
Comparative Evaluation of Metformin and Metronidazole Release from Oral Lyophilisates with Different Methods


 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of IPEC
2.3. IPEC Characterization
2.3.1. Dynamic Light Scattering
2.3.2. Zeta Potential Measurements
2.4. In Vitro Mucoadhesive Properties of IPEC
2.5. Preparation of Oral Lyophilisates
2.6. Characterization of Oral Lyophilisates
2.7. In Vitro Drug Release Test
2.7.1. USP 2 Apparatus (Paddle Method)
2.7.2. USP 4 Apparatus (Flow-through Cell Method)
2.7.3. Vertical Franz Cell
2.7.4. Calibration Curves for Drug Content Analysis
2.8. Statistical Analysis
2.9. Mathematical Modelling
3. Results and Discussion
3.1. Characterization of Drug-Free IPEC
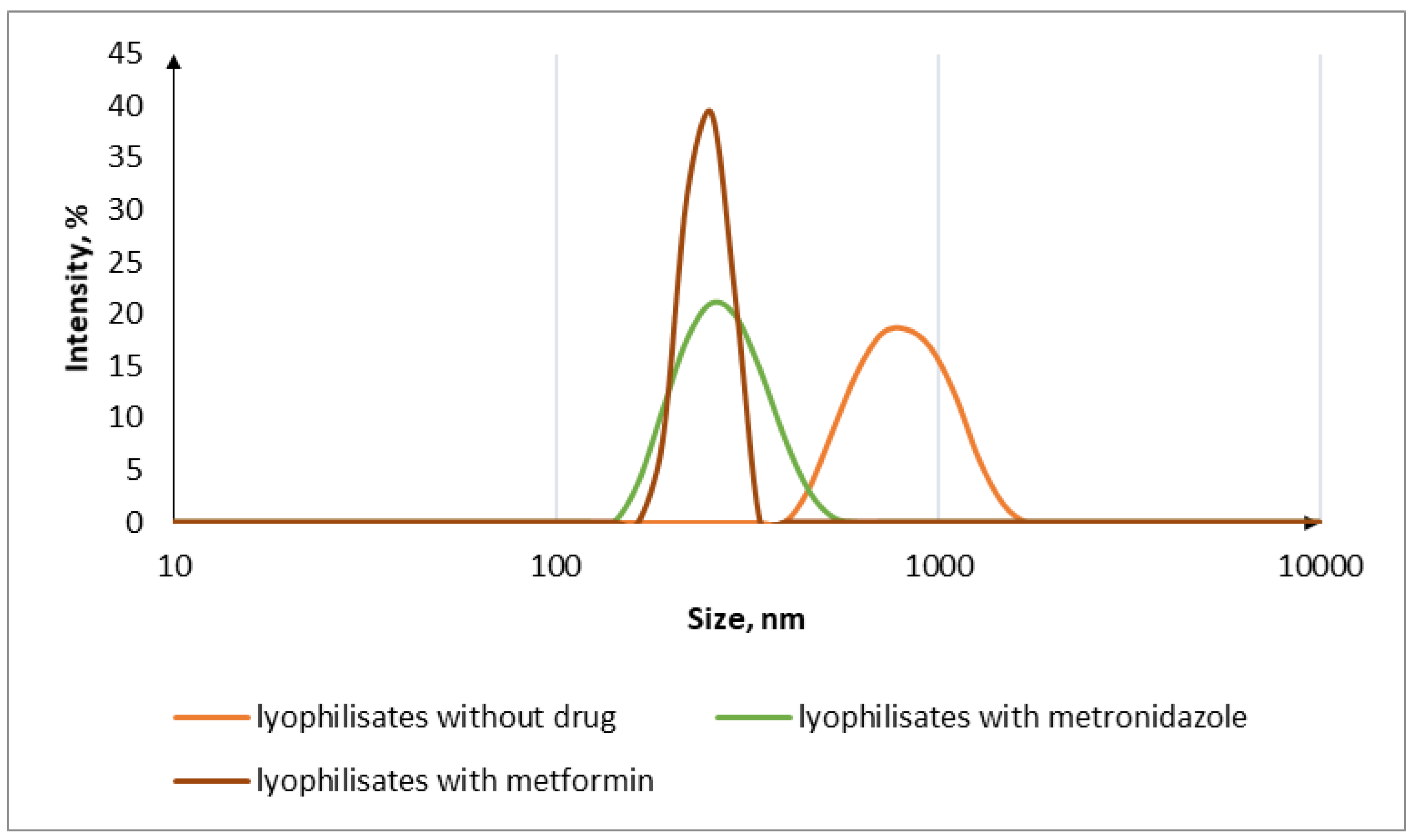
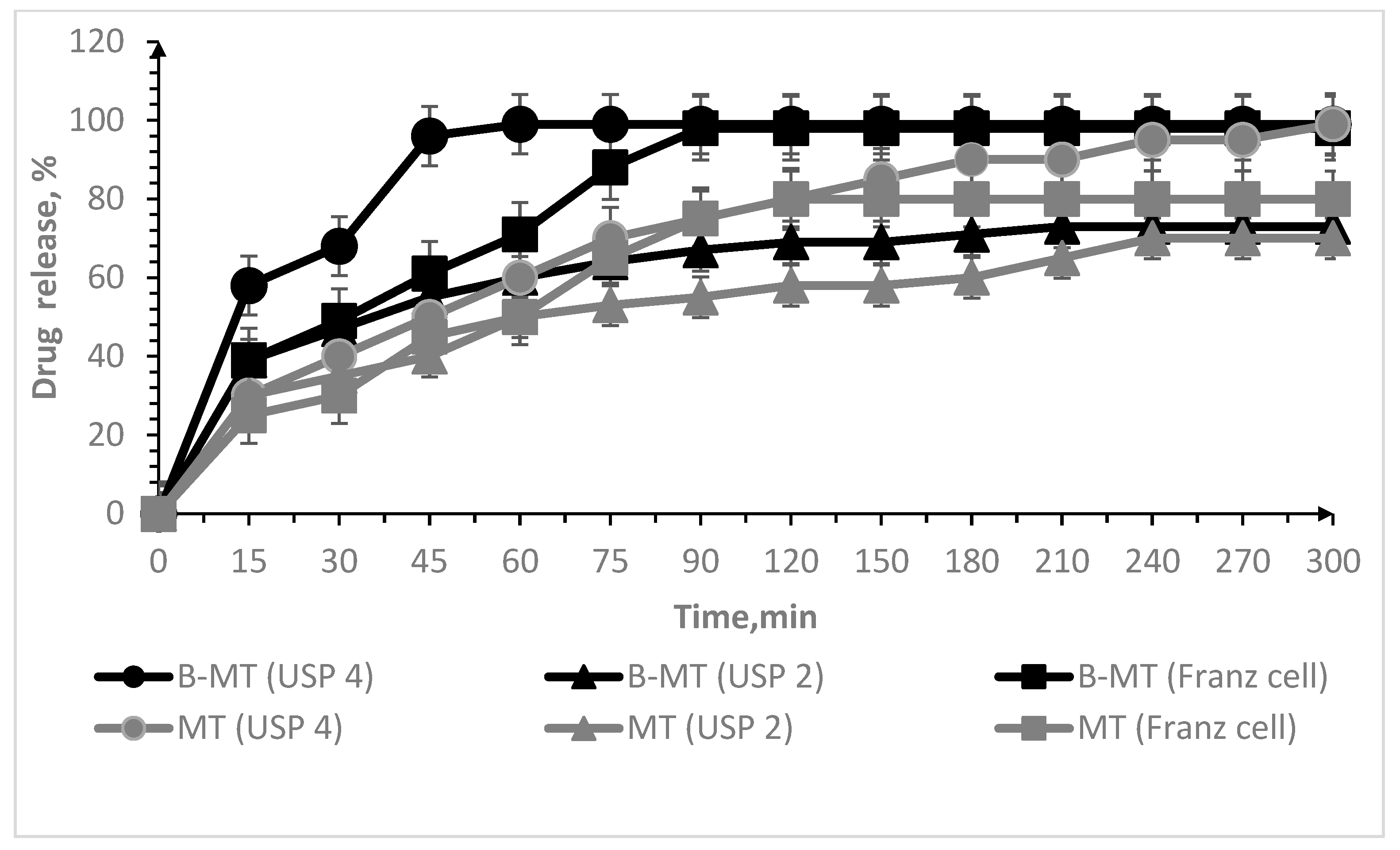
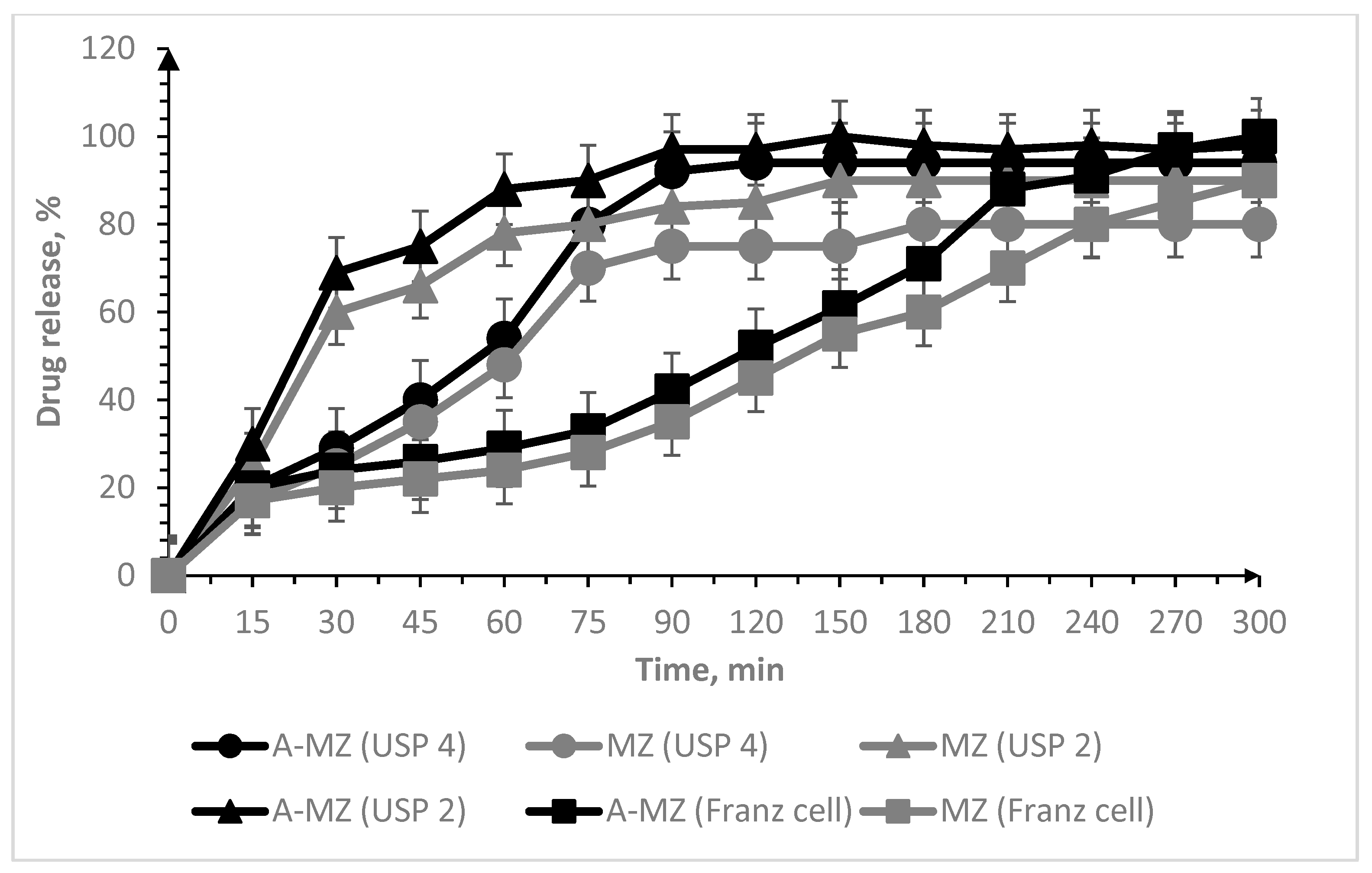
3.2. Oral Lyophilisates
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wiedey, R.; Kokott, M.; Breitkreutz, J. Orodispersible Tablets for Pediatric Drug Delivery: Current Challenges and Recent Advances. Expert Opin. Drug Deliv. 2021, 18, 1873–1890. [Google Scholar] [CrossRef] [PubMed]
- Stegemann, S.; Gosch, M.; Breitkreutz, J. Swallowing Dysfunction and Dysphagia Is an Unrecognized Challenge for Oral Drug Therapy. Int. J. Pharm. 2012, 430, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Musazzi, U.M.; Khalid, G.M.; Selmin, F.; Minghetti, P.; Cilurzo, F. Trends in the Production Methods of Orodispersible Films. Int. J. Pharm. 2019, 576, 118963. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Singh, K. Oral Disintegrating Tablets—An Updated Patent Perspective. Recent Pat. Drug Deliv. Formul. 2020, 14, 166–190. [Google Scholar] [CrossRef] [PubMed]
- Sachan, A.K.; Singh, S.; Kumar, V.; Kumari. Comparative Study of Natural and Synthetic Superdisintegrants In Orodispersible Metformin Tablet. Asian J. Pharm. Res. Dev. 2019, 7, 46–53. [Google Scholar] [CrossRef]
