
2D-QSAR and CoMFA Models for Antitubercular Activity of Scalarane-Type Sesterterpenes

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. General Procedures
2.2. Chemical Derivatizations
2.3. Antitubercular Activity
2.4. Compound Dataset
2.5. QSAR
2.5.1. 2D-QSAR Model
2.5.2. CoMFA Model
3. Results and Discussion
3.1. Derivatization of the Scalaranes
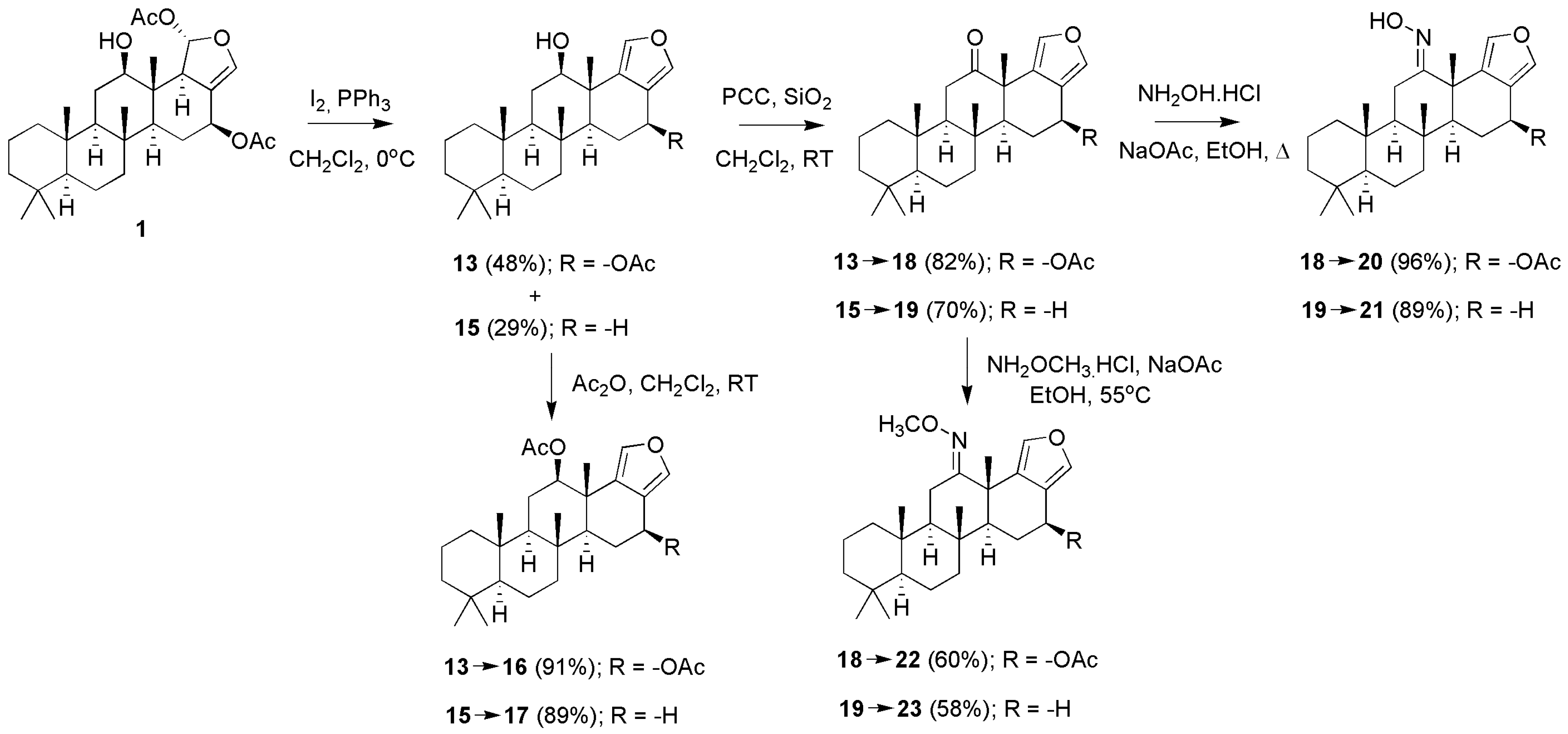
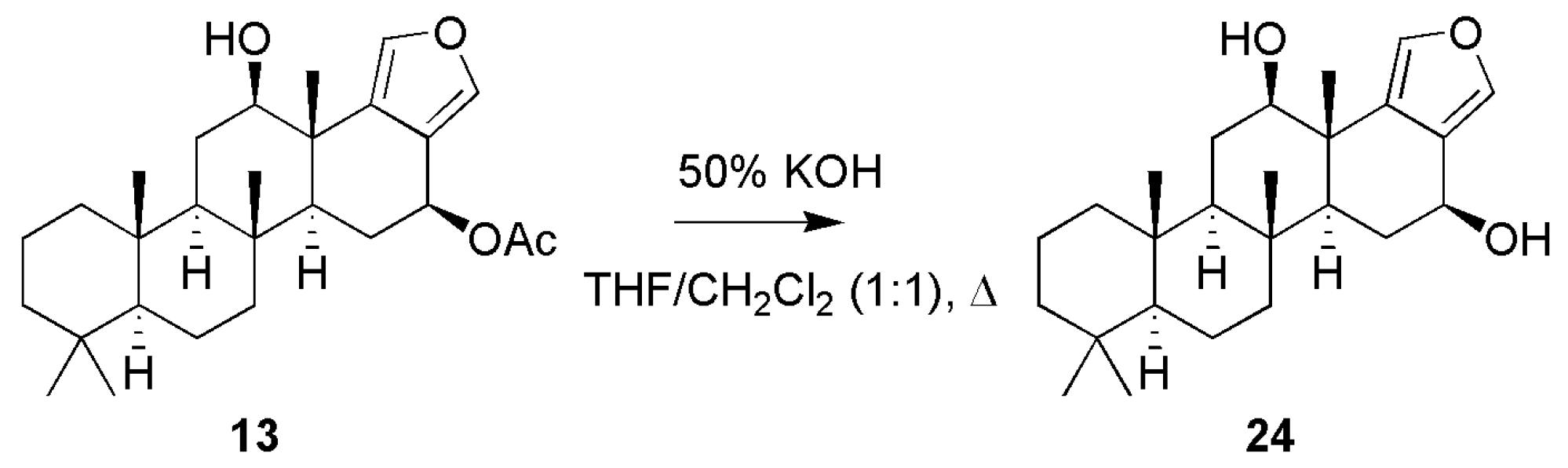
