
Oral Bioavailability Enhancement of Vancomycin Hydrochloride with Cationic Nanocarrier (Leciplex): Optimization, In Vitro, Ex Vivo, and In Vivo Studies


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of VAN-Loaded LPX
2.3. Characterization of VAN-Loaded LPX
2.3.1. Determination of Entrapment Efficiency (E.E.%)
2.3.2. Determination of Particle Size, Polydispersity Index, and Zeta Potential
2.3.3. In Vitro Drug Release Study
2.4. Optimization of VAN-Loaded LPX Formulations
2.5. Selecting the Optimized VAN-LPX Formula
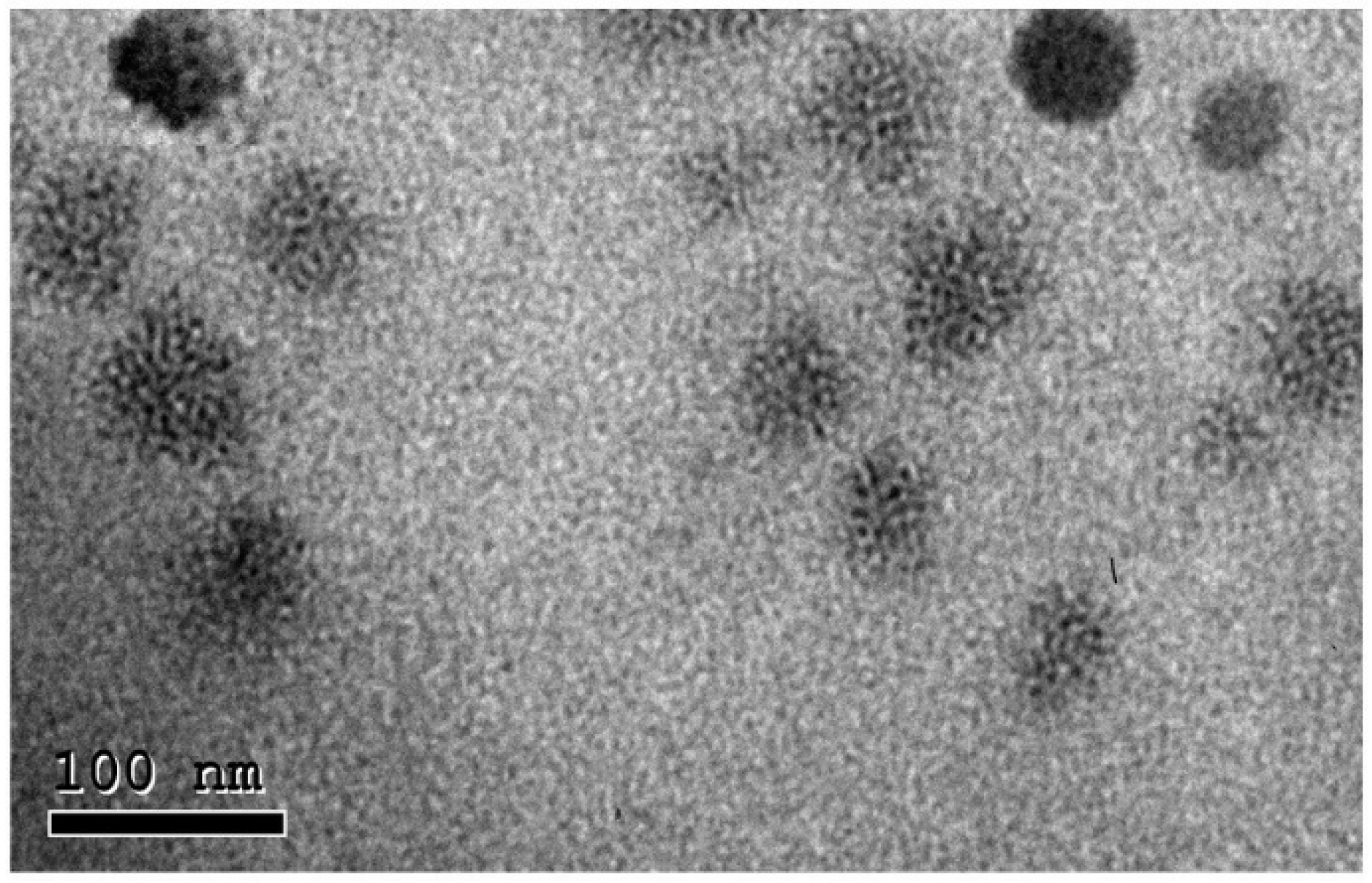
2.6. Morphology of the Optimized VAN-LPX Formula
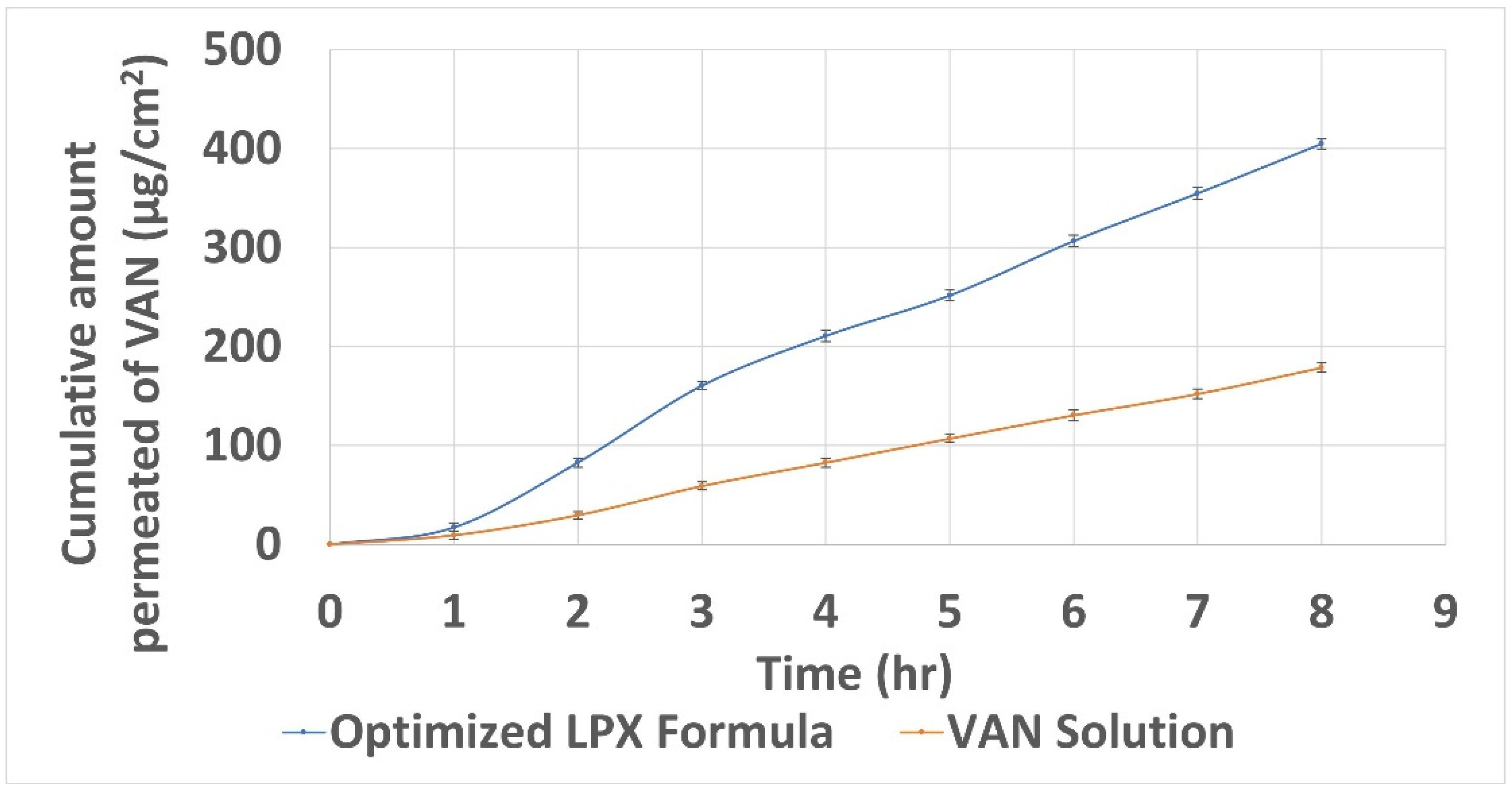
2.7. Ex Vivo Permeation Study via Non-Everted Intestinal Sac Model
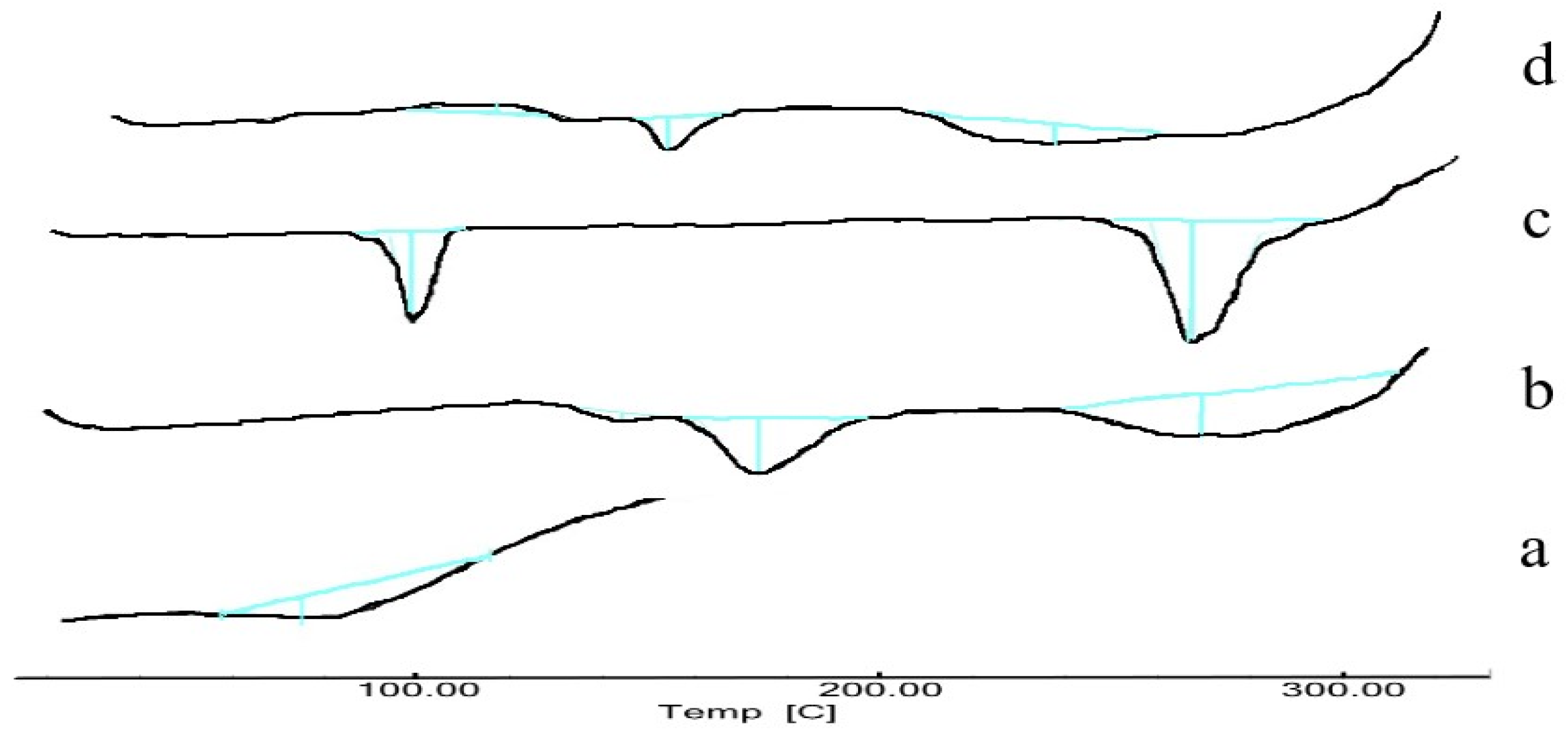
2.8. Differential Scanning Calorimetry
2.9. Effect of Storage on the Selected LPX Formula
2.10. Evaluation of Mucoadhesion Properties of the Selected Formula
2.11. Cytotoxicity Assay
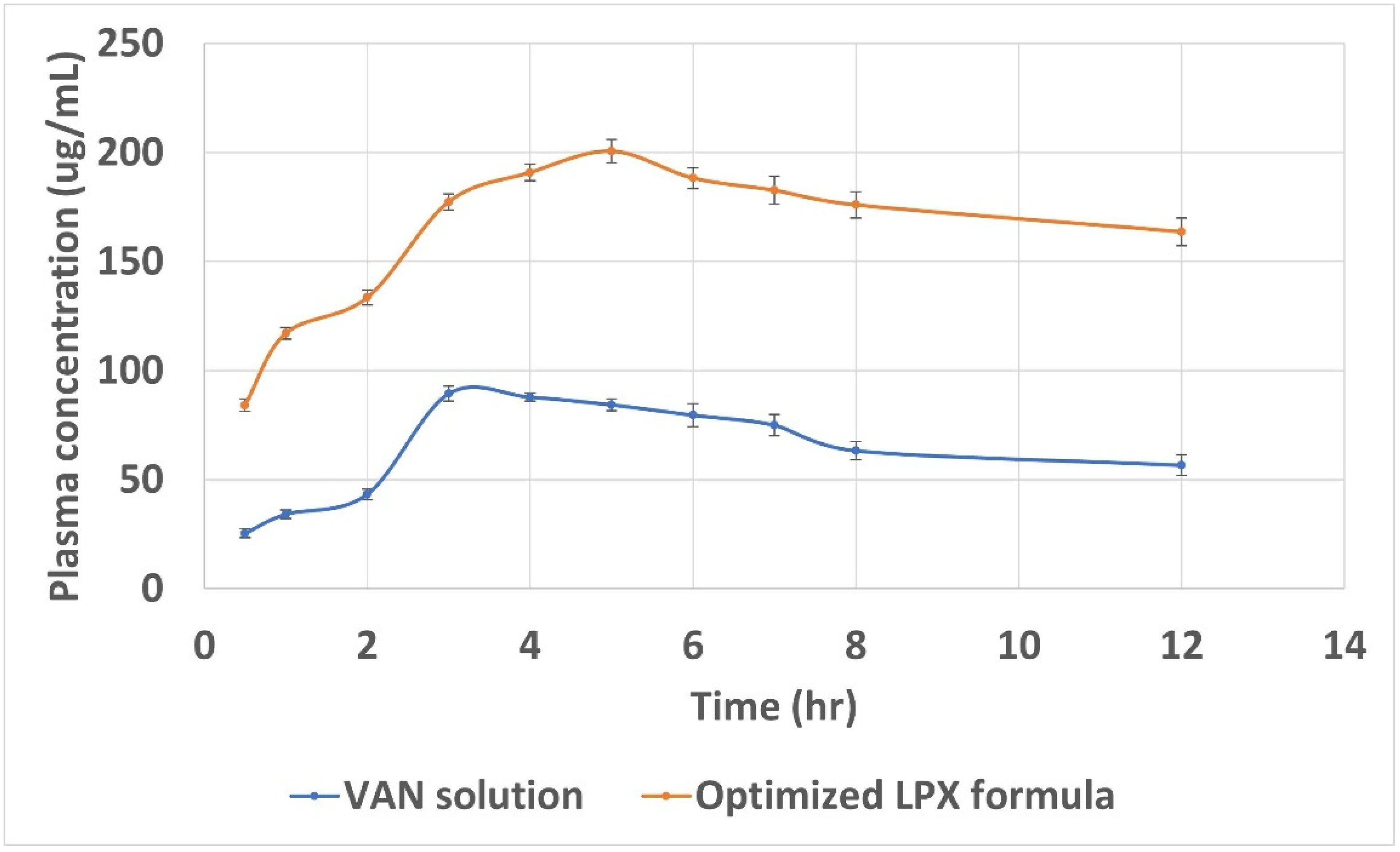
2.12. In Vivo Assessment of the Selected LPX Formula
Statistical Analysis of Data
3. Results and Discussion
3.1. Design Optimization
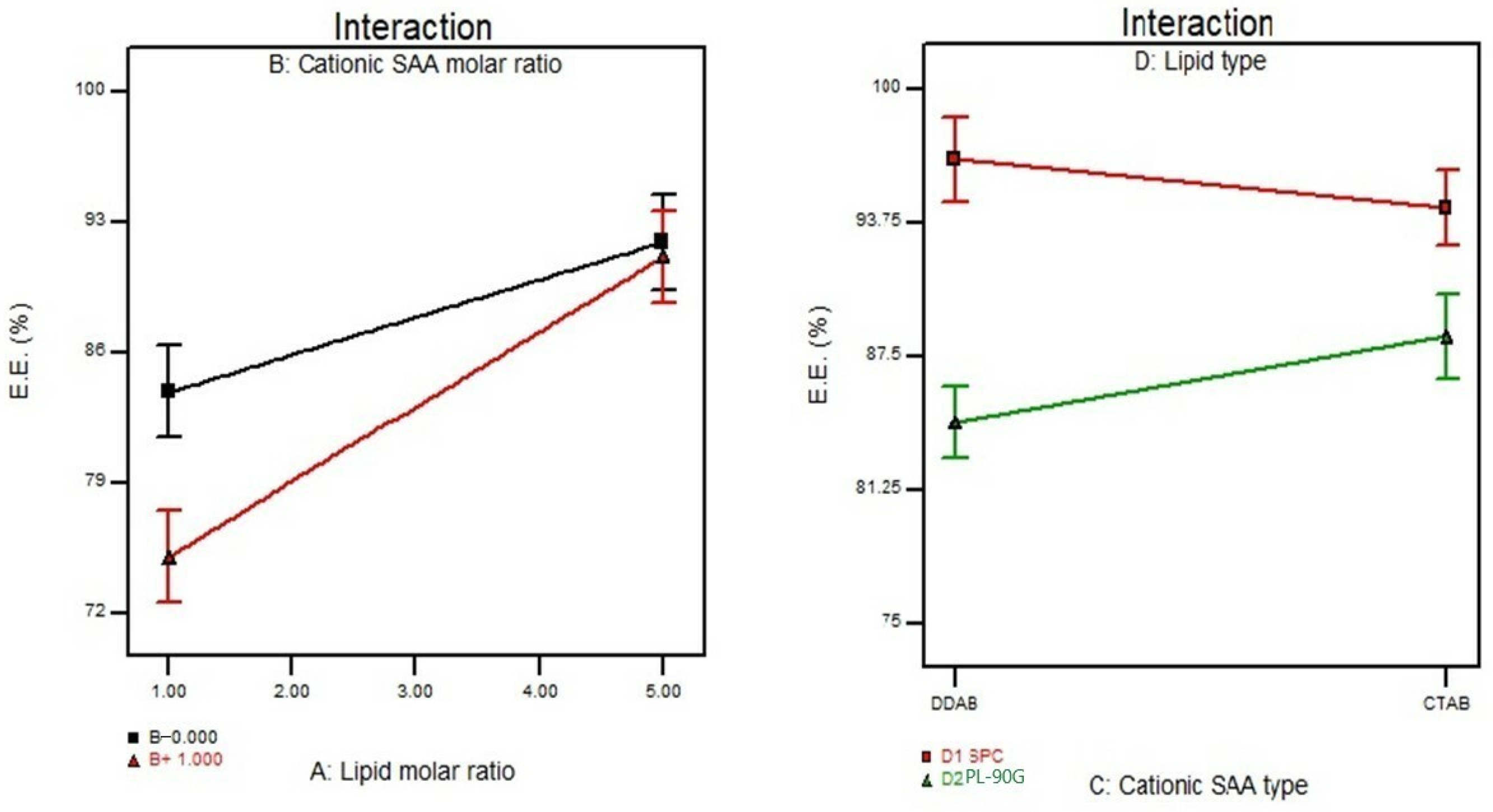
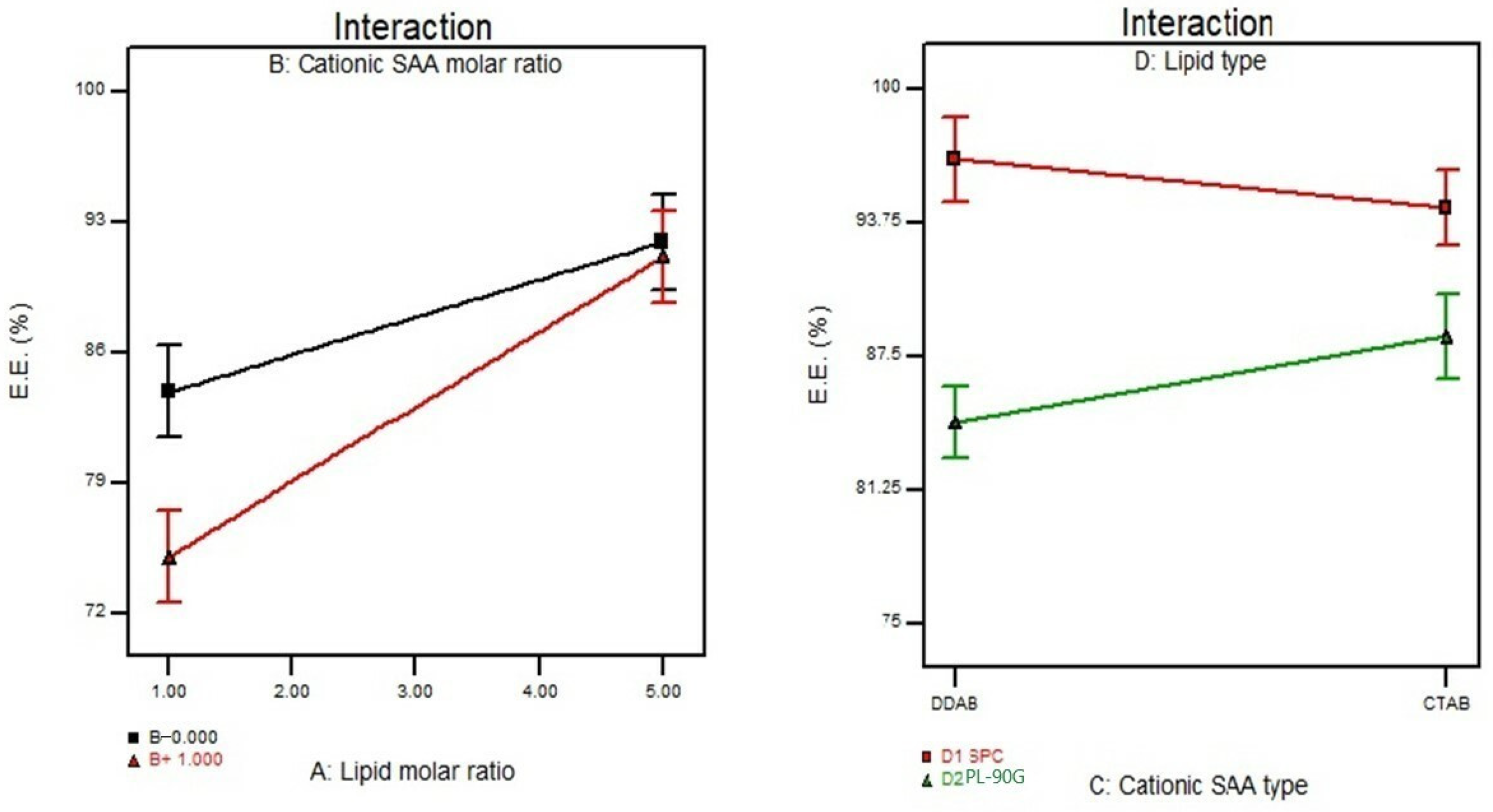
3.2. Effect of Formulation Variables on the Entrapment Efficiency%
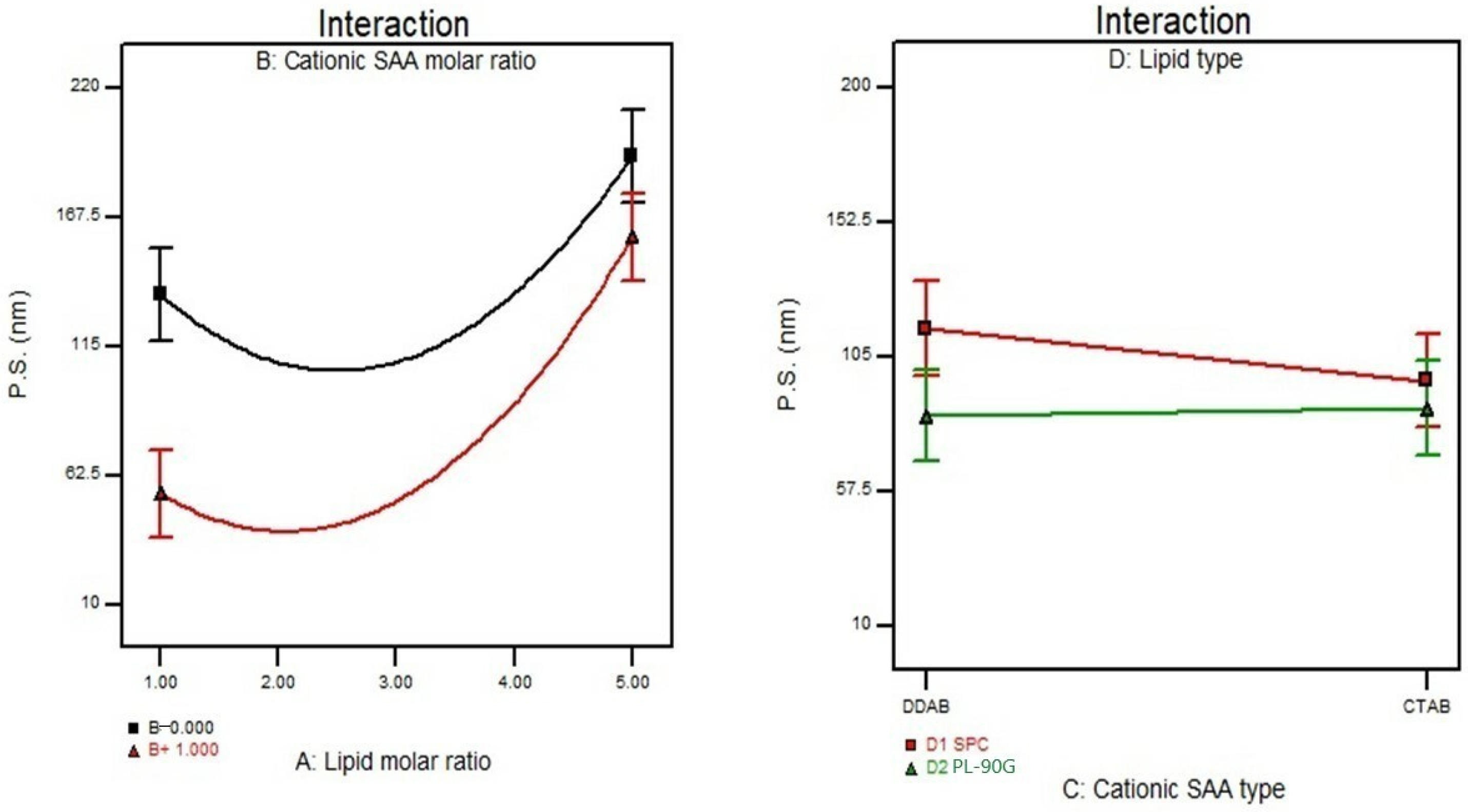
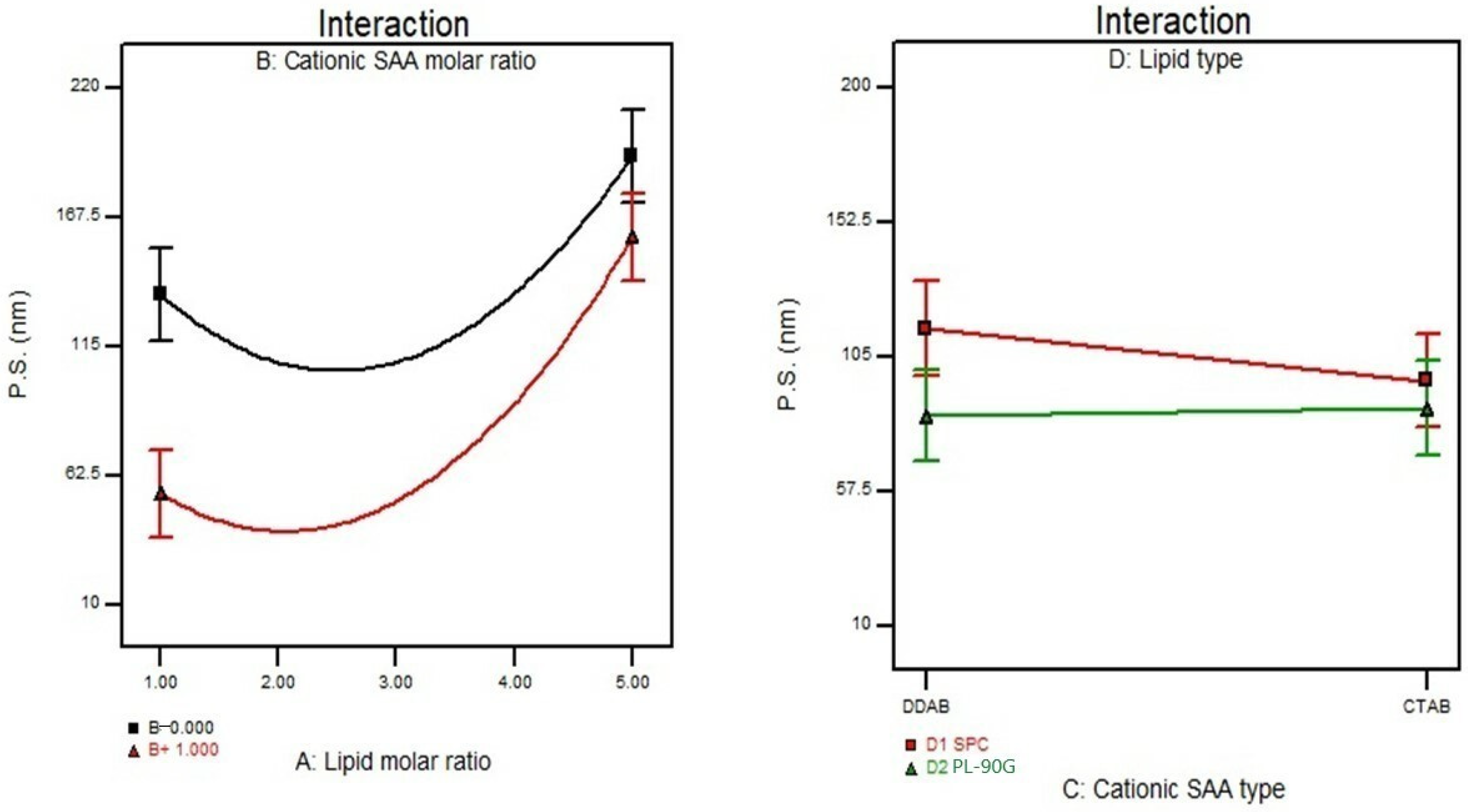
3.3. Effect of Formulation Variables on the Particle Size
3.4. Effect of Formulation Variables on the Polydispersity Index
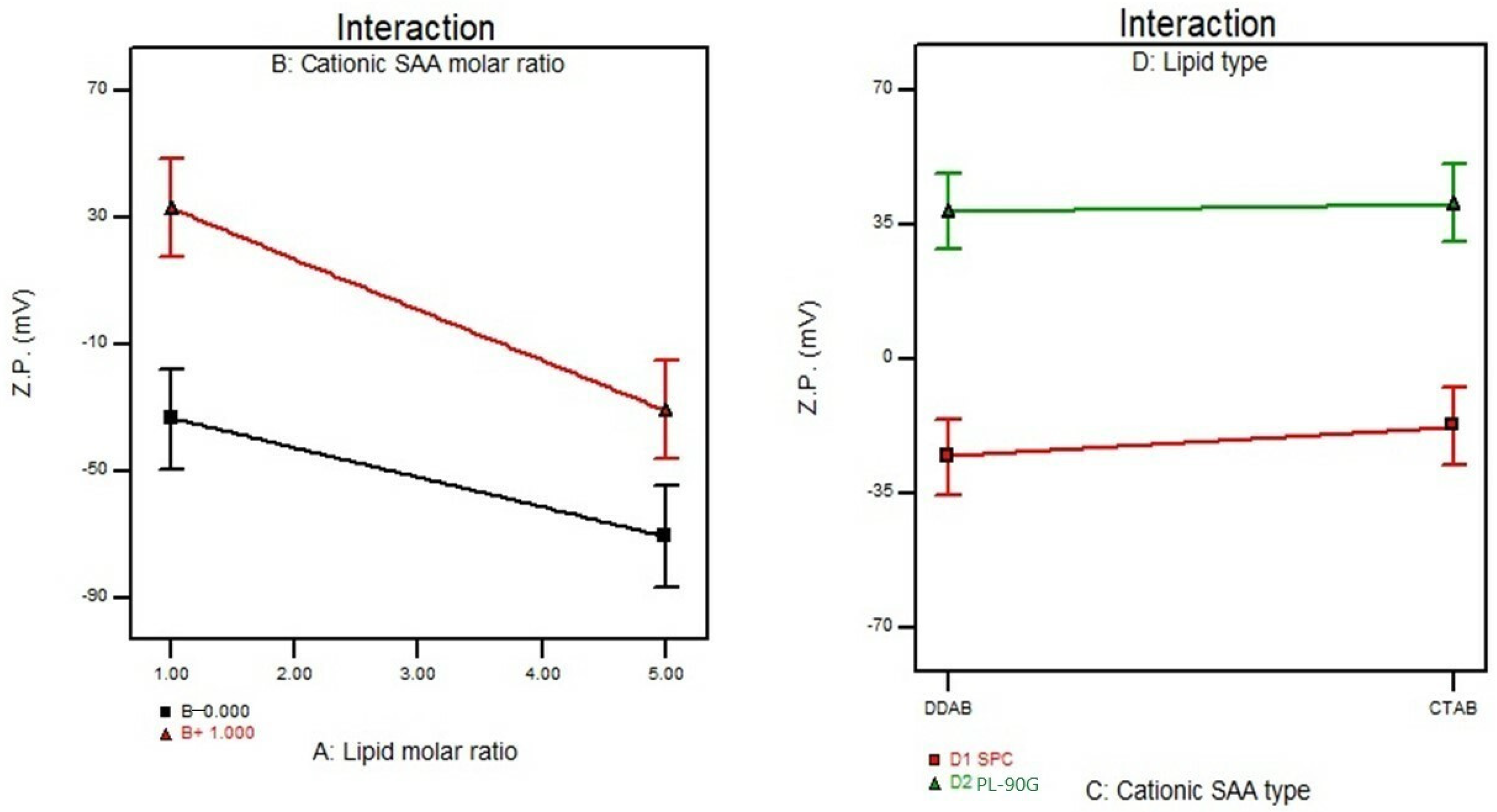
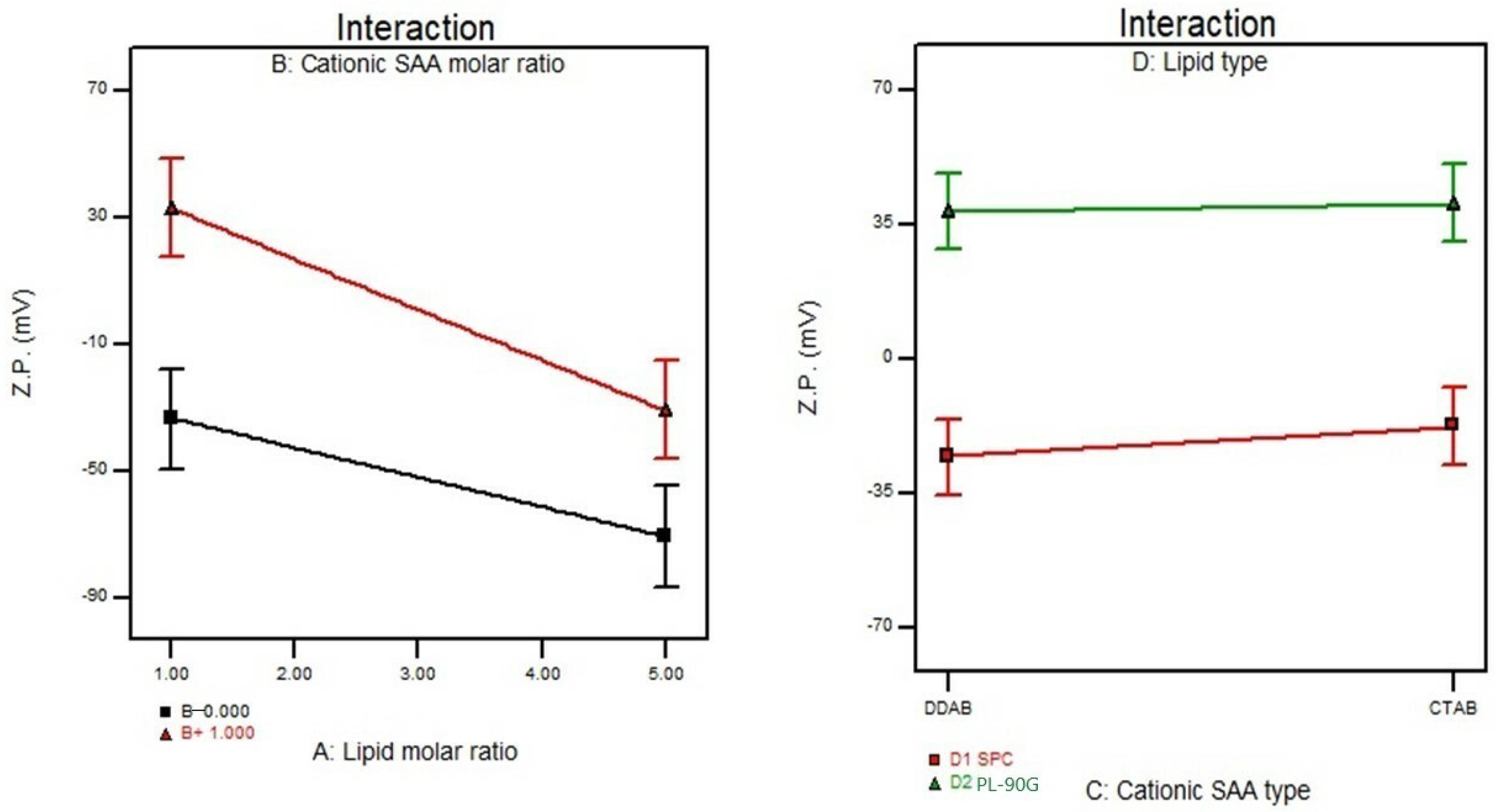
3.5. Effect of Formulation Variables on the Zeta Potential
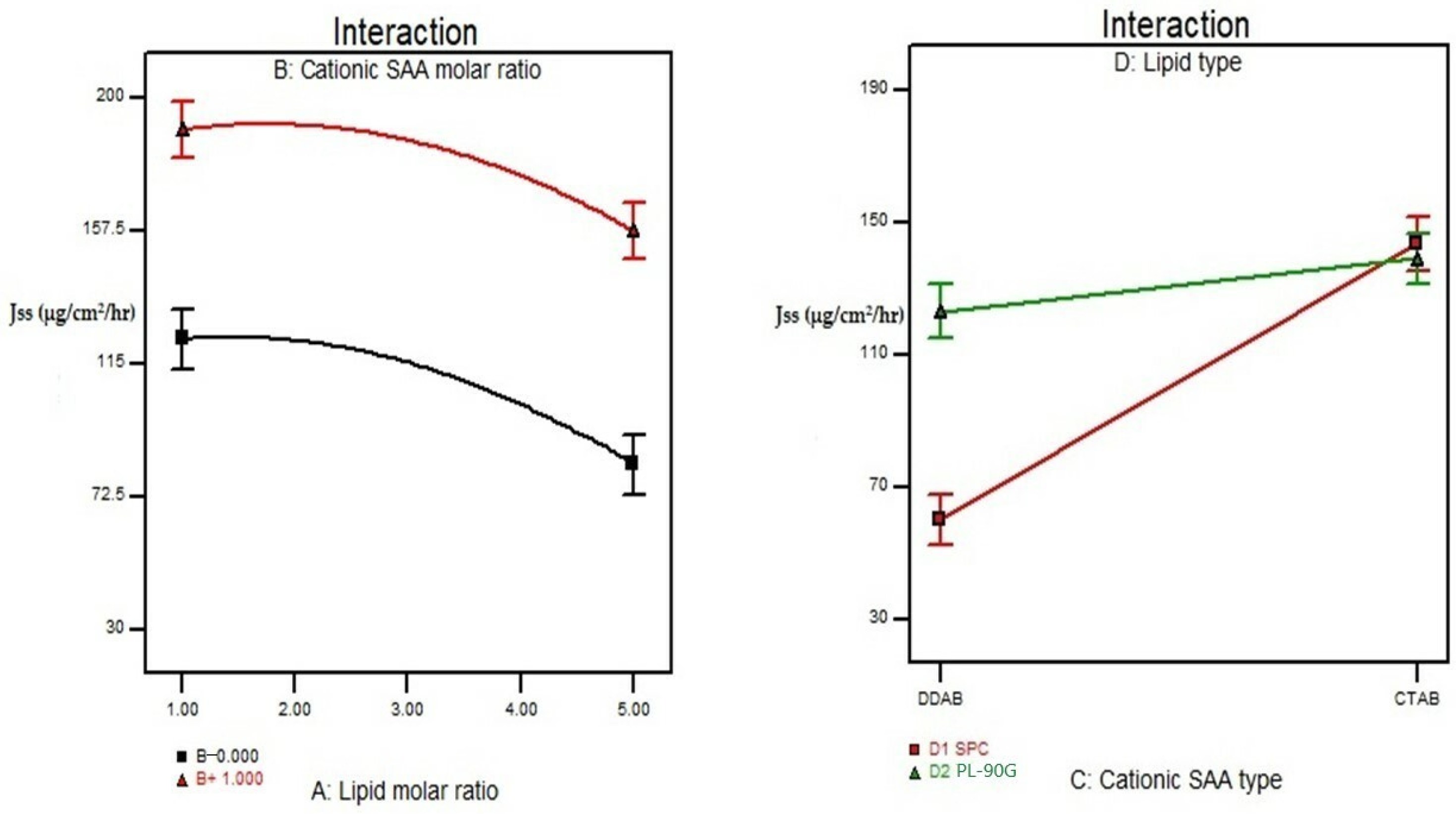
3.6. Effect of Formulation Variables on the In Vitro Drug Release
3.7. Selection of the Optimized LPX Formula
3.8. Morphology of the Optimized LPX Formula
3.9. Ex Vivo Permeation Study
3.10. Differential Scanning Calorimetry
3.11. Effect of Storage on the Selected LPX Formula
3.12. Evaluation of Mucoadhesion Properties
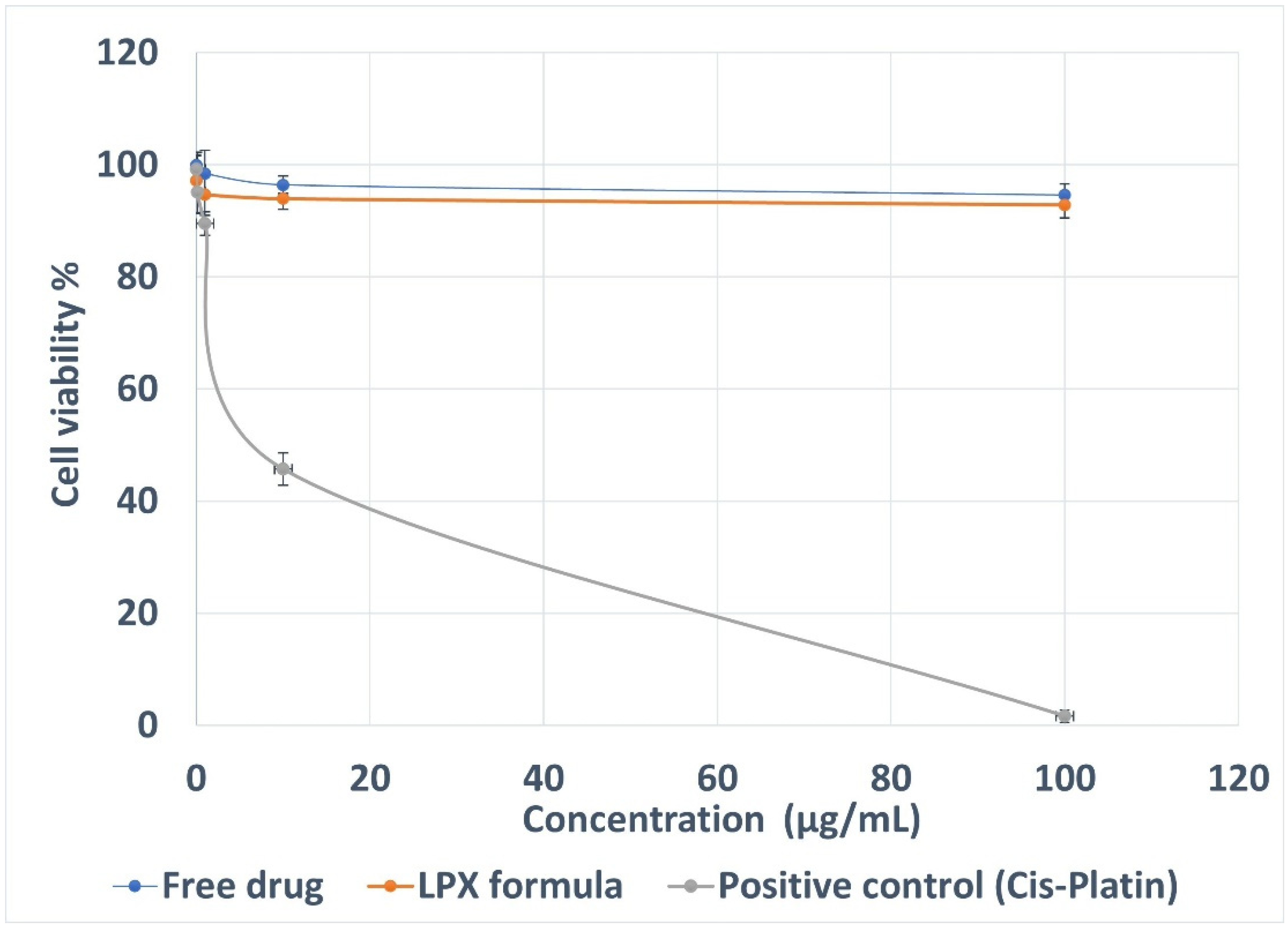
3.13. Cytotoxicity Assay
3.14. In Vivo Assessment of the Selected LPX Formula
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maher, S.; Geoghegan, C.; Brayden, D.J. Intestinal permeation enhancers to improve oral bioavailability of macromolecules: Reasons for low efficacy in humans. Expert Opin. Drug Deliv. 2021, 18, 273–300. [Google Scholar] [CrossRef]
- Gamboa, A.; Schüßler, N.; Soto-Bustamante, E.; Romero-Hasler, P.; Meinel, L.; Morales, J.O. Delivery of ionizable hydrophilic drugs based on pharmaceutical formulation of ion pairs and ionic liquids. Eur. J. Pharm. Biopharm. 2020, 156, 203–218. [Google Scholar] [CrossRef]