- Bhavna, B.G.; Bhushan, R.R.; Jain, A.S. Formulation and Evaluation of Orodispersible Tablet of Sulindac. Eur. J. Pharm. Res. 2022, 2, 11–19. [Google Scholar] [CrossRef]
- Khalid, G.M.; Selmin, F. Applications of Alginates in the Design and Preparation of Orodispersible Dosage Forms. In Properties and Applications of Alginates; IntechOpen: London, UK, 2021. [Google Scholar] [CrossRef]
- Kumar, R.; Sheela, M.A.; Sachdeva, M. Formulation and Evaluation of Orodispersible Tablets Containing CoCrystals of Modafinil. J. Drug Deliv. Ther. 2022, 12, 82–89. [Google Scholar] [CrossRef]
- Mohana, M.; Vijayalakshmi, S. Development and characterization of solid dispersion-based orodispersible tablets of cilnidipine. Beni Suef Univ. J. Basic Appl. Sci. 2022, 11, 83. [Google Scholar] [CrossRef]
- Kumari, P.V.K.; Srinivasa, Y.R. Formulation and evaluation of orodispersible tablets of donepezil hydrochloride. Int. J. Curr. Pharm. Res. 2020, 12, 45–51. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Mohanta, T.D.S.; Basak, R. Orodispersible Tablet in Treatment of Migraine: Opportunities, Challenges and Recent Advancements. J. Drug Deliv. Ther. 2021, 11, 149–156. [Google Scholar] [CrossRef]
- Manda, P.; Popescu, C.; Juluri, A.; Janga, K.; Kakulamarri, P.R.; Narishetty, S.; Narasimha Murthy, S.; Repka, M.A. Micronized Zaleplon Delivery via Orodispersible Film and Orodispersible Tablets. AAPS PharmSciTech 2018, 19, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Kuck, J.; Breitkreutz, J. Impact of lubrication on key properties of orodispersible minitablets in comparison to conventionally sized orodispersible tablets. Eur. J. Pharm. Biopharm. 2022, 180, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Mhetre, L.R.; Kadam, P.S.; Gadhire, P.H. Formulation and Evaluation of Naproxen Orodispersible Tablets. Int. J. Pharm. Sci. Nanotechnol. 2022, 15, 6055–6060. [Google Scholar] [CrossRef]
