Abstract
The design, development, and release kinetics of omeprazole (OME) from solid dosage forms have been investigated. These formulations were examined for their resilience in pH = 4.5 buffer solutions and their rate of disintegration in a small-intestine-like environment (pH = 6.8). The results were compared with those of the well-known brand product Losec®, where its use is accompanied by numerous benefits but drawbacks as well. Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) tests were conducted in order to examine the release kinetics of the various dosage forms and provide explanations based on the interactions between the excipients and the active substance.
1. Introduction
Omeprazole (OME) has a molecular formula of C17H19N3O3S and a molecular weight of 345.42 g/mol. It is a 5-methoxy-2-[(4-methoxy-3,5-dimethylpyridin-2-yl)methanesulfinyl]-1H-benzimidazole, comprising in its skeleton a substituted pyridine ring connected to a substituted benzimidazole via a -CH2S(=O)- chain. Containing tri-coordinated sulfonyl sulfur in a pyramidal configuration, it can exist as either an (R)- or (S)-enantiomer. However, omeprazole is a racemic mixture; in the acidic environment of gastric parietal cells, both enantiomers are converted to achiral sulfenic acid and/or sulfenamide derivatives, which react with H+/K+ ATPase, inhibiting the ability of parietal cells to secrete gastric acid. Thus, therapeutically, it belongs to medications widely known as proton-pump inhibitors (PPIs) [1,2,3]. Moreover, OME is used against peptic ulcer disease (PUD) and Zollinger–Ellison syndrome, among others [2,4]. PPIs are considered to be membrane-permeable, weak bases, with a pKa1 between 3.8 and 4.9 [5,6,7,8]. Each type of PPI creates disulfide bonds with specific cysteine residues of ATPases. As for omeprazole, these bonds concern cysteines 813 and 822 [6]. Omeprazole was first synthesized in 1979, encoded as compound H168/68 by Astra AB, now amalgamated with AstraZeneca, and later given the brand name Losec®. This name was then changed to Prilosec® in the U.S. market at the request of the Food and Drug Administration agency (FDA), to avoid confusion with the extant diuretic Lasix® [9,10].
To become fully functional and reach its full therapeutic potential, an encapsulated pharmaceutical compound must be released from the capsule and enter blood circulation. There are two main delivery systems through which this can take place, i.e., the Immediate-Release Drug Delivery System (IRDDS) and the Modified-Release Drug Delivery System (MRDDS) [11,12]. Drug products belonging to the IRDDS are able to disintegrate rapidly when entering the body orally and, as such, they release the active reagent immediately, resulting in both quick absorption of the compound and therapeutic action [11,12,13]. However, even though this system has many benefits, including cost efficiency, high drug loading, flexibility in the processing and packaging of the tablets, and improved stability, bioavailability, and solubility of the pharmaceutical composition, it is unable to properly deliver products that are administrated as pro-drugs or are considered lipophilic [12,14]. For these two categories, the Modified-Release System is considered to be more appropriate. In general, the term Modified-Release Drug Delivery System refers to pharmaceutical products that alter the timing and/or the release rate of a drug substance. These products belong to three main categories: (1) extended-release drug products, (2) delayed-release drug products, and (3) targeted-release drug products [11,12,15]. Regarding these three categories, delayed release refers to products where a discrete portion or portions of the drug are released at a time other than promptly after administration. However, an initial portion may be released promptly after. This method is highly useful for categories of drugs that can cause irritation of the gastric mucosa (e.g., NSAIDS) and which are very sensitive to quick degradation in the various acids of the gastrointestinal tract or need to become available in its lower portions, to treat local conditions like inflammatory bowel diseases. On top of this, delayed-release drugs are also used when considerable time needs to pass between the administration of the product and the initiation of the therapeutic action, like in the case of patients with rheumatic arthritis. Usually, this delay in action is achieved by the application of an enteric coating on dosage forms, such as tablets and capsules, which protects the active substance from the acidic conditions of the mucosa and allows for the release in the more neutral environment of the small intestine [11,12,15,16].
As previously mentioned, omeprazole, being a member of the proton-pump inhibitors, is very susceptible to rapid disintegration when meeting the highly acidic gastric fluids of the upper gastrointestinal tract. As a result, to enter blood circulation and become functional, omeprazole must not be immediately released when entering the body or the stomach, but must be released upon reaching the lower parts of the gastrointestinal tract (the small intestine), where the environment is practically neutral (pH = 6.8). This process is accomplished by a number of delayed-release products, mostly tablets and capsules coated with various gastroresistant excipients [3,4,17,18]. One of the most common compounds is Eudragit®. The brand name Eudragit® refers to a diverse range of highly thermoplastic, polymethacrylate-based copolymers, which can be of anionic, cationic, and neutral nature. The anionic forms of these three are widely used in delayed-release delivery systems because they are non-biodegradable, non-absorbable, and non-toxic and dissolve at a pH above 6, allowing them to safely transfer acid-labile drugs to the lower parts of the gastrointestinal tract [19,20,21].
In the current study, tablets containing omeprazole and the appropriate excipients were synthesized de novo on a laboratory scale and were coated with a layer of Eudragit®. Subsequently, their resistance in acidic buffers (pH = 4.5) and their dissolution rate in a simulated small intestine environment (pH = 6.8) were tested. The results were compared with those of a widely used brand product of a similar structure and function (Losec®), and the various advantages and disadvantages of the new drug forms are discussed herein. Furthermore, the release kinetics of the different dosage forms were evaluated, and explanations are given herein on the basis of the interactions of the excipients, as obtained from thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) studies.
