.; Gracia, I.; Plaza-Pedroche, R.; Rodríguez, J.F.; Pérez-Ortiz, J.M.; Rodríguez-López, J.; Ramos, M.J. In vitro antioxidant and pancreatic anticancer activity of novel 5-fluorouracil-coumarin conjugates. Pharmaceutics 2022, 14, 2152. [Google Scholar] [CrossRef] [PubMed]
- Nesaragi, A.R.; Algethami, J.S.; Alsaiari, M.; Alsareii, S.A.; Mathada, B.S.; Ningaiah, S.; Sasidhar, B.S.; Harraz, F.A.; Patil, S.A. A comprehensive overview of coumarinyl-triazole hybrids as anticancer agents. J. Mol. Struct. 2024, 1302, 137478. [Google Scholar] [CrossRef]
- Lipeeva, A.V.; Zakharov, D.O.; Burova, L.G.; Frolova, T.S.; Baev, D.S.; Shirokikh, I.V.; Evstropov, A.N.; Sinitsyna, O.I.; Tolstikova, T.G.; Shults, E.E. Design, synthesis and antibacterial activity of coumarin-1,2,3-triazole hybrids obtained from natural furocoumarin peucedanin. Molecules 2019, 24, 2126. [Google Scholar] [CrossRef] [PubMed]
- Lipeeva, A.V.; Khvostov, M.V.; Baev, D.S.; Shakirov, M.M.; Tolstikova, T.G.; Shults, E.E. Synthesis, in vivo anticoagulant evaluation and molecular docking studies of bicoumarins obtained from furocoumarin peucedanin. Med. Chem. 2016, 12, 674–683. [Google Scholar] [CrossRef]
- Ivanov, A.A.; Ukladov, E.A.; Kremis, S.A.; Sharapov, S.Z.; Baiborodin, S.I.; Lipeeva, A.V.; Shults, E.E.; Golubeva, T.S. Investigation of cytotoxic and antioxidative activity of 1,2,3-triazolyl-modified furocoumarins and 2,3-dihydrofurocoumarins. Protoplasma 2022, 259, 1321–1330. [Google Scholar] [CrossRef]
- Pramitha, P.; Bahulayan, D. Stereoselective synthesis of bio-hybrid amphiphiles of coumarin derivatives by Ugi–Mannich triazole randomization using copper catalyzed alkyne azide click chemistry. Bioorg. Med. Chem. Lett. 2012, 22, 2598–2603. [Google Scholar] [CrossRef]
- Raposo, C.D.; Conceição, C.A.; Barros, M.T. Nanoparticles based on novel carbohydrate-functionalized polymers. Molecules 2020, 25, 1744. [Google Scholar] [CrossRef]
- Jaabil, G.; Ranganathan, R.; Ponnuswamy, A.; Suresh, P.; Shanmugaiah, V.; Ravikumar, C.; Murugavel, S. A Green and efficient synthesis of bioactive 1,2,3-triazolyl-pyridine/cyanopyridine hybrids via one-pot multicomponent grinding protocol. ChemistrySelect 2018, 3, 10388–10393. [Google Scholar] [CrossRef]
- Berger, O.; Kaniti, A.; Tran van Ba, C.; Vial, H.; Ward, S.A.; Biagini, G.A.; Bray, P.G.; O’Neill, P.M. Synthesis and antimalarial activities of a diverse set of triazole-containing furamidine analogues. Chem. Med. Chem. 2011, 6, 2094–2108. [Google Scholar] [CrossRef]
- Mohamed, Z.H.; El-Koussi, N.A.; Mahfouz, N.M.; Youssef, A.F.; Jaleel, G.A.A.; Shouman, S.A. Cu (I) catalyzed alkyne-azide 1,3-dipolar cycloaddition (CuAAC): Synthesis of 17α-[1-(substituted phenyl)-1,2,3-triazol-4-yl]-19-nortestosterone-17β-yl acetates targeting progestational and antiproliferative activities. Eur. J. Med. Chem. 2015, 97, 75–82. [Google Scholar] [CrossRef]
- Bottaro, C.; Penwell, P.E.; Schmitt, R.J. Expedient synthesis of t-butyl azide. Synth. Commun. 1997, 27, 1465–1467. [Google Scholar] [CrossRef]
- Saikia, B.; Saikia, P.P.; Goswami, A.; Barua, N.C.; Saxena, A.K.; Suri, N. Synthesis of a novel series of 1,2,3-triazole-containing artemisinin dimers with potent anticancer activity involving Huisgen 1,3-dipolar cycloaddition reaction. Synthesis 2011, 2011, 3173–3179. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 16, 55–63. [Google Scholar] [CrossRef]