- Van Nguyen, K.; Dang, T.K.; Vu, L.T.; Ha, N.T.; Truong, H.D.; Tran, T.H. Orodispersible film incorporating nanoparticulate loratadine for an enhanced oral bioavailability. J. Pharm. Investig. 2023. [Google Scholar] [CrossRef]
- Jassem, N.A. Orodispersible Tablets: A Review on Recent Trends in Drug Delivery. Int. J. Drug Deliv. Technol. 2022, 12, 432–436. [Google Scholar]
- Sinha, S.; Sonali; Garg, V.; Thapa, S.; Singh, S.; Chauhan, M. Empagliflozin containing chitosan-alginate nanoparticles in orodispersible film: Preparation, characterization, pharmacokinetic evaluation and its in-vitro anticancer activity. Drug Dev. Ind. Pharm. 2022, 48, 279–291. [Google Scholar] [CrossRef]
- Petrovick, G.F.; Kleinebudde, P.; Breitkreutz, J. Orodispersible tablets containing taste-masked solid lipid pellets with metformin hydrochloride: Influence of process parameters on tablet properties. Eur. J. Pharm. Biopharm. 2018, 122, 137–145. [Google Scholar] [CrossRef]
- Oliveira, L.J.; Veiga, A.; Stofella, N.C.F.; Cunha, A.C.; Graça, M.; Toledo, T.; Andreazza, I.F.; Murakami, F.S. Development and Evaluation of Orodispersible Tablets Containing Ketoprofen. Curr. Drug Deliv. 2020, 17, 348–360. [Google Scholar] [CrossRef]
- Lew, M.F. Selegiline Orally Disintegrating Tablets for the Treatment of Parkinson’s Disease. Expert Rev. Neurother. 2005, 5, 705–712. [Google Scholar] [CrossRef]
- Hua, S. Advances in Nanoparticulate Drug Delivery Approaches for Sublingual and Buccal Administration (mini-review). Front. Pharmacol. 2019, 10, 1328. [Google Scholar] [CrossRef]