- Phan, T.N.Q.; Shahzadi, I.; Bernkop-Schnürch, A. Hydrophobic ion-pairs and lipid-based nanocarrier systems: The perfect match for delivery of BCS class 3 drugs. J. Control. Release 2019, 304, 146–155. [Google Scholar] [CrossRef]
- Deng, F.; Bae, Y.H. Bile acid transporter-mediated oral drug delivery. J. Control. Release 2020, 327, 100–116. [Google Scholar] [CrossRef]
- Homayun, B.; Lin, X.; Choi, H.-J. Challenges and Recent Progress in Oral Drug Delivery Systems for Biopharmaceuticals. Pharmaceutics 2019, 11, 129. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Shrestha, N.; Préat, V.; Beloqui, A. An overview of in vitro, ex vivo and in vivo models for studying the transport of drugs across intestinal barriers. Adv. Drug Deliv. Rev. 2021, 175, 113795. [Google Scholar] [CrossRef]
- Xu, Y.; Shrestha, N.; Préat, V.; Beloqui, A. Overcoming the intestinal barrier: A look into targeting approaches for improved oral drug delivery systems. J. Control. Release 2020, 322, 486–508. [Google Scholar] [CrossRef]
- Zhu, Q.; Chen, Z.; Paul, P.K.; Lu, Y.; Wu, W.; Qi, J. Oral delivery of proteins and peptides: Challenges, status quo and future perspectives. Acta Pharm. Sin. B 2021, 11, 2416–2448. [Google Scholar] [CrossRef]
- Dünnhaupt, S.; Kammona, O.; Waldner, C.; Kiparissides, C.; Bernkop-Schnürch, A. Nano-carrier systems: Strategies to overcome the mucus gel barrier. Eur. J. Pharm. Biopharm. 2015, 96, 447–453. [Google Scholar] [CrossRef]
- Ye, C.; Chi, H. A review of recent progress in drug and protein encapsulation: Approaches, applications and challenges. Mater. Sci. Eng. C 2018, 83, 233–246. [Google Scholar] [CrossRef]
- Parmentier, J.; Hofhaus, G.; Thomas, S.; Cuesta, L.C.; Gropp, F.; Schröder, R.; Hartmann, K.; Fricker, G. Improved Oral Bioavailability of Human Growth Hormone by a Combination of Liposomes Containing Bio-Enhancers and Tetraether Lipids and Omeprazole. J. Pharm. Sci. 2014, 103, 3985–3993. [Google Scholar] [CrossRef]
- Ndayishimiye, J.; Cao, Y.; Kumeria, T.; Blaskovich, M.A.; Falconer, J.R.; Popat, A. Engineering mesoporous silica nanoparticles towards oral delivery of vancomycin. J. Mater. Chem. B 2021, 9, 7145–7166. [Google Scholar] [CrossRef]
- Cerchiara, T.; Abruzzo, A.; Parolin, C.; Vitali, B.; Bigucci, F.; Gallucci, M.C.; Nicoletta, F.P.; Luppi, B. Microparticles based on chitosan/carboxymethylcellulose polyelectrolyte complexes for colon delivery of vancomycin. Carbohydr. Polym. 2016, 143, 124–130. [Google Scholar] [CrossRef]
- Loveymi, B.D.; Jelvehgari, M.; Zakeri-Milani, P.; Valizadeh, H. Design of vancomycin RS-100 nanoparticles in order to increase the intestinal permeability. Adv. Pharm. Bull. 2012, 2, 43–56. [Google Scholar] [CrossRef]