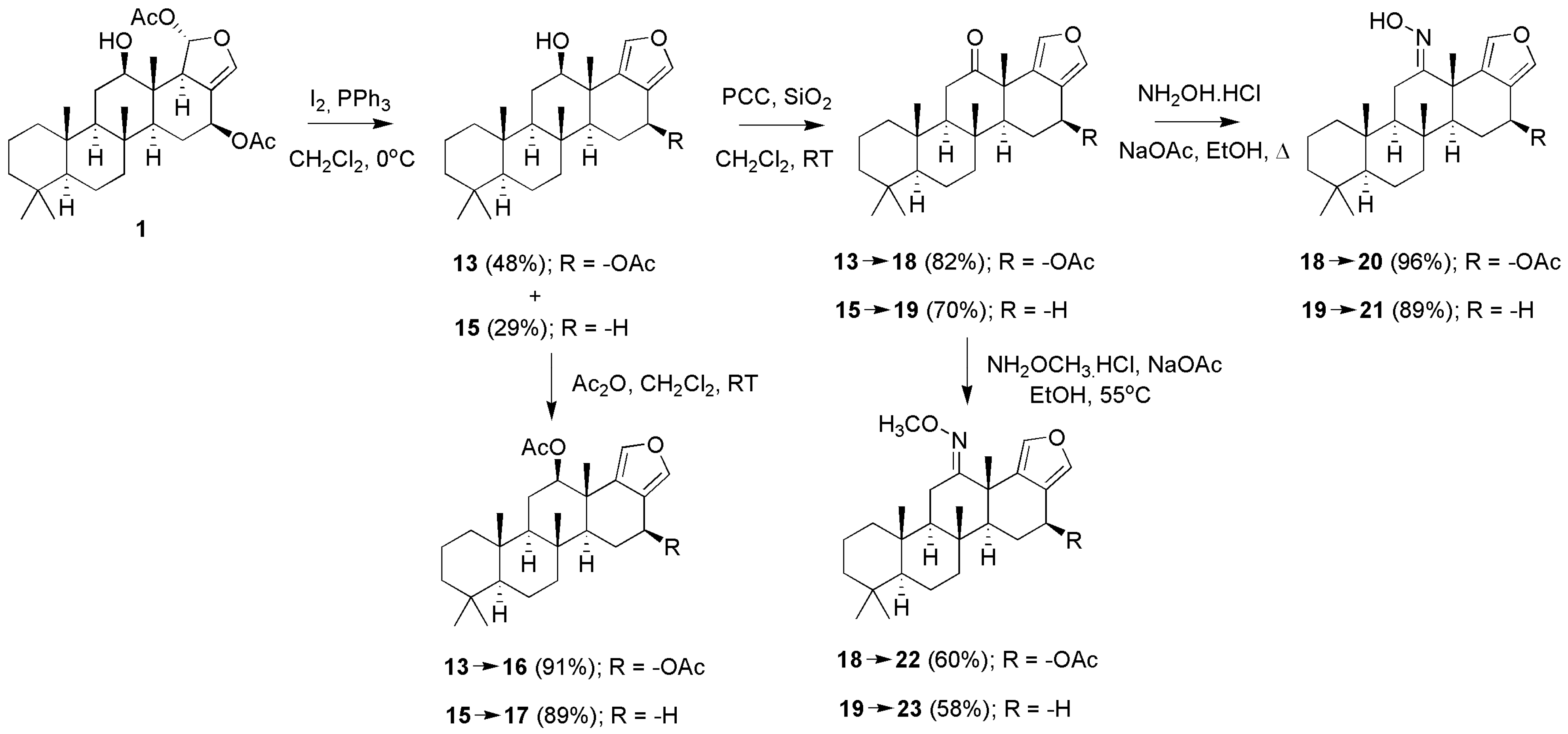
3.1.1. Scalarafuran Series
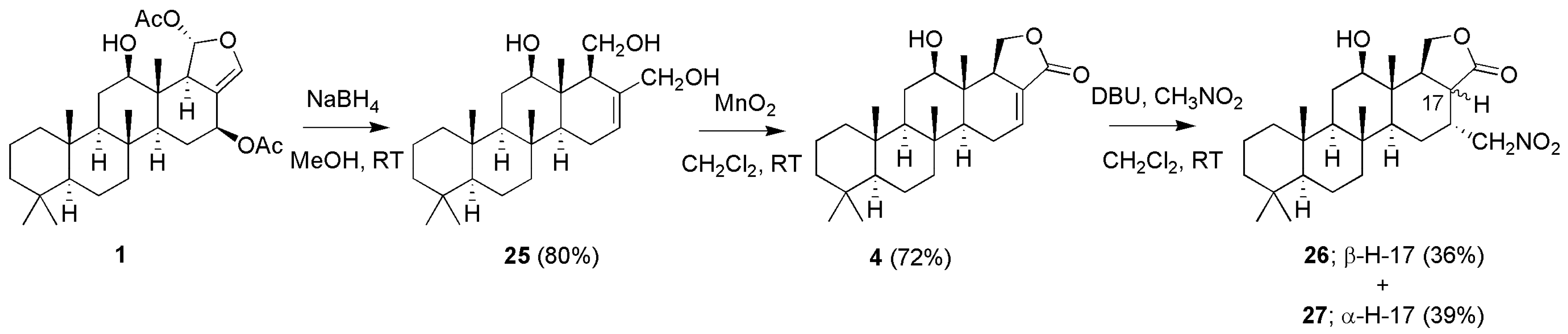
3.1.2. Deoxyscalarin Series
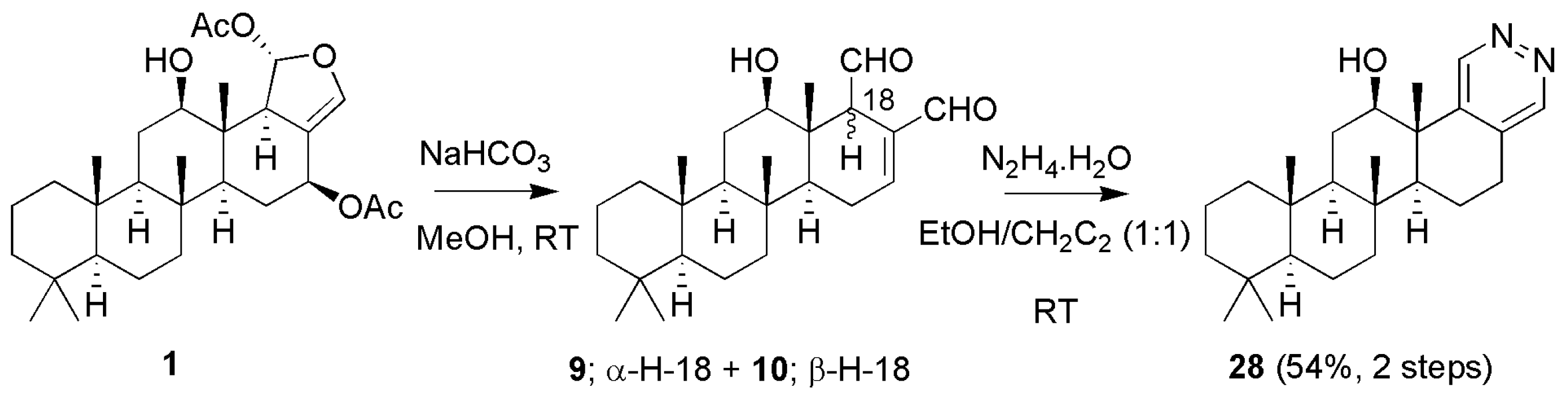
3.1.3. Scalaradials and Scalarapyridazine
3.2. Determination of Antitubercular Activity
3.3. Structure Activity Relationship and Computer Modeling
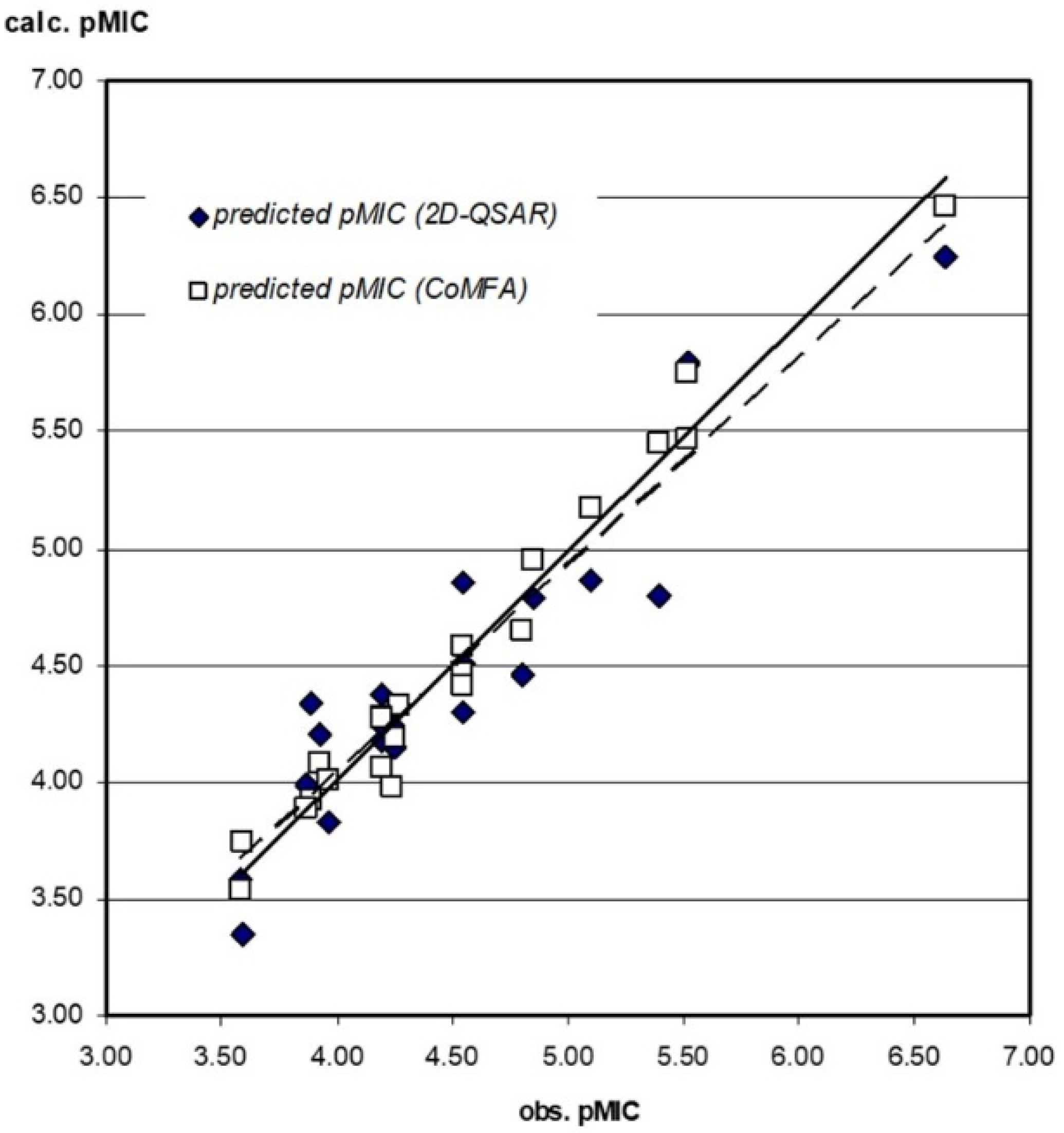
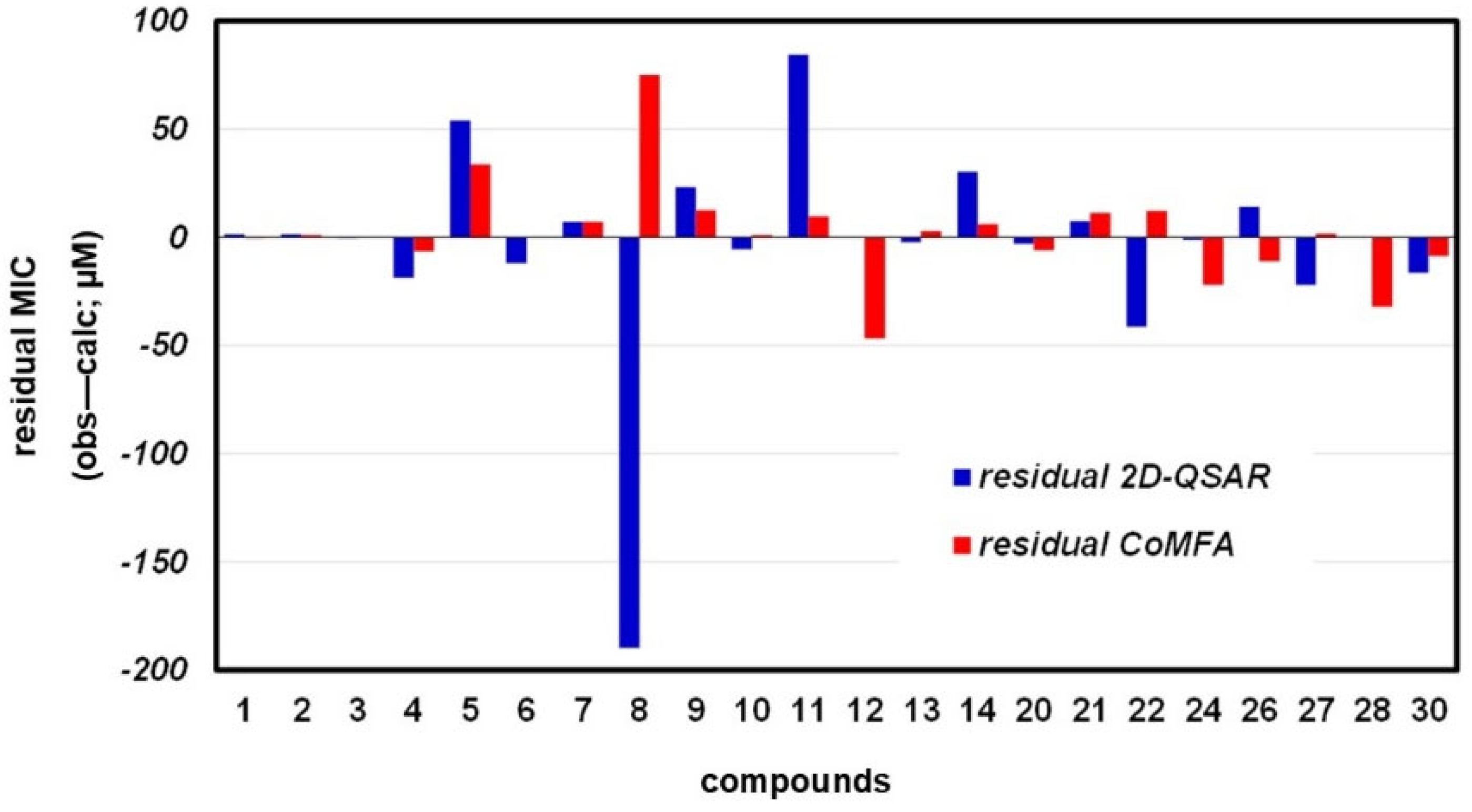