- Nurmaganbetov, Z.S.; Savelyev, V.A.; Gatilov, Y.V.; Nurkenov, O.A.; Seidakhmetova, R.B.; Shulgau, Z.T.; Mukusheva, G.K.; Fazylov, S.D.; Shults, E.E. Synthesis and analgesic activity of 1-[(1,2,3-triazol-1-yl)methyl]quinolizines based on the alkaloid lupinine. Chem. Heterocycl. Compd. 2021, 57, 911–919. [Google Scholar] [CrossRef]
- Finke, A.O.; Pavlova, A.V.; Morozova, E.A.; Tolstikova, T.G.; Shults, E.E. Synthesis of 1,2,3-triazolyl-substituted derivatives of the alkaloids sinomenine and tetrahydrothebaine on ring A and their analgesic activity. Chem. Nat. Compd. 2022, 58, 895–902. [Google Scholar] [CrossRef]
- Schepetkin, I.A.; Nurmaganbetov, Z.S.; Fazylov, S.D.; Nurkenov, O.A.; Khlebnikov, A.I.; Seilkhanov, T.M.; Kishkentaeva, A.S.; Shults, E.E.; Quinn, M.T. Inhibition of acetylcholinesterase by novel lupinine derivatives. Molecules 2023, 28, 3357. [Google Scholar] [CrossRef]
- Burlison, J.A.; Blagg, B.S. Synthesis and evaluation of coumermycin A1 analogues that inhibit the Hsp90 protein folding machinery. Org. Lett. 2006, 8, 4855–4858. [Google Scholar]
- Kusuma, B.R.; Peterson, L.B.; Zhao, H.; Vielhauer, G.; Holzbeierlein, J.; Blagg, B.S.J. Targeting the heat shock protein 90 dimer with dimeric inhibitors. J. Med. Chem. 2011, 54, 6234–6253. [Google Scholar] [CrossRef]
- Tan, G.; Yao, Y.; Gu, Y.; Li, S.; Lv, M.; Wang, K.; Chen, H.; Li, X. Cytotoxicity and DNA binding property of the dimers of triphenylethylene–coumarin hybrid with one amino side chain. Bioorg. Med. Chem. Lett. 2014, 24, 2825–2830. [Google Scholar] [CrossRef]
- Creary, X.; Anderson, A.; Brophy, C.; Crowell, F.; Funk, Z. Method for assigning structure of 1,2,3-triazoles. J. Org. Chem. 2012, 77, 8756–8761. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, S.; Singh, A.; Manhas, N.; Seboletswe, P.; Khubone, L.; Kumar, G.; Jonnalagadda, S.B.; Raza, A.; Sharma, A.K.; Singh, P. Exploring novel coumarin-tethered bis-triazoles: Apoptosis induction in human pancreatic cancer cells, antimicrobial effects, and molecular modelling investigations. ChemMedChem 2024, 19, e202400297. [Google Scholar] [CrossRef] [PubMed]
- Al-Warhi, T.; Sabt, A.; Elkaeed, E.B.; Eldehna, W.M. Recent advancements of coumarin-based anticancer agents: An up-to-date review. Bioorg. Chem. 2020, 103, 104163. [Google Scholar] [CrossRef]
- Yadav, A.K.; Shrestha, R.M.; Yadav, P.N. Anticancer mechanism of coumarin-based derivatives. Eur. J. Med. Chem. 2024, 267, 116179. [Google Scholar] [CrossRef]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A target for anticancer therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S.J. Apoptosis in cancer: From pathogenesis to treatment. J. Exper. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef]
- Nagy, N.; Kuipers, H.F.; Frymoyer, A.R.; Ishak, H.D.; Bollyky, J.B.; Wight, T.N.; Bollyky, P.L. 4-Methylumbelliferone treatment and hyaluronan inhibition as a therapeutic strategy in inflammation, autoimmunity, and cancer. Front. Immunol. 2015, 6, 123. [Google Scholar] [CrossRef]
- Martin, S.J. Cell death and inflammation: The case for IL-1 family cytokines as the canonical DAMPs of the immune system. FEBS J. 2016, 283, 2599–2615. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Growth Inhibition of Cells (GI50 ± SEM, μM) [a, b] | ||||||
|---|---|---|---|---|---|---|---|
 | C33 A (HPV- Negative) | CaSki (HPV-16) | HeLa (HPV-18) | MCF-7 | DU-145 | VERO | |
| 2a |  | >100 | 56 ± 3.6 | 29 ± 2.0 | 51 ± 2.9 | >100 | >100 |
| 2b | 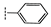 | 81 ± 0.2 | 38 ± 3.2 | 35 ± 4.0 | 41 ± 2.1 | >100 | >100 |
| 2c |  | 76 ± 4.1 | 33 ± 1.5 | 29 ± 1.8 | 62 ± 1.5 | >100 | >100 |