- Zakeri-Milani, P.; Loveymi, B.D.; Jelvehgari, M.; Valizadeh, H. The characteristics and improved intestinal permeability of vancomycin PLGA-nanoparticles as colloidal drug delivery system. Colloids Surf. B Biointerfaces 2013, 103, 174–181. [Google Scholar] [CrossRef]
- Zaichik, S.; Steinbring, C.; Caliskan, C.; Bernkop-Schnürch, A. Development and in vitro evaluation of a self-emulsifying drug delivery system (SEDDS) for oral vancomycin administration. Int. J. Pharm. 2019, 554, 125–133. [Google Scholar] [CrossRef]
- Efiana, N.A.; Dizdarević, A.; Huck, C.W.; Bernkop-Schnürch, A. Improved Intestinal Mucus Permeation of Vancomycin via Incorporation Into Nanocarrier Containing Papain-Palmitate. J. Pharm. Sci. 2019, 108, 3329–3339. [Google Scholar] [CrossRef]
- Uhl, P.; Sauter, M.; Hertlein, T.; Witzigmann, D.; Laffleur, F.; Hofhaus, G.; Fidelj, V.; Tursch, A.; Özbek, S.; Hopke, E. Overcoming the Mucosal Barrier: Tetraether Lipid-Stabilized Liposomal Nanocarriers Decorated with Cell-Penetrating Peptides Enable Oral Delivery of Vancomycin. Adv. Ther. 2021, 4, 2000247. [Google Scholar] [CrossRef]
- Maher, S.; Mrsny, R.J.; Brayden, D.J. Intestinal permeation enhancers for oral peptide delivery. Adv. Drug Deliv. Rev. 2016, 106, 277–319. [Google Scholar] [CrossRef]
- Velpula, A.; Jukanti, R.; Janga, K.Y.; Sunkavalli, S.; Bandari, S.; Kandadi, P.; Veerareddy, P.R. Proliposome powders for enhanced intestinal absorption and bioavailability of raloxifene hydrochloride: Effect of surface charge. Drug Dev. Ind. Pharm. 2013, 39, 1895–1906. [Google Scholar] [CrossRef]
- Babadi, D.; Dadashzadeh, S.; Osouli, M.; Daryabari, M.S.; Haeri, A. Nanoformulation strategies for improving intestinal permeability of drugs: A more precise look at permeability assessment methods and pharmacokinetic properties changes. J. Control. Release 2020, 321, 669–709. [Google Scholar] [CrossRef] [PubMed]
- Kamel, R.; El-Deeb, N.M.; Abbas, H. Development of a potential anti-cancer pulmonary nanosystem consisted of chitosan-doped LeciPlex loaded with resveratrol using a machine learning method. J. Drug Deliv. Sci. Technol. 2022, 70, 103259. [Google Scholar] [CrossRef]
- Date, A.A.; Nagarsenker, M.S.; Patere, S.; Dhawan, V.; Gude, R.P.; Hassan, P.A.; Aswal, V.; Steiniger, F.; Thamm, J.; Fahr, A. Lecithin-Based Novel Cationic Nanocarriers (Leciplex) II: Improving Therapeutic Efficacy of Quercetin on Oral Administration. Mol. Pharm. 2011, 8, 716–726. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Lu, Y.; Qi, J.; Zhu, Q.; Chen, Z.; Wu, W. Adapting liposomes for oral drug delivery. Acta Pharm. Sin. B 2019, 9, 36–48. [Google Scholar] [CrossRef]
- Cuomo, F.; Ceglie, S.; Miguel, M.; Lindman, B.; Lopez, F. Oral delivery of all-trans retinoic acid mediated by liposome carriers. Colloids Surf. B Biointerfaces 2021, 201, 111655. [Google Scholar] [CrossRef]