2. Materials and Methods
2.1. Materials
For the preparation of the tablets and capsules, omeprazole pellets (Cipla Ltd., Mumbai, India), according to the British Pharmacopeia formula, offered for free by Pharma Data SA (Lavrion, Greece), were used. The pellets consisted of 8.5% w/w omeprazole and various excipients (Batch No. YXX4086, CIPLA). Along with them, the following excipients were used: No 0 hard gelatin capsules (Syndesmos, Athens, Greece), alginic acid sodium salt of medium viscosity (Alfa Aesar GmbH & Co KG, Schwerte, Germany), Avicel® PH-101 (Alfa Aesar GmbH & Co KG, Schwerte, Germany), Avicel® PH-102 (Sigma-Aldrich, Darmstadt, Germany), lactose monohydrate (Merck, Darmstadt, Germany), magnesium stearate (Riedel-De Haen, Seelze, Germany), and PVP K-30 (Sigma-Aldrich, Darmstadt, Germany). All the excipients were pharmaceutical-grade.
The reagents used for the preparation of the buffer solutions for the dissolution testing (KH2PO4 and NaOH) were of analytical grade and were both provided by Sigma-Aldrich (Darmstadt, Germany).
For the experimental procedure, the following laboratory equipment was used: ATX224® high-precision analytical balance (Shimadzu, Kyoto, Japan), Turbula® T2F blender mixer (WAB, Nidderau, Germany), AR® 402 twin screw mixer (ERWEKA GmbH, Langen, Germany), Binder® F115 oven (Carbolite Gero GmbH & Co KG, Derbyshire, UK), AR® 400 cubic mixer (ERWEKA GmbH, Langen, Germany), IKA® RCT standard magnetic stirrer (LLG Labware, Meckenheim, Germany), 10 mm tableting matrix (Maassen GmbH, Möglingen, Germany), dry granulator (ERWEKA GmbH, Langen, Germany), THB 28 hardness tester (ERWEKA GmbH, Langen, Germany), PT-DT7 dissolution apparatus (Pharma Test, Hainburg, Germany), Multi 3410 Set 1 pH meter (Wissenschaftlich-Technische Werkstätten GmbH, Weilheim, Germany), MP 150 hydraulic press (Maasen GmbH, Möglingen, Germany), uniSPEC 2 UV-Vis Spectrophotometer (LLG Labware, Meckenheim, Germany), Titan3™ 0.45 μm regenerated cellulose syringe filters (Thermo Fisher Scientific Inc. Rockwood, TN, USA), and Q500IW Image Analysis system (Leica Microsystems, Wetzlar, Germany).
2.2. Methods
2.2.1. Granules’ Production
Lactose monohydrate and microcrystalline cellulose (Avicel® PH-101) were first passed through a No. 20 sieve and then were blended into a cubical mixer for 15 min. The high-shear mixer granulator was then loaded with the mixture and the binder solution (0.135% w/w PVP K30 in distilled water) was added. The wet mass produced was placed in furnace beds at 55 °C for 12 h. Then, the resulting dry granules were pushed through the regular sieve No. 12 sieve [22].
2.2.2. Formulation Production (Capsule Filling and Tablet Production)
For all formulations, the same amount (235 mg) of omeprazole pellets was used, and for formulations 2 to 7, the same amount (4.70 mg) of magnesium stearate was used. In order to achieve content uniformity by blending omeprazole pellets with the excipient to form tablets, we used a regular sieve. Additionally, the pellets exhibited a high degree of uniformity, which is crucial for the next steps of the formulation process. The ratios of the rest of the excipients in each formulation are given in the table below (Table 1). The rationale of the formulations of OME is to investigate how different excipients can affect the interactions between the API, its dissolution, and its release from the prepared tablets.

Table 1.
Composition of OME formulations.
All the formulations were prepared by direct compression, except formulation F6, which was prepared by wet granulation. For the production of the capsules (formulation 1), omeprazole was simply embedded within the gelatinous casing. For the rest of the formulations, the following procedure was followed, with the only difference being that in the case of formulation 6, the three main excipients (Avicel® PH-101, lactose monohydrate, and PVP K-30) underwent dry granulation before their use.
Initially, the required amount of excipients was weighed using an ATX224® high-precision analytical balance and transferred into a small, sterile glass jar. Then, the jar was placed in a Turbula® T2F blender mixer for 10 min. Next, omeprazole pellets were added to the excipients’ mixture and blending was continued for another 5 min. Then, the mixture of omeprazole and the excipients was transferred into a 10 mm tableting matrix and compressed using an MP 150 hydraulic press. This step led to the formation of uniform tablets without cracks or other visible weak points.
2.2.3. Tablet Characterization (Tablets’ Weight, Thickness, and Crushing Strength Uniformity)
Before the dissolution testing, tablets from each of the formulations 2 to 7 were used for the evaluation of two main physical characteristics (weight and dimensions) of each batch and to evaluate the hardness. Weight and hardness were calculated using an ATX224® high-precision analytical balance and a THB 28 hardometer, respectively, and the dimensions (diameter and thickness) were recorded with a vernier caliper scale.
2.2.4. Image Analysis
The differences in the characteristics of the tablets (containing granules and physical mixtures) were assessed by image analysis. Image taking and processing were performed in a Leica Q500IW Image Analysis system, equipped with a JVCTK-C 1381 color camera, combined with a Century Precision Optics C15838 (+7) lens for better image magnification. The light system consisted of a fluorescent tube fitted around the microscope glass, which was covered to prevent external light interference.
2.2.5. Dissolution Testing
The dissolution studies for each formulation were carried out in a PT-DT7 dissolution apparatus with the use of baskets (USP, 2007, Apparatus type I). The procedure was the same for every formulation and the temperature remained constant at 37 ± 0.5 °C throughout the experimental procedure. Initially, 3 capsules/tablets were placed in one basket each, immersed within glass vessels containing 900 mL of KH2PO4/NaOH buffer solution (pH = 4.5). Every 15 min and up to 45 min, 5 mL of solution was taken from each vessel and replaced with an equal volume of buffer solution and the absorption was measured at 295 nm, using a uniSPEC 2 UV-Vis Spectrophotometer. After 45 min, the baskets were transferred into vessels containing 900 mL of KH2PO4/NaOH buffer solution (pH = 6.8). Aliquots were withdrawn every 15 min for a maximum of 285 min from the start of the experiment and the absorption was measured at 301 nm.