- Watchorn, J.; Clasky, A.J.; Prakash, G.; Johnston, I.A.E.; Chen, P.Z.; Gu, F.X. Untangling Mucosal Drug Delivery: Engineering, Designing, and Testing Nanoparticles to Overcome the Mucus Barrier. ACS Biomater. Sci. Eng. 2022, 8, 1396–1426. [Google Scholar] [CrossRef] [PubMed]
- Mehanna, M.M.; Mneimneh, A.T.; Domiati, S.; Allam, A.N. Tadalafil-Loaded Limonene-Based Orodispersible Tablets: Formulation, in vitro Characterization and in vivo Appraisal of Gastroprotective Activity. Int. J. Nanomed. 2020, 15, 10099–10112. [Google Scholar] [CrossRef] [PubMed]
- Garipova, V.R.; Gennari, C.G.M.; Selmin, F.; Cilurzo, F.; Moustafine, R.I. Mucoadhesive Interpolyelectrolyte Complexes for the Buccal Delivery of Clobetasol. Polymers 2018, 10, 85. [Google Scholar] [CrossRef]
- Russo, E.; Selmin, F.; Baldassari, S.; Gennari, C.G.M.; Caviglioli, G.; Cilurzo, F.; Minghetti, P.; Parodi, B. A focus on mucoadhesive polymers and their application in buccal dosage forms. J. Drug Deliv. Sci. Technol. 2016, 32, 113–125. [Google Scholar] [CrossRef]
- Ruiz-Rubio, L.; Alonso, M.L.; Pérez-Álvarez, L.; Alonso, R.M.; Vilas, J.L.; Khutoryanskiy, V.V. Formulation of Carbopol®/poly(2-ethyl-2-oxazoline)s mucoadhesive tablets for buccal delivery of hydrocortisone. Polymers 2018, 10, 175. [Google Scholar] [CrossRef]
- Khutoryanskiy, V.V. Advances in mucoadhesion and mucoadhesive polymers. Macromol. Biosci. 2011, 11, 748–764. [Google Scholar] [CrossRef]
- Gupta, M.S.; Kumar, T.P. Characterization of Orodispersible Films: An Overview of Methods and Introduction to a New Disintegration Test Apparatus Using LDR-LED Sensors. J. Pharm. Sci. 2020, 109, 2925–2942. [Google Scholar] [CrossRef]
- Desai, N.; Redfearn, A.; MacLeod, G.; Tuleu, C.; Hanson, B.; Orlu, M. How Do Orodispersible Tablets Behave in an In Vitro Oral Cavity Model: A Pilot Study. Pharmaceutics 2020, 12, 651. [Google Scholar] [CrossRef]