- Dave, V.S.; Gupta, D.; Yu, M.; Nguyen, P.; Varghese Gupta, S. Current and evolving approaches for improving the oral permeability of BCS Class III or analogous molecules. Drug Dev. Ind. Pharm. 2017, 43, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Romes, N.B.; Wahab, R.A.; Abdul Hamid, M.; Hashim, S.E. D-optimal design-assisted Elaeis guineensis leaves extract in olive oil-sunflower seed nanoemulsions: Development, characterization, and physical stability. J. Dispers. Sci. Technol. 2022, 43, 289–301. [Google Scholar] [CrossRef]
- Bhattacharya, S. Design and development of docetaxel solid Self-Microemulsifying drug delivery system using principal component analysis and D-Optimal design. Asian J. Pharm. 2018, 12, S122–S144. [Google Scholar]
- Hassan, D.H.; Abdelmonem, R.; Abdellatif, M.M. Formulation and Characterization of Carvedilol Leciplex for Glaucoma Treatment: In-Vitro, Ex-Vivo and In-Vivo Study. Pharmaceutics 2018, 10, 197. [Google Scholar] [CrossRef] [Green Version]
- Albash, R.; Abdellatif, M.M.; Hassan, M.; Badawi, N.M. Tailoring Terpesomes and Leciplex for the Effective Ocular Conveyance of Moxifloxacin Hydrochloride (Comparative Assessment): In-vitro, Ex-vivo, and In-vivo Evaluation. Int. J. Nanomed. 2021, 16, 5247–5263. [Google Scholar] [CrossRef]
- Abdellatif, M.M.; Josef, M.; El-Nabarawi, M.A.; Teaima, M. Sertaconazole-Nitrate-Loaded Leciplex for Treating Keratomycosis: Optimization Using D-Optimal Design and In Vitro, Ex Vivo, and In Vivo Studies. Pharmaceutics 2022, 14, 2215. [Google Scholar] [CrossRef] [PubMed]
- Fong, S.Y.K.; Martins, S.M.; Brandl, M.; Bauer-Brandl, A. Solid Phospholipid Dispersions for Oral Delivery of Poorly Soluble Drugs: Investigation Into Celecoxib Incorporation and Solubility-In Vitro Permeability Enhancement. J. Pharm. Sci. 2016, 105, 1113–1123. [Google Scholar] [CrossRef]
- Lavanya, N.; Muzib, Y.I.; Aukunuru, J.; Balekari, U. Preparation and evaluation of a novel oral delivery system for low molecular weight heparin. Int. J. Pharm. Investig. 2016, 6, 148–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allam, R.M.; Al-Abd, A.M.; Khedr, A.; Sharaf, O.A.; Nofal, S.M.; Khalifa, A.E.; Mosli, H.A.; Abdel-Naim, A.B. Fingolimod interrupts the cross talk between estrogen metabolism and sphingolipid metabolism within prostate cancer cells. Toxicol. Lett. 2018, 291, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Prasad, Y.V.R.; Puthli, S.P.; Eaimtrakarn, S.; Ishida, M.; Yoshikawa, Y.; Shibata, N.; Takada, K. Enhanced intestinal absorption of vancomycin with Labrasol and d-α-tocopheryl PEG 1000 succinate in rats. Int. J. Pharm. 2003, 250, 181–190. [Google Scholar] [CrossRef]