2.2.6. Dissolution Profile Comparison
In order to compare the dissolution profiles of the formulations and Losec®, the respective graphs of % omeprazole release vs. time were constructed. The values of t20%, t50%, and t90% were also calculated for each product, with tx% corresponding to the time that x% of the active substance is released from the pharmaceutical formulation.
In addition to tx%, the mean dissolution time values (MDT) were also calculated, using the following equation (Equation (1)) [23]:
where W∞ refers to the maximum amount of dissolved omeprazole, and ABC is the area between the drug dissolution curve and its asymptote.
2.2.7. Data Analysis
For the determination and comparison of the kinetics of the dissolution tests, the in vitro release data, for each formulation, were calculated using the zero order, first order, Higuchi square root, and Korsmeyer–Peppas equation.
2.2.8. Model-Independent Analysis of Dissolution Profiles
For the model-independent comparison of the dissolution profiles of each formulation, the difference (f1) and similarity (f2) factors were calculated. The f1 factor refers to the average relative difference between two curves, across all the time points, and is given by the following equation (Equation (2)) [24]:
where Rt refers to the mean % reference drug dissolved at time t after initiation of the study, Tt refers to the mean % test drug dissolved at time t after initiation of the study, and n is the number of sampling time points.
In contrast to f1, the f2 factor is used in order to compare the cumulative curves of percentage dissolution between two samples. It is calculated using the following equation (Equation (3)) [24]:
where, similar to f1, Rt refers to the mean % reference drug dissolved at time t, after initiation of the study; Tt refers to the mean % test drug dissolved at time t, after initiation of the study; and n is the number of sampling time points.
The f2 factor can take values from 0 to 100. The larger the difference between the test and the reference profiles, the smaller the value f2 takes, approaching even zero. On the other hand, a value of 100 shows that the two profiles have absolute similarity. The two dissolution curves are generally considered similar when 0 < f1 < 15 and 50 ≤ f2 < 100.
2.2.9. Thermal Analysis Techniques
Thermogravimetric analysis (TGA) was performed using a TGA 55 Thermogravimetric Analyzer (TA Instruments, New Castle, DE, USA) at a heating rate of 10 °C/min from 26 to 600 °C under a 25 mL/min nitrogen flow.
Differential scanning calorimetry (DSC) analysis was performed using a DSC 25 differential scanning calorimeter (TA Instruments, New Castle, DE, USA). Samples of 7–8 mg sealed in aluminum pans were heated from 40 to 240 °C at a constant rate of 10 °C/min under a 50 mL/min nitrogen flow.
3. Results
3.1. Release Kinetics of OME from Different Formulations
Table 2 shows that the tablets were within the acceptable range for all evaluation parameters (tablet weight, thickness, and breaking force), as defined by international standards [25]. Figure 1 depicts the image analysis of two OME formulations containing the same ingredients; formulation F6 was prepared with wet granulation while formulation F7 was prepared with direct compression. The size of the pellets was the same, as visualized and measured in Figure 2. The pellet distribution was quite uniform, taking into account the size of the pellets and the formulation processes. In any case, we cannot claim that our pellets exhibited a high degree of uniformity. We would like to underline that in all cases, tabletability was achieved without observing any visual defects, i.e., capping or lamination. The shape of the tablets after compression and during storage was decided.
Table 2.
Results of tablets’ weight, thickness, and crushing strength uniformity.

Figure 1.
Image analysis results from formulations F6 (A) and F7 (B); scale-magnification ×50.
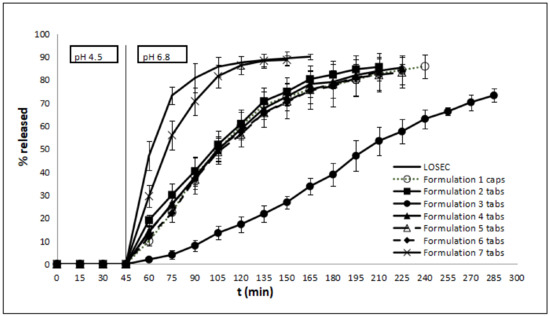
Figure 2.
% Omeprazole release (mean ± SD) (n = 3) vs. time (min) for Losec® and formulations 1–7 (pH 4.5 for 45 min and pH 6.8 thereafter).
The dissolution curves of Losec® and the produced formulations are depicted in Figure 2. The dissolution test results are shown in Table 3, Table 4 and Table 5. More in particular, Table 3 denotes the t20%, t50%, and t90%, and MDT for each formulation. In order to probe the kinetic behavior, the results of the in vitro release from the formulations were fitted to the zero order, first order, Higuchi square root, and Korsmeyer–Peppas equation (Table 4).
Table 3.
Kinetic release properties of the OME formulations.
Table 4.
R2, release rate constants, and n values of the OME formulations.
Table 5.
f1 and f2 index.
In relation to Figure 1, it can be stated that all formulations did not exhibit any release at pH 4.5, implying that the tableting force used did not damage the omeprazole pellets and their coating. Also, the ratio of the excipients used for the pellets’ preparation was appropriate for maintaining pellet integrity and drug content. The active substance from the commercially available Losec® is rapidly released at pH = 6.8, reaching a plateau almost 2 h later. The release from formulation F7 can be compared to that of Losec® (see identical values of the kinetic release profiles in Table 3 and also the f1 and f2 index values in Table 4). Likewise, formulations F1 (capsules containing OME pellets) and F2 (tablets containing a mixture of lactose monohydrate and OME pellets) also show comparable drug release results (see identical values of the kinetic release profiles in Table 3, and also the f1 and f2 index values in Table 4). On the other hand, formulation F3, containing sodium alginate, reveals a more extended-release profile when compared with all the other formulations. The substitution of Avicel® PH-101 for Avicel® PH-102 (formulations F5 and F4, respectively) revealed no significant difference in drug release. Compared to formulations F6 and F7 that have the same excipients, in F6, the excipients are mixed with the pellets of OME in the form of granules, while in F7, the excipients are in powder form, which one could argue favors a more direct and immediate drug release. As it was observed from the dissolution studies, all formulations released the total amount of the encapsulated API. For this reason, the prepared formulations did not exhibit drug uniformity problems, either.