| 2d | 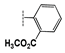 | 21 ± 1.7 | 19 ± 0.9 | 27 ± 1.7 | 40 ± 2.7 | 34 ± 2.0 | >100 |
| 2e |  | 31 ± 3.1 | 25 ± 1.4 | 32 ± 2.5 | 36 ± 1.1 | 41 ± 2.9 | >100 |
| 2f | 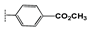 | 25 ± 1.1 | 22 ± 1.8 | 23 ± 0.9 | 30 ± 1.7 | 48 ± 1.6 | >100 |
| 2g | 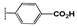 | 4.4 ± 0.5 | 18 ± 2.5 | 22 ± 1.4 | 24 ± 1.6 | 19 ± 1.0 | >100 |
| 2h | 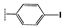 | 17 ± 1.1 | 20 ± 0.2 | 18 ± 1.3 | 31 ± 0.9 | 35 ± 1.1 | >100 |
| 2i | R = C4H9 | 36 ± 2.5 | 31 ± 1.6 | 29 ± 1.1 | 34 ± 1.5 | 56 ± 3.6 | >100 |
| 3 | 7.0 ± 2.6 | 15 ± 1.6 | 20 ± 1.2 | 25 ± 2.0 | 22 ± 1.2 | >100 | |
| Doxorubicin | 2.5 ± 0.8 | 10.1 ± 0.7 | 6.1 ± 0.7 | 5.2 ± 0.8 | 15 ± 0.3 | 8 ± 1.8 | |
| Compound | Concentration, μM | Population (% Cell Distribution) [b] | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 24 h | 48 h | ||||||||
| G1 | S | G2 | SubG1 | G1 | S | G2 | SubG1 | ||
| 2g | 4.4 (GI50) | 55.9 | 16.7 | 24.1 | 3.3 | 50.5 | 16.5 | 27.2 | 5.8 |
| 3 | 7.0 (GI50) | 21.8 | 15.9 | 49.9 | 12.3 | 4.2 | 5.5 | 9.4 | 80.9 |
| DOX | 2.5 (GI50) | 8.3 | 21.5 | 67.7 | 2.5 | 9.4 | 24.3 | 45.8 | 20.5 |
| Control + DMSO [a] | 57.6 | 19.3 | 20.7 | 2.4 | 61.4 | 18.8 | 17.7 | 2.1 | |
| Control [a] | 59.3 | 14.7 | 24.2 | 1.8 | 61.7 | 17.5 | 18.4 | 2.4 | |
| Compound | Concentration, μM | Population (% Cell Distribution) [b] | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 24 h | 48 h | ||||||||
| G1 | S | G2 | SubG1 | G1 | S | G2 | SubG1 | ||
| 2g | 18.0 (GI50) | 46.7 | 19.4 | 29.8 | 4.1 | 43.7 | 16.7 | 28.3 | 11.3 |
| 3 | 15.0 (GI50) | 42.5 | 19.9 | 28.5 | 9.1 | 42.4 | 17.9 | 29.7 | 10.0 |
| DOX | 10.1 (GI50) | 24.8 | 5.6 | 5.8 | 63.8 | 24.6 | 8.0 | 4.7 | 62.7 |
| Control + DMSO [a] | 60.3 | 17.7 | 19.6 | 1.4 | 59.7 | 15.7 | 19.1 | 2.2 | |
| Control [a] | 61.8 | 16.0 | 21.0 | 1.2 | 61.1 | 17.6 | 19.5 | 1.8 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Published by MDPI on behalf of the Österreichische Pharmazeutische Gesellschaft. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kishkentayeva, A.S.; Hamad, M.S.; Pokrovsky, M.A.; Shaimerdenova, Z.R.; Adekenova, A.S.; Mambeterzina, G.K.; Savelyev, V.A.; Pokrovsky, A.G.; Shults, E.E. Synthesis and Biological Evaluation of Some Coumarin–Triazole Conjugates as Potential Anticancer Agents. Sci. Pharm. 2025, 93, 16. https://doi.org/10.3390/scipharm93020016
Kishkentayeva AS, Hamad MS, Pokrovsky MA, Shaimerdenova ZR, Adekenova AS, Mambeterzina GK, Savelyev VA, Pokrovsky AG, Shults EE. Synthesis and Biological Evaluation of Some Coumarin–Triazole Conjugates as Potential Anticancer Agents. Scientia Pharmaceutica. 2025; 93(2):16. https://doi.org/10.3390/scipharm93020016
Chicago/Turabian StyleKishkentayeva, Anarkul S., Mohammad Saleh Hamad, Mikhail A. Pokrovsky, Zhanar R. Shaimerdenova, Aigerim S. Adekenova, Gulnara K. Mambeterzina, Victor A. Savelyev, Andrey G. Pokrovsky, and Elvira E. Shults. 2025. "Synthesis and Biological Evaluation of Some Coumarin–Triazole Conjugates as Potential Anticancer Agents" Scientia Pharmaceutica 93, no. 2: 16. https://doi.org/10.3390/scipharm93020016
APA StyleKishkentayeva, A. S., Hamad, M. S., Pokrovsky, M. A., Shaimerdenova, Z. R., Adekenova, A. S., Mambeterzina, G. K., Savelyev, V. A., Pokrovsky, A. G., & Shults, E. E. (2025). Synthesis and Biological Evaluation of Some Coumarin–Triazole Conjugates as Potential Anticancer Agents. Scientia Pharmaceutica, 93(2), 16. https://doi.org/10.3390/scipharm93020016