- Date, A.A.; Srivastava, D.; Nagarsenker, M.S.; Mulherkar, R.; Panicker, L.; Aswal, V.; Hassan, P.A.; Steiniger, F.; Thamm, J.; Fahr, A. Lecithin-based novel cationic nanocarriers (LeciPlex) I: Fabrication, characterization and evaluation. Nanomedicine 2011, 6, 1309–1325. [Google Scholar] [CrossRef]
- Salama, A.; Badran, M.; Elmowafy, M.; Soliman, G.M. Spironolactone-Loaded LeciPlexes as Potential Topical Delivery Systems for Female Acne: In Vitro Appraisal and Ex Vivo Skin Permeability Studies. Pharmaceutics 2020, 12, 25. [Google Scholar] [CrossRef] [Green Version]
- Eroğlu, İ.; Azizoğlu, E.; Özyazıcı, M.; Nenni, M.; Gürer Orhan, H.; Özbal, S.; Tekmen, I.; Ertam, İ.; Ünal, İ.; Özer, Ö. Effective topical delivery systems for corticosteroids: Dermatological and histological evaluations. Drug Deliv. 2016, 23, 1502–1513. [Google Scholar] [CrossRef]
- Therdphapiyanak, N.; Jaturanpinyo, M.; Waranuch, N.; Kongkaneramit, L.; Sarisuta, N. Development and assessment of tyrosinase inhibitory activity of liposomes of Asparagus racemosus extracts. Asian J. Pharm. Sci. 2013, 8, 134–142. [Google Scholar] [CrossRef] [Green Version]
- Khatoon, K.; Rizwanullah, M.; Amin, S.; Mir, S.R.; Akhter, S. Cilnidipine loaded transfersomes for transdermal application: Formulation optimization, in-vitro and in-vivo study. J. Drug Deliv. Sci. Technol. 2019, 54, 101303. [Google Scholar] [CrossRef]
- Ammar, H.O.; Tadros, M.I.; Salama, N.M.; Ghoneim, A.M. Ethosome-Derived Invasomes as a Potential Transdermal Delivery System for Vardenafil Hydrochloride: Development, Optimization and Application of Physiologically Based Pharmacokinetic Modeling in Adults and Geriatrics. Int. J. Nanomed. 2020, 15, 5671–5685. [Google Scholar] [CrossRef] [PubMed]
- Parmar, J.J.; Singh, D.J.; Hegde, D.D.; Lohade, A.A.; Soni, P.S.; Samad, A.; Menon, M.D. Development and evaluation of inhalational liposomal system of budesonide for better management of asthma. Indian J. Pharm. Sci. 2010, 72, 442–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, S.-j.; Xu, S.; Wang, H.-m.; Ling, Y.; Dong, J.; Xia, R.-d.; Sun, X.-h. Nanoparticles: Oral Delivery for Protein and Peptide Drugs. AAPS PharmSciTech 2019, 20, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beg, S.; Raza, K.; Kumar, R.; Chadha, R.; Katare, O.; Singh, B. Improved intestinal lymphatic drug targeting via phospholipid complex-loaded nanolipospheres of rosuvastatin calcium. RSC Adv. 2016, 6, 8173–8187. [Google Scholar] [CrossRef]
- Uhl, P.; Pantze, S.; Storck, P.; Parmentier, J.; Witzigmann, D.; Hofhaus, G.; Huwyler, J.; Mier, W.; Fricker, G. Oral delivery of vancomycin by tetraether lipid liposomes. Eur. J. Pharm. Sci. 2017, 108, 111–118. [Google Scholar] [CrossRef]