- Khalid, G.M.; Selmin, F.; Musazzi, U.M.; Gennari, C.G.M.; Minghetti, P.; Cilurzo, F. Trends in the Characterization Methods of Orodispersible Films. Curr. Drug Deliv. 2021, 18, 935–946. [Google Scholar] [CrossRef]
- Guhmann, M.; Preis, M.; Gerber, F.; Pöllinger, N.; Breitkreutz, J.; Weitschies, W. Design, Development and in-Vitro Evaluation of Diclofenac Taste-Masked Orodispersible Tablet Formulations. Drug Dev. Ind. Pharm. 2015, 41, 540–551. [Google Scholar] [CrossRef]
- Wasilewska, K.; Winnicka, K. How to Assess Orodispersible Film Quality? A Review of Applied Methods and Their Modifications. Acta Pharm. 2019, 69, 155–176. [Google Scholar] [CrossRef] [PubMed]
- Cilurzo, F.; Musazzi, U.M.; Franzé, S.; Selmin, F.; Minghetti, P. Orodispersible dosage forms: Biopharmaceutical improvements and regulatory requirements. Drug Discov. Today 2018, 23, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Medina, J.R.; Salazar, D.K.; Hurtado, M.; Cortés, A.R.; Domínguez-Ramírez, A.M. Comparative in Vitro Dissolution Study of Carbamazepine Immediate-Release Products Using the USP Paddles Method and the Flow-through Cell System. Saudi Pharm. J. 2014, 22, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Selmin, F.; Khalid, G.M.; Musazzi, U.M.; Demartin, F.; Minghetti, P.; Cilurzo, F. Relevance of Production Method on the Physical Stability and in Vitro Biopharmaceutical Performances of Olanzapine Orodispersible Film. Int. J. Pharm. 2021, 603, 120697. [Google Scholar] [CrossRef]
- Perioli, L.; Ambrogi, V.; Rubini, D.; Giovagnoli, S.; Ricci, M.; Blasi, P.; Rossi, C. Novel Mucoadhesive Buccal Formulation Containing Metronidazole for the Treatment of Periodontal Disease. J. Control. Release 2004, 95, 521–533. [Google Scholar] [CrossRef]