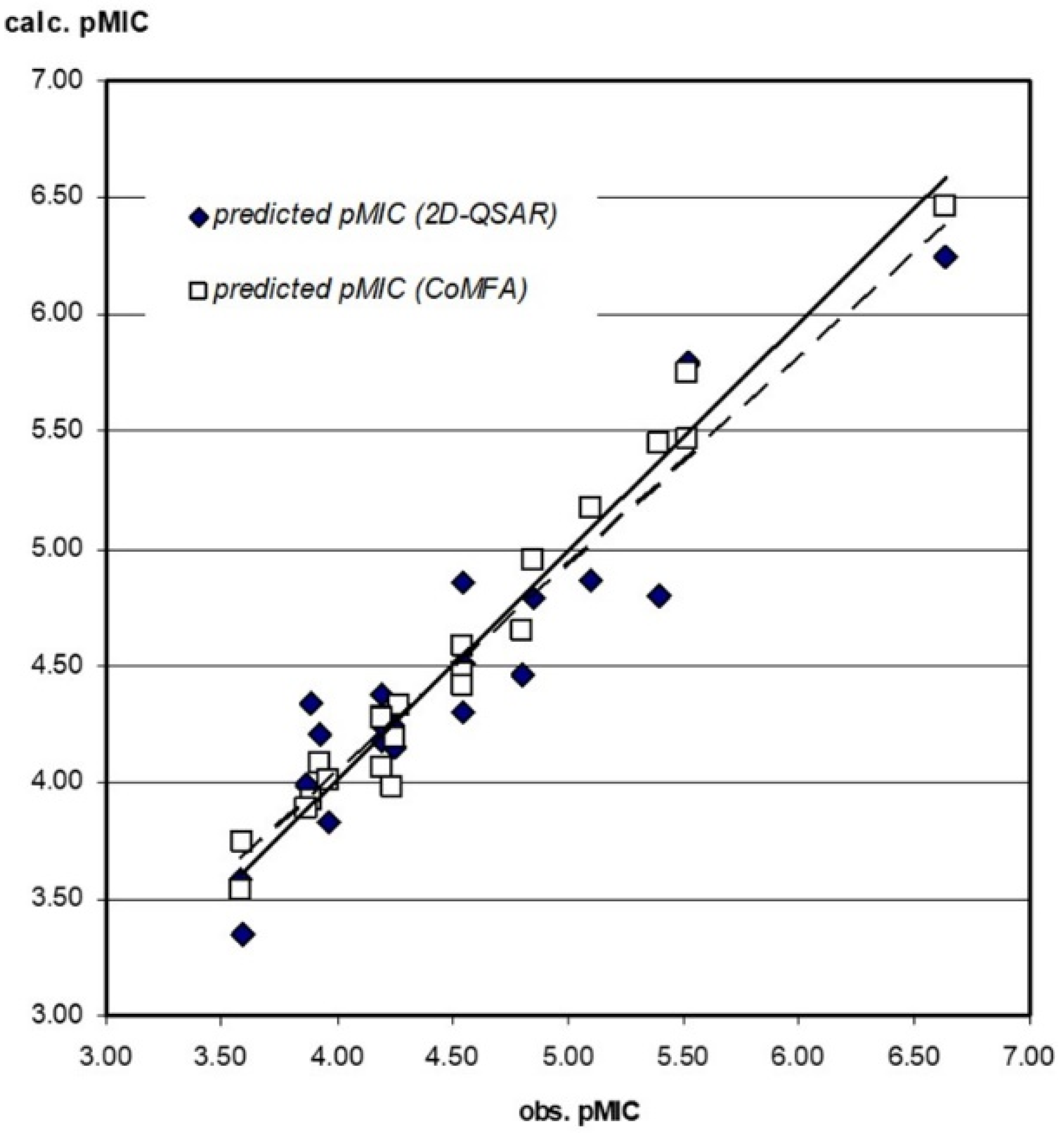
3.3.1. 2D-QSAR
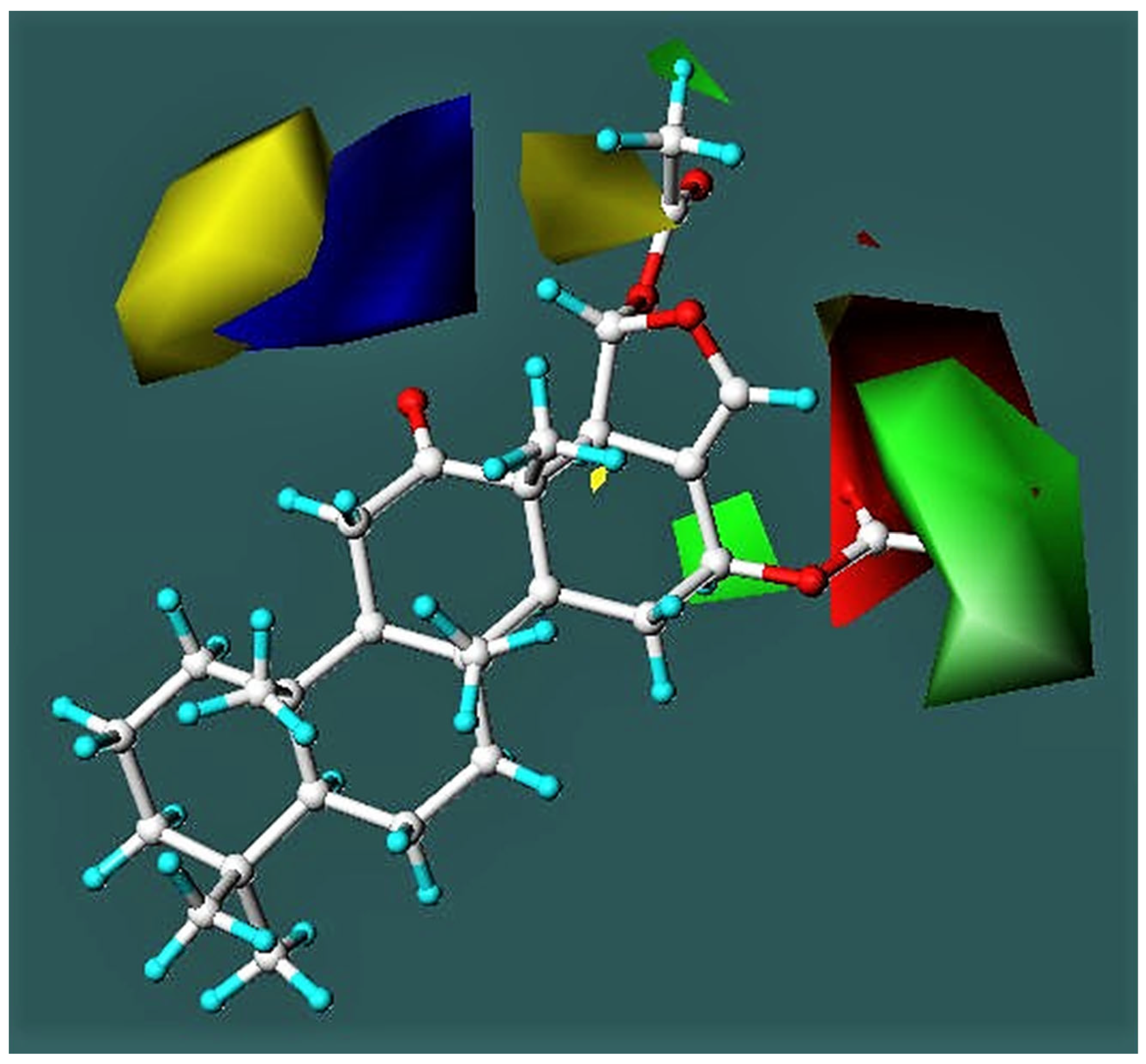
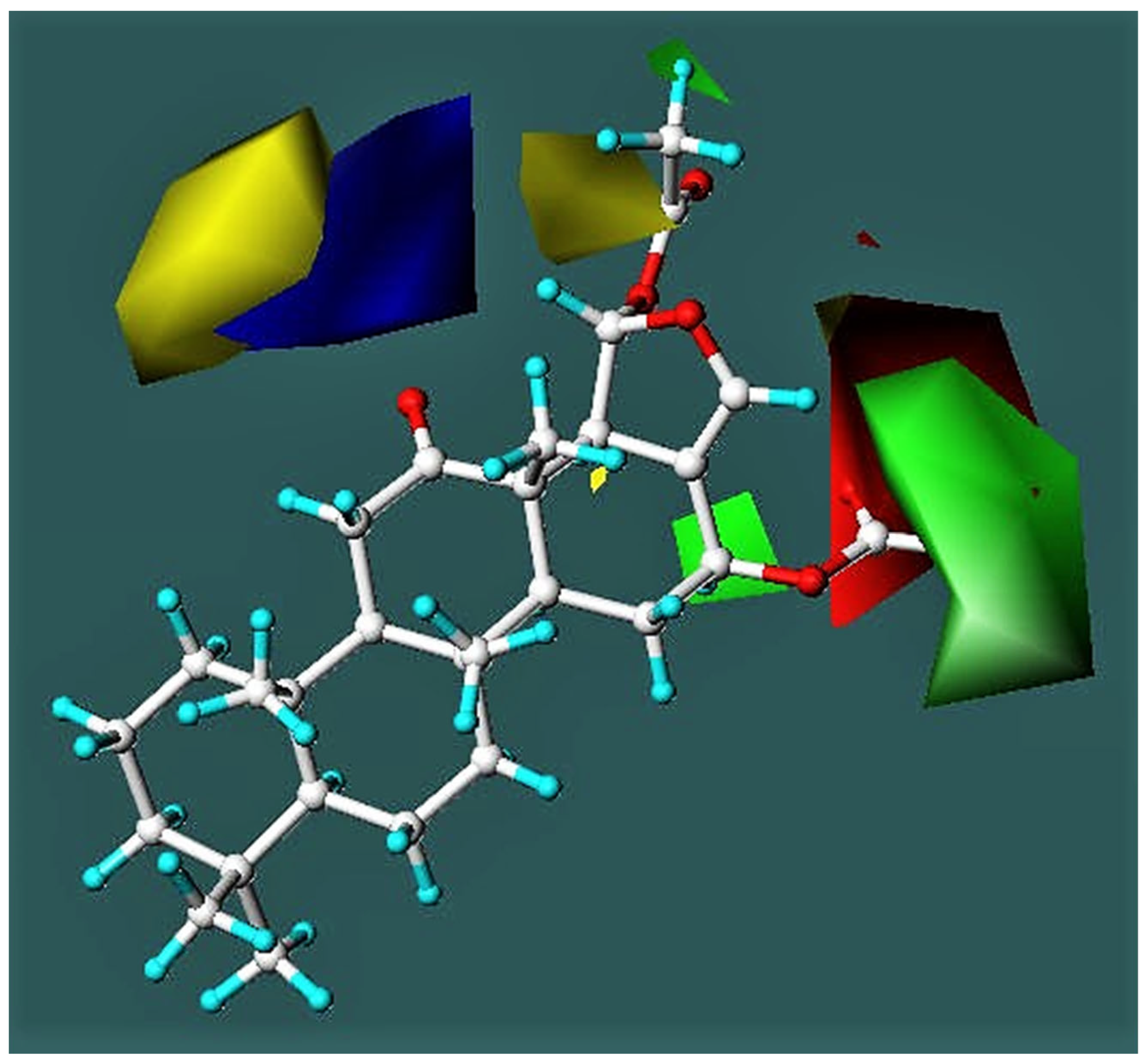
3.3.2. CoMFA Model
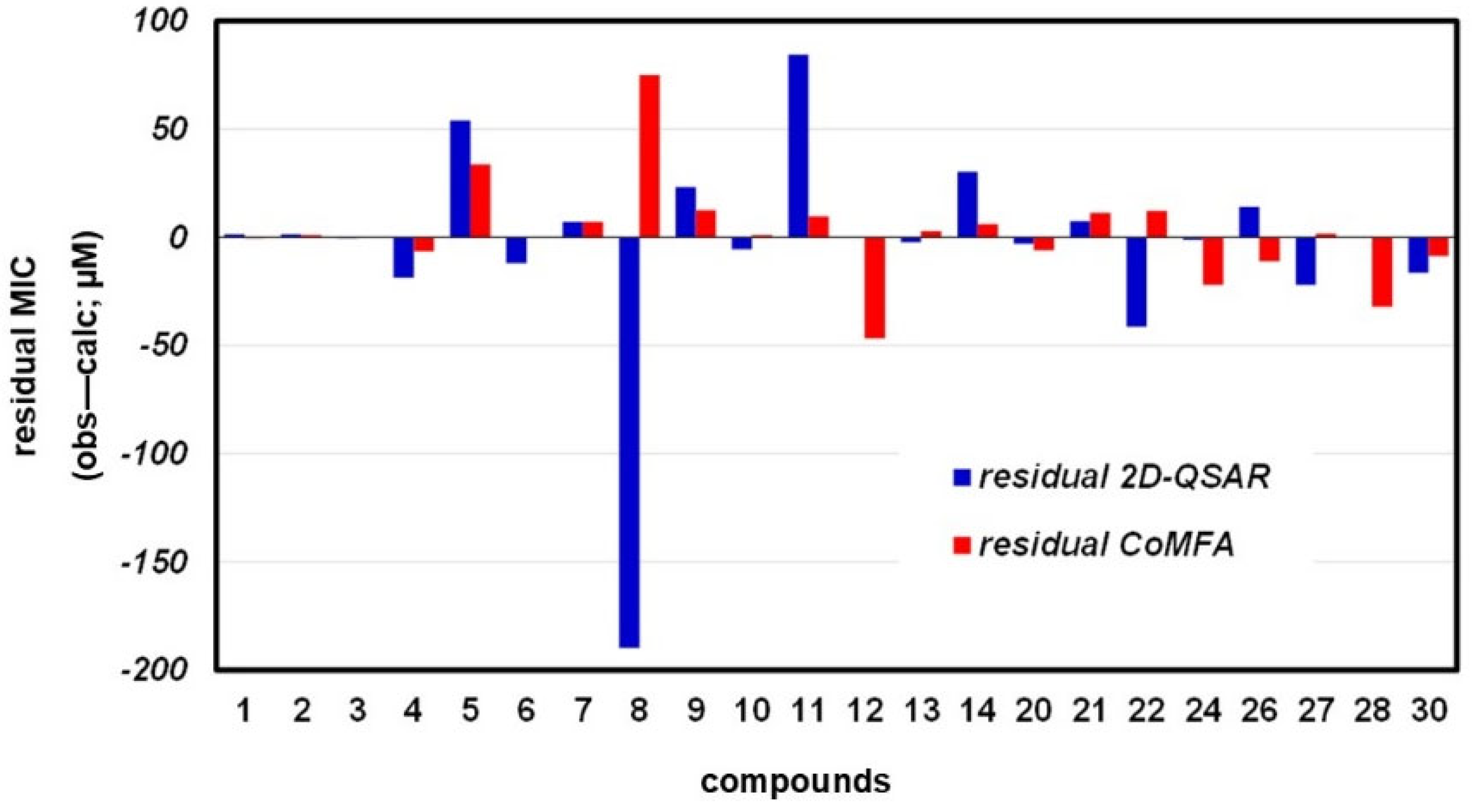
3.3.3. Compound Test Set
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2021; World Health Organization: Geneva, Switzerland, 2021; pp. 4–10.
- Wonganuchitmeta, S.; Yuenyongsawad, S.; Keawpradub, N.; Plubrukarn, A. Antitubercular