- Lin, Y.-J.; Shatkin, J.A.; Kong, F. Evaluating mucoadhesion properties of three types of nanocellulose in the gastrointestinal tract in vitro and ex vivo. Carbohydr. Polym. 2019, 210, 157–166. [Google Scholar] [CrossRef]
- Sheikholeslami, B.; Lam, N.W.; Dua, K.; Haghi, M. Exploring the impact of physicochemical properties of liposomal formulations on their in vivo fate. Life Sci. 2022, 300, 120574. [Google Scholar] [CrossRef]
- Elnaggar, Y.S.R.; Omran, S.; Hazzah, H.A.; Abdallah, O.Y. Anionic versus cationic bilosomes as oral nanocarriers for enhanced delivery of the hydrophilic drug risedronate. Int. J. Pharm. 2019, 564, 410–425. [Google Scholar] [CrossRef]
- Parmentier, J.; Hartmann, F.J.; Fricker, G. In vitro evaluation of liposomes containing bio-enhancers for the oral delivery of macromolecules. Eur. J. Pharm. Biopharm. 2010, 76, 394–403. [Google Scholar] [CrossRef]
- Yi, X.; Shi, X.; Gao, H. Cellular Uptake of Elastic Nanoparticles. Phys. Rev. Lett. 2011, 107, 098101. [Google Scholar] [CrossRef] [Green Version]
- Xu, A.; Yao, M.; Xu, G.; Ying, J.; Ma, W.; Li, B.; Jin, Y. A physical model for the size-dependent cellular uptake of nanoparticles modified with cationic surfactants. Int. J. Nanomed. 2012, 7, 3547–3554. [Google Scholar] [CrossRef]
- Gossmann, R.; Langer, K.; Mulac, D. New perspective in the formulation and characterization of didodecyldimethylammonium bromide (DMAB) stabilized poly (lactic-co-glycolic acid)(PLGA) nanoparticles. PLoS ONE 2015, 10, e0127532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delon, L.; Gibson, R.J.; Prestidge, C.A.; Thierry, B. Mechanisms of uptake and transport of particulate formula-tions in the small intestine. J. Control. Release 2022, 343, 584–599. [Google Scholar] [CrossRef] [PubMed]
- Bajka, B.H.; Rigby, N.M.; Cross, K.L.; Macierzanka, A.; Mackie, A.R. The influence of small intestinal mucus structure on particle transport ex vivo. Colloids Surf. B Biointerfaces 2015, 135, 73–80. [Google Scholar] [CrossRef]
- Daeihamed, M.; Haeri, A.; Ostad, S.N.; Akhlaghi, M.F.; Dadashzadeh, S. Doxorubicin-loaded lipo-somes: Enhancing the oral bioavailability by modulation of physicochemical characteristics. Nanomedicine 2017, 12, 1187–1202. [Google Scholar] [CrossRef]
- Sheue Nee Ling, S.; Magosso, E.; Abdul Karim Khan, N.; Hay Yuen, K.; Anne Barker, S. Enhanced Oral Bioavailability and Intestinal Lymphatic Transport of a Hydrophilic Drug Using Liposomes. Drug Dev. Ind. Pharm. 2006, 32, 335–345. [Google Scholar] [CrossRef]
- Palassi, S.; Valizadeh, H.; Allahyari, S.; Zakeri-Milani, P. Preparation and In Vitro Characterization of Enoxaparin Nano-liposomes through Different Methods. Adv. Pharm. Bull. 2021, 11, 295–300. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Independent Variables for LPX Design | Levels | |