- Proctor, W.R.; Bourdet, D.L.; Thakker, D.R. Mechanisms Underlying Saturable Intestinal Absorption of Metformin. Drug Metab. Dispos. 2008, 36, 1650–1658. [Google Scholar] [CrossRef]
- Sander, C.; Nielsen, H.M.; Jacobsen, J. Buccal Delivery of Metformin: TR146 Cell Culture Model Evaluating the Use of Bioadhesive Chitosan Discs for Drug Permeability Enhancement. Int. J. Pharm. 2013, 458, 254–261. [Google Scholar] [CrossRef]
- Sander, C.; Madsen, K.D.; Hyrup, B.; Nielsen, H.M.; Rantanen, J.; Jacobsen, J. Characterization of spray dried bioadhesive metformin microparticles for oromucosal administration. Eur. J. Pharm. Biopharm. 2013, 85, 682–688. [Google Scholar] [CrossRef]
- Belayneh, A.; Molla, F.; Kahsay, G. Formulation and Optimization of Monolithic Fixed-Dose Combination of Metformin HCl and Glibenclamide Orodispersible Tablets. Adv. Pharmacol. Pharm. Sci. 2020, 2020, 3546597. [Google Scholar] [CrossRef]
- Li, M.; Tan, H. Technical Note: Comparison of USP Apparatus 5 and 7 for In Vitro Drug Release from Nicotine Transdermal Systems. Dissolut. Technol. 2019, 26, 68–71. [Google Scholar] [CrossRef]
- Timergalieva, V.R.; Khusnutdinov, R.R.; Musina, R.R.; Elizarova, E.S.; Alsynbaev, R.R.; Nasibullin, S.F.; Moustafine, R.I. Development of orodispersible ibuprofen tablets based on a polymer-drug complex. Drug Dev. Regist. 2022, 11, 113–120. [Google Scholar] [CrossRef]
- Viktorova, A.S.; Elizarova, E.S.; Romanova, R.S.; Timergalieva, V.R.; Khutoryanskiy, V.V.; Moustafine, R.I. Interpolymer complexes based on Carbopol® and poly(2-ethyl-2-oxazoline) as carriers for buccal delivery of metformin. Drug Dev. Regist. 2021, 10, 48–55. [Google Scholar] [CrossRef]
- Dash, S.; Murthy, P.N.; Nath, L.; Chowdhury, P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol. Pharm. Drug Res. 2010, 67, 217–223. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Sample Code | IPEC (mol/mol) | Oral Lyophilisates | MT | MZ | Dh (nm) | ζ (mV) | MDF (kPa) | WA (mJ) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| EPO | C10 | NAA-1 | ||||||||