Therefore, it can be concluded that formulations F1, F2, F4, F5, and F6 have comparable drug release profiles, implying that the drug release is controlled by the pellets and not by the other excipients present in the tablet formulations, which most likely play the role of fillers. This phenomenology is expected, as the excipients are simple fillers with no specific functionality. Once the Eudragit coating dissolves, the tablets disintegrate relatively quickly (depending on their hardness).
3.2. Thermotropic Behavior of OME Formulations
The thermal behavior of OME is widely studied in the literature [26,27,28]. We studied the thermal behavior and stability of omeprazole when none of the manufacturing steps included any thermal processing of the API because it is well established in the literature that the tableting processes affect the thermotropic properties of the APIs due to the interactions between the formulation components and the process due to the tablets or granule formulations. Additionally, the investigation of the thermal behavior of pure omeprazole is very useful for comparison reasons. The aim of the published works was to identify the most stable state of OME in different forms (granules or powders), temperatures, and pH environments using DSC. Furthermore, with TGA at 135 °C, decomposition began and was influenced by the rate of heating and the eutectic behavior of breakdown products, causing omeprazole’s melting point to decrease with gradual heating [26,27,28]. In this study, we investigated the thermal stability and behavior of OME in different formulations using TGA and DSC, respectively. For comparison reasons, we also tested the behavior of mixtures of excipients with the same techniques. The DSC and TGA analyses for the pellets are included in Figure 1 (for F1). F1 are capsules of OME pellets, and TGA and DSA are performed for the content of the capsules, which is the pellets.
From TGA studies (Figure 3), all the excipients showed an initial decomposition step starting approximately at 139 °C, which corresponds to the loss of the water molecules present in the excipients’ mixtures, followed by the main decomposition step starting at 199 °C for LMSA and approximately at 219 °C for the rest of the excipients’ mixtures. For the LMSA, this decomposition ends at 249 °C. The presence of Avicel® PH-101 or Avicel® PH-102 leads to the decomposition ending at 338 °C and 339 °C, respectively. On the other hand, the presence of PVP leads to the decomposition ending at 343 °C. This decomposition step corresponds to the loss of the water molecules that are present in the mixtures of excipients. The main difference between Avicel® PH-101 and Avicel® PH-102, which are both microcrystalline celluloses, is the degree of agglomeration of the particles, which is higher in 102 [29]. For this reason, we did not observe any significant differences at the endpoint of the decomposition process.
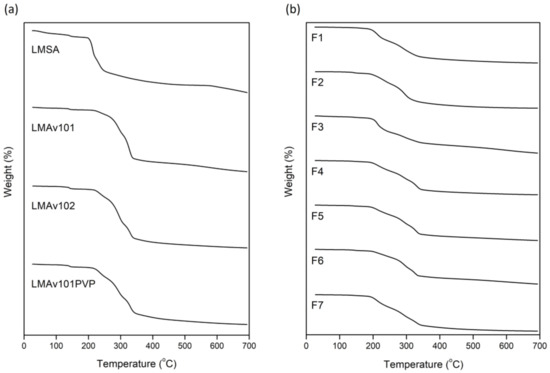
Figure 3.
TGA curves of (a) excipients and (b) formulations of OME (LMSA: lactose monohydrate/sodium alginate; LMAv101: lactose monohydrate/Avicel 101; LMAv102: lactose monohydrate/Avicel 102; LMAv101PVP: granules).
For the OME formulations, the capsules exhibited a different decomposition behavior in comparison to all the other formulations. Namely, for F1, the decomposition started at 190 °C and ended at 336 °C with no dehydration step observed, while for F2–F7, the main decomposition started at 191–196 °C with an initial step at 138–140 °C attributed to the dehydration process, as mentioned above.
For the tablet formulations, the temperature range of the decomposition phenomenon is strongly dependent on the excipients that are used. For F2, which is composed of lactose monohydrate, the main decomposition ended at 323 °C, while the presence of sodium alginate or Avicel® PH-101 or Avicel® PH-102 extended the end of this decomposition process by approximately 20 °C (Figure 3b).
According to our previous studies, sodium alginate exhibits a characteristic endothermic peak in the temperature range of 120–190 °C, with a center around 163 °C [30]. Additionally, lactose monohydrate exhibited two characteristic endotherms at 147 °C and 210 °C (Figure 4a). The mixture of these excipients exhibited an endothermic peak around 150 °C, closer to the characteristic endothermic peak of lactose monohydrate with this result strongly dependent on the synergistic thermal events of the two ingredients. In our opinion, this result is strongly dependent on the dehydration phenomenon of sodium alginate and the hydrophilic groups in the backbone of this biopolymer [31,32]. In other words, the mixture of these excipients exhibited the synergistic effect of the thermal events of the two ingredients with a broad endotherm up to 140 °C due to the dehydration of sodium alginate followed by a sharp endothermic peak at around 150 °C, corresponding to the characteristic dehydration peak of lactose monohydrate.
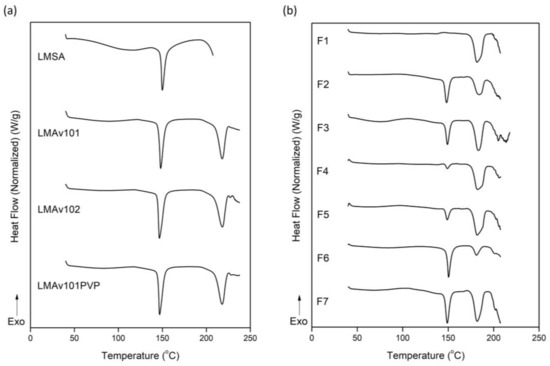
Figure 4.