|---|---|---|
| Low | High | |
| X1: lipid molar ratio | 0 | 1 |
| X2: cationic surfactant molar ratio | 1 | 5 |
| X3: cationic surfactant type | SPC | PL-90 G |
| X4: lipid type | CTAB | DDAB |
| Formulation Code | Lipid Molar Ratio (X1) | Surfactant Molar Ratio (X2) | Surfactant Type (X3) | Lipid Type (X4) | E.E.% (Y1) | P.S. (nm) (Y2) | P.I. (Y3) | Z.P. (mV) (Y4) | Jss (µg/cm2/h) (Y5) |
|---|---|---|---|---|---|---|---|---|---|
| F1 | 5 | 0 | DDAB | SPC | 99.33 ± 1.5 | 194.5 ± 1.75 | 0.26 ± 0.008 | −64.4 ± 0.55 | 36.05 ± 0.49 |
| F2 | 2 | 0.5 | DDAB | PG90 | 81.6 ± 1.8 | 28.32 ± 1.15 | 0.29 ± 0.002 | 57.1 ± 1.1 | 166.23 ± 0.55 |
| F3 | 1 | 1 | DDAB | PG90 | 75.6 ± 1.2 | 18.51 ± 0.77 | 0.19 ± 0.005 | 64.9 ± 0.45 | 187.41 ± 1.20 |
| F4 | 5 | 1 | CTAB | PG90 | 94.0 ± 1.5 | 156.0 ± 1.59 | 0.40 ± 0.025 | 55.9 ± 1.35 | 102.61 ± 0.42 |
| F5 | 3 | 0 | CTAB | SPC | 93.3 ± 1.1 | 96.48 ± 1.24 | 0.26 ± 0.005 | −52.9 ± 0.89 | 136.08 ± 0.05 |
| F6 | 5 | 1 | DDAB | PG90 | 90.9 ± 0.78 | 147.0 ± 0.94 | 0.23 ± 0.043 | 52.3 ± 0.96 | 156.0 ± 0.96 |
| F7 | 3 | 0.5 | DDAB | SPC | 94.4 ± 0.60 | 68.47 ± 0.85 | 0.24 ± 0.010 | −29.3 ± 0.35 | 102.15 ± 1.10 |
| F8 | 3 | 0 | DDAB | PG90 | 88.2 ± 1.0 | 103.0 ± 0.55 | 0.51 ± 0.020 | 10.5 ± 1.20 | 113.28 ± 1.19 |
| F9 | 5 | 0 | DDAB | SPC | 96.99 ± 1.7 | 189.3 ± 1.75 | 0.24 ± 0.018 | −66.8 ± 0.41 | 37.72 ± 0.89 |
| F10 | 1 | 0 | CTAB | PG90 | 92.0 ± 1.7 | 102.2 ± 2.1 | 0.31 ± 0.028 | 19.8 ± 0.25 | 142.98 ± 0.69 |
| F11 | 1 | 1 | CTAB | SPC | 91.2 ± 1.4 | 49.17 ± 0.28 | 0.28 ± 0.018 | 33.6 ± 0.30 | 178.44 ± 0.31 |
| F12 | 5 | 1 | CTAB | SPC | 96.8 ± 1.41 | 121.2 ± 0.66 | 0.26 ± 0.051 | −33.2 ± 1.4 | 141.25 ± 0.17 |
| F13 | 1 | 1 | DDAB | SPC | 91.9 ± 0.81 | 73.85 ± 1.35 | 0.33 ± 0.030 | 42.3 ± 0.95 | 133.72 ± 0.5 |
| F14 | 1 | 0 | DDAB | SPC | 95.6 ± 1.3 | 148.4 ± 0.22 | 0.26 ± 0.002 | −49.4 ± 0.6 | 58.37 ± 0.26 |
| F15 | 2 | 0.5 | CTAB | SPC | 95.0 ± 1.1 | 83.54 ± 1.23 | 0.34 ± 0.008 | 22.1 ± 1.05 | 160.69 ± 0.48 |
| F16 | 5 | 0 | CTAB | PG90 | 93.4 ± 1.2 | 162.4 ± 1.30 | 0.41 ± 0.046 | 3.05 ± 0.19 | 76.63 ± 0.45 |
| F17 | 1 | 0 | CTAB | PG90 | 89.6 ± 1.9 | 102.2 ± 1.69 | 0.31 ± 0.022 | 19.8 ± 0.15 | 142.98 ± 0.69 |
| F18 | 3 | 0.5 | CTAB | PG90 | 87.2 ± 1.3 | 61.7 ± 1.35 | 0.38 ± 0.005 | 38.2 ± 0.65 | 149.63 ± 0.43 |
| F19 | 5 | 1 | DDAB | PG90 | 91.4 ± 1.9 | 140 ± 1.52 | 0.35 ± 0.001 | 50.6 ± 0.25 | 161.24 ± 0.16 |
| Source | E.E.% (Y1) | P.S. (nm) (Y2) | P.I. (Y3) | Z.P. (mV) (Y4) | Jss (µg/cm2/h) (Y5) |
|---|---|---|---|---|---|
| p-value | <0.0001 | 0.0016 | 0.1982 | <0.0001 | <0.0001 |
| Model | 2 FI | Quadratic | - | Linear | Quadratic |