| A-IPEC | 1 | 1 | -- | -- | -- | -- | 294 ± 20 | −14.40 | 103.5 ± 0.7 | 2154 ± 66 |
| B-IPEC | 2 | -- | 1 | -- | -- | -- | 298 ± 30 | 15.00 | 111.0 ± 9.5 | 2611 ± 413 |
| A-lyo | 1 | 1 | -- | + | -- | -- | 921 ± 34 | −48.86 | -- | -- |
| B-lyo | 2 | -- | 1 | + | -- | -- | 921 ± 34 | −20.00 | -- | -- |
| A-MZ | 1 | 1 | -- | + | - | + | 272 ± 35 | −10.10 | -- | -- |
| B-MT | 2 | -- | 1 | + | + | - | 465 ± 50 | −19.00 | -- | -- |
| PE | - | - | - | - | - | - | - | 13.4 ± 0.7 | 407 ± 175 | |
| Chitosan | - | - | - | - | - | - | - | 62.0 ± 10.0 | 1682 ± 162 | |
| Oral Lyophilisates Code | Oral Lyophilisates Composition (%, w/w) | Disint. Time (s) | ||||
|---|---|---|---|---|---|---|
| IPEC A | IPEC B | Drug | MDX | S80 | ||
| A-lyo | 9.95 | - | - | 89.56 | 0.49 | 23 |
| B-lyo | - | 9.95 | - | 89.56 | 0.49 | 23 |
| A-MZ | 2.76 | - | 46.10 | 49.76 | 1.38 | 60 |
| B-MT | - | 2.76 | 46.10 | 49.76 | 1.38 | 50 |
| MZ | - | - | 46.10 | 52.52 | 1.38 | 50 |
| MT | - | - | 46.10 | 52.52 | 1.38 | 50 |
| Parameters | |||
|---|---|---|---|
| B-MT USP 2 | B-MT USP 4 | B-MT Franz Cell | |
| Exponential release (n) | 0.157 ± 0.023 | 0.124 ± 0.036 | 0.106 ± 0.069 |
| Constant release (k) | 31.094 ± 3.691 | 51.832 ± 9.467 | 45.605 ± 15.972 |
| Correlation coefficient (R2) | 0.85228 | 0.49391 | 0.16469 |
| A-MZ USP 2 | A-MZ USP 4 | A-MZ Franz cell | |
| Exponential release (n) | 0.113 ± 0.029 | 0.390 ± 0.116 | 0.713 ± 0.056 |
| Constant release (k) | 53.672 ± 8.020 | 12.448 ± 7.303 | 1.787 ± 0.540 |
| Correlation coefficient (R2) | 0.63839 | 0.68902 | 0.96691 |
| Parameters | |||
|---|---|---|---|
| B-MT USP 2 | B-MT USP 4 | B-MT Franz cell | |