DSC curves of (a) excipients and (b) formulations of OME (LMSA: lactose monohydrate/sodium alginate; LMAv101: lactose monohydrate/Avicel 101; LMAv102: lactose monohydrate/Avicel 102; LMAv101PVP: granules).
The characteristic endothermic peak of lactose monohydrate remains unaffected in the presence of the other excipients, but the enthalpy of the melting is higher (Figure 4a, Table 6). The presence of Avicel® PH-101 or Avicel® PH-102 leads to the formation of a new endothermic peak at 218 °C (Figure 4a). The enthalpy of the second peak at 218 °C is higher for Avicel® PH-102 in comparison to the peak of Avicel® PH-101, meaning that the agglomeration process needs a higher amount of heating for melting (Figure 4a, Table 6).
Table 6.
DSC calorimetric values of excipients and formulations of OME.
All the formulations except F1 exhibited an endothermic peak between 148.3 °C and 150.4 °C. The enthalpy of this peak is quite different between the formulations (Figure 4b, Table 6). Furthermore, all the formulations exhibited a second endothermic peak around 180 °C without differences in enthalpy (Figure 4b, Table 6). This phenomenology is discussed in depth in the next section, leading to an outcome that explains the release profile of OME.
4. Discussion
Mechanistic Explanation of OME Release Kinetics: The Role of the Excipients
The multi-unit pellet tablet, which uses coated pellets to deliver drugs with modified release, is a potent therapeutic substitute for immediate-release dose forms. The three key benefits are that they are simple to swallow, can be divided without affecting the way that each unit releases the medicine, and have a uniform distribution of multiparticulates throughout the gastrointestinal tract. Tablets of the multi-unit pellet system can be produced more affordably than pellet-filled capsules due to the tableting method’s substantially higher manufacturing rate. Despite their superiority, manufacturing issues, such as impaired coated pellet integrity and uneven content, have made their implementation difficult [33,34,35].
Several excipients needed to be used to aid the compaction process and prevent coating pellet damage during the tableting process. As previously mentioned, the ideal materials for pellet tableting should prevent direct contact between the pellets acting protectively during compression [36,37]. It has been stated that 29% of excipients are required to fill the void space between densely packed spheres, but tablet variations are acceptable at a pellet content of ~50%, even with nongranulated excipients [37]. In our case, almost 50% of excipients were used to form a layer between the coated pellets to prevent adhering and to produce hard tablets at relatively low compression forces while having no or, in other cases, some effect on drug release. The perfect cooperativity of our excipients is also shown in the DSC studies of both the pure excipient mixture and the OME formulations (Figure 4, Table 6). Cooperativity is a thermal property that corresponds to how uniform the transition is and is equal to half the width at half the peak height of the transition. The thermal property is linked to the compaction properties because this formulation process is strongly dependent on the interactions between the different components. Apart from their compaction properties, the excipients needed to achieve a uniform pellet–excipient mixture, avoiding segregation and, as a result, weight variation in the resulting tablets; for this reason, larger granules were also used for comparison purposes.
Sodium alginate is a straight-chain, hydrophilic, colloidal, polyuronic acid composed of guluronic and mannuronic acid residues that can form gels with different structures at acidic and neutral pH. At low pH values (pH = 3), hydrogen bonds are formed between the sodium alginate chains, rapidly converting it into alginic acid, which swells and becomes insoluble. As a result, drug release is entirely dependent on diffusion through the polymer. On the contrary, at neutral pH, the alginic acid chains form a gel due to cross-links formed between their carboxyl groups and various cations such as calcium ions. The ability to selectively bind alginic acid to various cations is proportional to the percentage of guluronic acid monomers present. It has been demonstrated that the higher the concentration of alginic acid in guluronic acid monomers, the stronger the bond with cations and thus the mechanical stiffness of the gel [38,39,40,41,42,43,44,45]. Therefore, the formation of a viscous gel had a significant impact on drug release, prolonging the OME release at neutral pH, as seen in F3.
Microcrystalline cellulose (MCC) is a common excipient used in pharmaceutics as a binder, disintegrant, filler, and anti-adherent. Avicel® PH-101 and Avicel® PH-102 are the most widely used grades; the former has a nominal mean particle size of 50 pm and is used in the wet granulation process, while the latter has a nominal mean particle size of 100 pm and is used in the direct compression process [46]. Avicel® PH-101 is the original MCC grade, whereas Avicel® PH-102 is a partially agglomerated product with a larger particle size distribution and slightly better fluidity. Both of them exhibited more or less the same thermotropic behavior, as TGA and DSC studies revealed (Figure 3 and Figure 4 and Table 6). Researchers who studied their differences in packing and flow properties concluded that they were due to differences in moisture content, particle shape, and particle size distribution, whereas neither grade shows a significant difference in compressibility [29,47]. In the present study, no significant difference in drug release was noticed when Avicel® PH-101 was substituted for Avicel® PH-102 (formulations F5 and F4, respectively). In our opinion, this phenomenology is strongly dependent on the same interactions of both forms with the other excipients and OME, as DSC studies showed (Table 6) [48].
Polyvinylpyrrolidone (PVP), also called polyvidone or povidone, is a biodegradable, water-soluble polymer derived from its monomer N-vinylpyrrolidone. In addition to being a hydrophilic polymer, PVP has excellent solubility properties in solvents of different polarities and good binding properties. The solubility of the PVP K-30 (35,000–51,000 Daltons) used in this study had a positive influence on drug dissolution. Moreover, its viscosity strongly affected the release of the poorly water-soluble drug OME [49,50]. In particular, the low-viscosity gel created during the tablets’ dissolution allowed the necessary suspension of OME inside the polymer matrix, thus explaining the higher release rate that was observed in formulation F7.