sesterterpenes from the Thai sponge Brachiaster sp. J. Nat. Prod. 2004, 67, 1767–1770. [Google Scholar] [CrossRef] [PubMed]
- Jaisamut, S.; Thengyai, S.; Yuenyongsawad, S.; Karalai, C.; Plubrukarn, A.; Suwanborirux, K. Structure-activity relationships of antitubercular scalaranes: Heteronemin revisited. Pure Appl. Chem. 2009, 81, 1019–1026. [Google Scholar] [CrossRef]
- Kazlauskas, R.; Murphy, P.T.; Quinn, R.J.; Wells, R.J. Heteronemin, a new scalarin type sesterterpene from the sponge Heteronema erecta. Tetrahedron Lett. 1976, 17, 2631–2634. [Google Scholar] [CrossRef]
- Yashman, Y.; Rudi, A. The 13C-NMR spectrum and stereochemistry of heteronemin. Tetrahedron 1977, 33, 2997–2998. [Google Scholar] [CrossRef]
- Karuso, P.; Cambie, R.C.; Bowden, B.F.; Bergquist, P.R. Chemistry of sponges, VI. Scalarane sesterterpenes from Hyatella intestinalis. J. Nat. Prod. 1989, 52, 289–293. [Google Scholar] [CrossRef]