| X1 = A = Lipid molar ratio | <0.0001 | 0.0004 | 0.0962 | 0.0008 | <0.0001 |
| X2 = B = surfactant molar ratio | 0.0381 | 0.0043 | 0.2669 | <0.0001 | <0.0001 |
| X3 = C = surfactant type | 0.3271 | 0.8949 | 0.9691 | 0.5983 | <0.0001 |
| X4 = D = Lipid type | <0.0001 | 0.1166 | 0.1360 | <0.0001 | <0.0001 |
| Adequate precision R2 | 19.724 0.9636 | 10.43 0.84 | 4.53 0.33 | 19.791 0.9132 | 42.568 0.9969 |
| Adjusted R2 | 0.9226 | 0.76 | 0.14 | 0.8884 | 0.9907 |
| Predicted R2 | 0.8496 | 0.6 | −0.26 | 0.8332 | 0.9044 |
| Significant factors | X1, X2, X4 | X1, X2 | - | X1, X2, X4 | X1, X2, X3, X4 |
| Predicted value of the selected formula | 87.38 | 50.98 | 0.22 | 65.1 | 178.41 |
| Observed value of the selected formula | 85.20 ± 0.95 | 52.74 ± 0.91 | 0.21 ± 0.02 | 60.8 ± 1.75 | 175.03 ± 1.68 |
| The regression equation of the fitted model | +91.50 + 3.47\1 A−1.23 × B + 0.44 × C−3.84 × D + 1.92 × A\1 B−1.48\1 A\1 C + 1.34\1 A\1 D + 0.27\1 B\1 C 0.95\1 B\1 D + 1.61 × C × D | +106.80 + 43.44\1 A−21.6\1 B−2.16\1 C−6.84\1 D | - | +9.3017.03\1 A + 29.73\1 B + 1.67 × C + 28.76 × D | +126.49−20.74\1 A + 23.12\1 B + 8.84 \1 C + 7.92\1 D + 1.58\1 A\1 B6.16\1 A\1 C−5.24\1 A\1 D−12.03\1 B \1 C−3.62\1 B\1 D−20.32 1 C\1 D |
| Intestinal Permeability Parameters | LPX | VAN Solution |
|---|---|---|
| The total amount of VAN permeated per unit area after 8 h (μg/cm2) | 404.62 ± 1.95 | 178.74 ± 1.75 |
| Apparent permeability (cm/h) | 0.2240 ± 0.0007 | 0.00970 ± 0.0004 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdellatif, M.M.; Ahmed, S.M.; El-Nabarawi, M.A.; Teaima, M. Oral Bioavailability Enhancement of Vancomycin Hydrochloride with Cationic Nanocarrier (Leciplex): Optimization, In Vitro, Ex Vivo, and In Vivo Studies. Sci. Pharm. 2023, 91, 1. https://doi.org/10.3390/scipharm91010001
Abdellatif MM, Ahmed SM, El-Nabarawi MA, Teaima M. Oral Bioavailability Enhancement of Vancomycin Hydrochloride with Cationic Nanocarrier (Leciplex): Optimization, In Vitro, Ex Vivo, and In Vivo Studies. Scientia Pharmaceutica. 2023; 91(1):1. https://doi.org/10.3390/scipharm91010001
Chicago/Turabian StyleAbdellatif, Menna M., Sara Mohamed Ahmed, Mohamed A. El-Nabarawi, and Mahmoud Teaima. 2023. "Oral Bioavailability Enhancement of Vancomycin Hydrochloride with Cationic Nanocarrier (Leciplex): Optimization, In Vitro, Ex Vivo, and In Vivo Studies" Scientia Pharmaceutica 91, no. 1: 1. https://doi.org/10.3390/scipharm91010001
APA StyleAbdellatif, M. M., Ahmed, S. M., El-Nabarawi, M. A., & Teaima, M. (2023). Oral Bioavailability Enhancement of Vancomycin Hydrochloride with Cationic Nanocarrier (Leciplex): Optimization, In Vitro, Ex Vivo, and In Vivo Studies. Scientia Pharmaceutica, 91(1), 1. https://doi.org/10.3390/scipharm91010001