Both the wet granulation and the direct compression method were used successfully for developing OME tablet formulations. As mentioned in the literature, the granules showed better flowability, compressibility, and compactability compared to the direct compression formulation [51]. It is known that, by the wet granulation method, harder tablets are formed compared to those prepared by the direct compression method, as tablet hardness is a function of compression load, and granule or crystal hardness [51]. The creation of liquid bridges with subsequent crystallization and hardening of the adhesives by drying can also be associated with higher hardness values of wet granulation tablets [52]. For these reasons, the formulation with wet granulation (F6) showed a more prolonged release profile than the formulation with direct compression (F7). Additionally, the second endothermic peak of F7 needs higher energy for the melting and could facilitate the OME release from formulation F7 in comparison to F6 (Figure 4) because the dissolution rate and drug release are phenomena that are strongly dependent on the melting points of the excipients, as well as the interactions between excipients and APIs [53]. Furthermore, in addition to the formation of stronger granules, the particle size is completely different in comparison to the bulk excipients, and hence the tablet hardness will not be the same. We provided data for each tablet’s hardness, but those of F6 and F7 are completely different with a 20N difference. In our opinion, this phenomenology is strongly dependent on the hardness of these formulations and affects the release profile of the OME, too. As mentioned above, all the formulations were prepared by direct compression, except formulation F6, which was prepared by wet granulation. The different processes may also play a key role in the interactions between the excipients and the OME and/or between the excipients because they have an impact on the thermal history of the materials used. Last but not least, this thermal history and the presence of the binding liquid in the process of wet granulation may be responsible for the release of OME in the dissolution studies, as indicated in the literature [53,54,55].
5. Conclusions
The main objective of this work was the study and evaluation of the ability of specific excipients to create effective, sustained-release systems of omeprazole. For this purpose, six different excipients were selected (lactose monohydrate, alginic acid medium viscosity, Avicel® PH-101, Avicel® PH-102, PVP K-30, and magnesium stearate). The choice was made based on criteria based on the physicochemical properties of these excipients (e.g., low toxicity) as well as their low cost. For the preparation of the tablets, we used two excipients using different techniques, which are characterized by convenience, low cost (simple mixing, compression, and wet granulation), and time. The results of the experiments showed that the prepared systems have had a very good response, both regarding the characteristics of the tablets (hardness, size, homogeneity of composition) and the ability to release the omeprazole in a delayed interval after administration. In particular, one of the formulations (F7) displayed a similar release rate to the commercially available formulation omeprazole Losec® (80% release within two hours), while formulation prescription F3 showed a slow but relatively steady release for a longer period (at least four hours) and can therefore be considered a candidate for use as a background in the preparation of long-lasting and stable-release delivery systems (e.g., 24 h release) of medicinal substances. The remaining formulations showed a similar, intermediate-duration release profile. All release profiles were also evaluated, taking into account the interactions between the excipients and/or the excipients and OME, as quantified by thermal analysis techniques. In summary, the systems that were studied are characterized by the relatively low cost of production, their high performance, and the variety of release rates of omeprazole they showed. Hence, they can be viewed as of high interest for future use in other active substances, with the corresponding modifications. The use of thermal analysis techniques is of paramount importance for the study of the interactions between the formulation’s components and plays a key role in the mechanistic explanation of the release profile of the encapsulated APIs when different processes are involved.
Author Contributions
Conceptualization, A.S., M.V. and N.P.; methodology, G.A., A.S., S.K. and N.P.; validation, G.A., A.S. and N.P.; formal analysis, G.A., A.S., S.K. and N.P.; investigation, G.A., A.S. and N.P.; data curation, G.A., A.S., S.K. and N.P.; writing—original draft preparation, G.A., A.S. and N.P.; writing—review and editing, M.V. and N.P.; supervision, M.V. and N.P.; project administration, M.V. and N.P.; funding acquisition, M.V. and N.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data generated during this study are included in this published article.
Acknowledgments