| Maximum dissolution (a) | 70.275 ± 1.792 | 98.931 ± 1.748 | 98.732 ± 2.235 |
| Dissolution rate (xc) | 12.820 ± 2.689 | 11.104 ± 1.610 | 21.335 ± 2.591 |
| Undissolved proportion (k) | 0.052 ± 0.009 | 0.082 ± 0.013 | 0.037 ± 0.004 |
| Correlation coefficient (R2) | 0.94573 | 0.96812 | 0.97017 |
| A-MZ USP 2 | A-MZ USP 4 | A-MZ Franz cell | |
| Maximum dissolution (a) | 96.894 ± 1.296 | 95.704 ± 2.249 | 119.621 ± 10.359 |
| Dissolution rate (xc) | 17.357 ± 1.294 | 35.900 ± 2.604 | 98.052 ± 12.697 |
| Undissolved proportion (k) | 0.063 ± 0.006 | 0.035 ± 0.004 | 0.008 ± 0.001 |
| Correlation coefficient (R2) | 0.98660 | 0.97784 | 0.98352 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timergalieva, V.R.; Gennari, C.G.M.; Cilurzo, F.; Selmin, F.; Moustafine, R.I. Comparative Evaluation of Metformin and Metronidazole Release from Oral Lyophilisates with Different Methods. Sci. Pharm. 2023, 91, 23. https://doi.org/10.3390/scipharm91020023
Timergalieva VR, Gennari CGM, Cilurzo F, Selmin F, Moustafine RI. Comparative Evaluation of Metformin and Metronidazole Release from Oral Lyophilisates with Different Methods. Scientia Pharmaceutica. 2023; 91(2):23. https://doi.org/10.3390/scipharm91020023
Chicago/Turabian StyleTimergalieva, Venera R., Chiara G. M. Gennari, Francesco Cilurzo, Francesca Selmin, and Rouslan I. Moustafine. 2023. "Comparative Evaluation of Metformin and Metronidazole Release from Oral Lyophilisates with Different Methods" Scientia Pharmaceutica 91, no. 2: 23. https://doi.org/10.3390/scipharm91020023
APA StyleTimergalieva, V. R., Gennari, C. G. M., Cilurzo, F., Selmin, F., & Moustafine, R. I. (2023). Comparative Evaluation of Metformin and Metronidazole Release from Oral Lyophilisates with Different Methods. Scientia Pharmaceutica, 91(2), 23. https://doi.org/10.3390/scipharm91020023