The authors wish to thank Vassilios Roussis for the provision of facilities for the TGA and DSC measurements. The authors would also like to thank Pharmadata S.A. for providing us with omeprazole beads for this investigation.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Brandstrom, A.; Lindberg, P.; Junggren, U. Structure-activity relationships of substituted benzimidazoles. Scand. J. Gastroenterol. Suppl. 1985, 108, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Massoomi, F.; Savage, J.; Destache, C.J. Omeprazole: A Comprehensive Review. Pharmacotherapy 1993, 13, 46–59. [Google Scholar] [CrossRef]
- Filho, V.J.T.; Andreazza, I.F.; Sato, M.E.O.; Murakami, F.S. Development of a multiparticulate system containing enteric release mini-tablets of omeprazole. Braz. J. Pharm. Sci. 2014, 50, 505–511. [Google Scholar] [CrossRef]
- El-Sayed, A.; Boraie, N.A.; Ismail, F.A.; El-Khordagui, L.K.; Khalil, S.A. Assessment of the pharmaceutical quality of omeprazole capsule brands marketed in Egypt. East. Mediterr. Health J. 2007, 13, 1427–1437. [Google Scholar] [CrossRef] [PubMed]
- Sachs, G.; Shin, J.M.; Howden, C.W. Review article: The clinical pharmacology of proton pump inhibitors. Aliment. Pharmacol. Ther. 2006, 23, 2–8. [Google Scholar] [CrossRef]
- Shin, J.M.; Sachs, G. Pharmacology of Proton Pump Inhibitors. Curr. Gastroenterol. Rep. 2008, 10, 528–534. [Google Scholar] [CrossRef]
- Strand, D.S.; Kim, D.; Peura, D.A. 25 Years of Proton Pump Inhibitors: A Comprehensive Review. Gut Liver 2017, 11, 27–37. [Google Scholar] [CrossRef]
- El-Rouby, N.; Lima, J.J.; Johnson, J.A. Proton pump inhibitors: From CYP2C19 pharmacogenetics to precision medicine. Expert Opin. Drug Metab. Toxicol. 2018, 14, 447–460. [Google Scholar] [CrossRef]
- Farley, D. Making it easier to read prescriptions. FDA Consum. 1995, 29, 25–27. [Google Scholar]
- Lundell, L. The physiological background behind and course of development of the first proton pump inhibitor. Scand. J. Gastroenterol. 2015, 50, 680–684. [Google Scholar] [CrossRef]
- Perrie, Y.; Rades, T. FASTtrack: Pharmaceutics—Drug Delivery and Targeting, 2nd ed.; Pharmaceutical Press: London, UK, 2012; pp. 1–24. [Google Scholar]
- Shargel, L.; Wu-Pong, S.; Yu, A.B.C. Applied Biopharmaceutics & Pharmacokinetics, 6th ed.; McGraw-Hill Education: New York, NY, USA, 2012; 5p. [Google Scholar]
- Saha, S. Immediate Release Drug Delivery Systems: A Current Update. J. Appl. Pharm. Res. 2018, 6, 1–9. [Google Scholar] [CrossRef]
- Nyol, S.; Gupta, M.M. Immediate Drug Release Dosage Form: A Review. J. Drug Deliv. Ther. 2013, 3, 155–161. [Google Scholar] [CrossRef]
- Trenfield, S.J.; Basit, A.W. Nanotechnology for Oral Drug Delivery: From Concept to Applications, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 177–197. [Google Scholar]
- Montero Mirabet, M.; Skalsky, B. Advanced approaches for delayed-release formulations. ONdrugDelivery Mag. 2017, 77, 4–9. [Google Scholar]
- Chakravarthy, K.K.; Younus, M.; Shaik, S.; Pisipati, S.V.V. Formulation and Evaluation of Enteric Coated Pellets of Omeprazole. Int. J. Drug Dev. Res. 2012, 4, 257–264. [Google Scholar]
- Ronchi, F.; Sereno, A.; Paide, M.; Sacre, P.; Guillaume, G.; Stephenne, V.; Goole, J.; Amighi, K. Development and evaluation of an omeprazole-based delayed-release liquid oral dosage form. Int. J. Pharm. 2019, 567, 118416. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.T.; Porter, S.C. Coatings for Controlled-Release Drug Delivery Systems. Drug Dev. Ind. Pharm. 1998, 24, 1139–1154. [Google Scholar] [CrossRef]
- Thakral, S.; Thakral, N.K.; Majumdar, D.K. Eudragit®: A technology evaluation. Expert Opin. Drug Deliv. 2013, 10, 131–149. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, J.; Da Silva, G.S.; Velho, M.C.; Beck, R.C.R. Eudragit®: A Versatile Family of Polymers for Hot Melt Extrusion and 3D Printing Processes in Pharmaceutics. Pharmaceutics 2021, 13, 1424. [Google Scholar] [CrossRef]
- Dallas, P. Handbook of Laboratory Exercises for the Undergraduate Pharmacy Students in National and Kapodistrian University of Athens; National and Kapodistrian University of Athens: Athens, Greece, 2023. [Google Scholar]
- Brockmeier, D.; Voegele, D.; Von Hattingberg, H. In vitro-in vivo correlation, a time scaling problem? Basic techniques for testing equivalence. Arzneimittelforschung 1983, 33, 598–601. [Google Scholar]
- Moore, J.W.; Flanner, H.H. Mathematical comparison of dissolution profiles. Pharm. Technol. 1996, 20, 64–74. [Google Scholar] [CrossRef]
- USP 29-NF24; Pharmacopoeia US. USP: Rockville, MD, USA, 2005.
- Ruiz, M.A.; Reyes, I.; Parera, A.; Gallardo, V. Determination of the stability of omeprazole by means of differential scanning calorimetry. J. Therm. Anal. Calorim. 1998, 51, 29–35. [Google Scholar] [CrossRef]
- Markovic, N.; Agotonovic-Kustrin, S.; Glass, B.; Prestidge, C.A. Physical and thermal characterisation of chiral omeprazole sodium salts. J. Pharm. Biomed. Anal. 2006, 42, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Murakami, F.S.; Lang, K.L.; Mendes, C.; Cruz, A.P.; Carvalho-Filho, M.A.; Silva, M.A. Physico-chemical solid-state characterization of omeprazole sodium: Thermal, spectroscopic and crystallinity studies. J. Pharm. Biomed. Anal. 2009, 49, 72–80. [Google Scholar] [CrossRef]
- Yohana Chaerunisaa, A.; Sriwidodo, S.; Abdassah, M. Microcrystalline Cellulose as Pharmaceutical Excipient. In Pharmaceutical Formulation Design—Recent Practices, 1st ed.; Ahmad, U., Akhtar, J., Eds.; IntechOpen: London, UK, 2020; 21p. [Google Scholar]
- Vlachou, Μ.; Pippa, Ν.; Siamidi, A.; Kyrili, A. Thermal analysis studies on the compatibility of furosemide with solid state and liquid crystalline excipients. Hem. Ind. 2020, 74, 15–23. [Google Scholar] [CrossRef]
- Soares, J.P.; Santos, J.E.; Chierice, G.O.; Cavalheiro, E.T.G. Thermal behavior of alginic acid and its sodium salt. Eclét. Quim. 2004, 29, 57–63. [Google Scholar] [CrossRef]
- Chadha, R.; Bhandari, S. Drug-excipient compatibility screening-Role of thermoanalytical and spectroscopic techniques. J. Pharm. Biom. Anal. 2014, 87, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Li, J.; Chen, T.; Sun, C.C.; Zheng, Y. Tablets of multi-unit pellet system for controlled drug delivery. J. Control. Release 2017, 262, 222–231. [Google Scholar] [CrossRef]
- Murthy Dwibhashyam, V.S.; Ratna, J.V. Key formulation variables in tableting of coated pellets. Indian J. Pharm. Sci. 2008, 70, 555–564. [Google Scholar] [CrossRef]
- Türkoğlu, M.; Varol, H.; Çelikok, M. Tableting and stability evaluation of enteric-coated omeprazole pellets. Eur. J. Pharm. Biopharm. 2004, 57, 279–286. [Google Scholar] [CrossRef]
- Tan, X.; Hu, J. Investigation for the quality factors on the tablets containing medicated pellets. Saudi Pharm. J. 2016, 24, 507–514. [Google Scholar] [CrossRef]
- Bodmeier, R. Tableting of coated pellets. Eur. J. Pharm. Biopharm. 1997, 43, 1–8. [Google Scholar] [CrossRef]
- Efentakis, M.; Buckton, G. The effect of erosion and swelling on the dissolution of theophylline from low and high viscosity sodium alginate matrices. Pharm. Dev. Technol. 2002, 7, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Holte, Ø.; Onsøyen, E.; Myrvold, R.; Karlsen, J. Sustained release of watersoluble drug from directly compressed alginate tablets. Eur. J. Pharm. Sci. 2003, 20, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Ghahramanpoor, M.K.; Najafabadi, S.A.H.; Abdouss, M.; Bagheri, F.; Eslaminejad, M.B. A hydrophobically-modified alginate gel system: Utility in the repair of articular cartilage defects. J. Mater. Sci. Mater. Med. 2011, 22, 2365–2375. [Google Scholar] [CrossRef]
- Draget, K.I.; Skjåk-Bræek, G.; Smidsrød, O. Alginate based new materials. Int. J. Biol. Macromol. 1997, 21, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Liew, C.V.; Chan, L.W.; Ching, A.L.; Heng, P.W.S. Evaluation of sodium alginate as drug release modifier in matrix tablets. Int. J. Pharm. 2006, 309, 25–37. [Google Scholar] [CrossRef]
- Enobakhare, B.; Bader, D.L.; Lee, D.A. Concentration and M/G ratio influence the physiochemical and mechanical properties of alginate constructs for tissue engineering. J. Appl. Biomech. 2006, 4, 87–96. [Google Scholar] [CrossRef]
- Martinsen, A.; Storra, I.; Skajak-Braek, G. Alginate as immobilization material: III. Diffusion properties. Biotechnol. Bioeng. 1992, 39, 186–194. [Google Scholar] [CrossRef]
- Yamagiwa, K.; Kozawa, T.; Ohkawa, A. Effects of alginate composition and gelling conditions on diffusional and mechanical properties of calcium alginate gel beads. J. Chem. Eng. Jpn. 1992, 28, 462–467. [Google Scholar] [CrossRef][Green Version]
- Rowe, R.C.; Sheskey, P.J.; Quinn, M.E. Handbook of Pharmaceutical Excipients, 6th ed.; RPS Publishing: London, UK, 2009; 917p. [Google Scholar]
- Vlachou, M.; Tsiakoulia, A.; Eikosipentaki, A. Controlled release of the pineal hormone melatonin from hydroxypropylmethylcellulose/sodium alginate matrices in aqueous media containing dioctyl sulfosuccinate. Curr. Drug. Discov. Technol. 2007, 4, 31–38. [Google Scholar] [CrossRef][Green Version]
- Narang, A.S.; Desai, D.; Badawy, S. Impact of excipient interactions on solid dosage form stability. Pharm. Res. 2012, 29, 2660–2683. [Google Scholar] [CrossRef]
- Franco, P.; De Marco, I. The Use of Poly(N-vinyl pyrrolidone) in the Delivery of Drugs: A Review. Polymers 2020, 12, 1114. [Google Scholar] [CrossRef]
- Mandal, S.; Basu, S.K.; Sa, B. Sustained release of a water-soluble drug from alginate matrix tablets prepared by wet granulation method. AAPS PharmSciTech 2009, 10, 1348–1356. [Google Scholar] [CrossRef] [PubMed]
- Patra, C.H.N.; Pandit, H.K.; Singh, S.P.; Devi, M.V. Applicability and comparative evaluation of wet granulation and direct compression technology to Rauwolfia serpentina root powder: A technical note. AAPS PharmSciTech 2008, 9, 100–104. [Google Scholar] [CrossRef]
- Reza, M.M.M.; Bhuiyan, M.A.; Quadir, M.A. Comparative Evaluation of Wet Granulation and Direct Compression methods for preparation of Compressed tablets using Avicel pH 101. Bangladesh Pharm. J. 2002, 12, 19–22. [Google Scholar]
- Park, J.H.; Kwon, D.Y.; Heo, J.Y.; Park, S.H.; Park, J.Y.; Lee, B.; Kim, J.H.; Kim, M.S. Effect of Drug Carrier melting points on drug release of dexamethasone-loaded microspheres. Tissue Eng. Regen. Med. 2017, 14, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kang, H.J.; Kwon, D.Y.; Lee, B.K.; Lee, B.; Jang, J.W.; Chun, H.J.; Kim, J.H.; Kim, M.S. Biodegradable poly(lactide-co-glycolide-co-ε-caprolactone) block copolymers—Evaluation as drug carriers for a localized and sustained delivery system. J. Mater. Chem. B 2015, 3, 8143–8153. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, B.K.; Park, S.H.; Kim, M.G.; Lee, J.W.; Lee, H.Y.; Lee, H.B.; Kim, J.H.; Kim, M.S. Preparation of Biodegradable and Elastic Poly(ε-caprolactone-co-lactide) Copolymers and Evaluation as a Localized and Sustained Drug Delivery Carrier. Int. J. Mol. Sci. 2017, 18, 671. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).