- Crews, P.; Bescansa, P. Sesterterpenes from a common marine sponge, Hyrtios erecta. J. Nat. Prod. 1986, 49, 1041–1052. [Google Scholar] [CrossRef]
- Kobayashi, M.; Okamoto, T.; Hayashi, K.; Yokoyama, N.; Sasaki, T.; Kitagawa, I. Marine natural products. XXXII. Absolute configurations of C-4 of the manoalide family, biologically active sesterterpenes from the marine sponge Hyrtios erecta. Chem. Pharm. Bull. 1994, 42, 265–270. [Google Scholar] [CrossRef]
- Ledroit, V.; Debitus, C.; Ausseil, F.; Raux, R.; Menou, J.-L.; Hill, B. Heteronemin as a protein farnesyl transferase inhibitor. Pharm. Biol. 2004, 42, 454–456. [Google Scholar] [CrossRef]
- Kamel, H.N.; Kim, Y.B.; Rimoldi, J.M.; Fronczek, F.R.; Ferreira, D.; Slattery, M. Scalarane sesterterpenoids: Semisynthesis and biological activity. J. Nat. Prod. 2009, 72, 1492–1496. [Google Scholar] [CrossRef]
- Fontana, A.; Cavaliere, P.; Ungur, N.; D’Souza, L.; Parameswaram, P.S.; Cimino, G. New scaralanes from the nudibranch Glossodoris atromarginata and its sponge prey. J. Nat. Prod. 1999, 62, 1367–1370. [Google Scholar] [CrossRef]
- El Sayed, K.A.; Bartyzel, P.; Shen, X.; Perry, T.L.; Zjawiony, J.K.; Hamann, M.T. Marine natural products as antituberculosis agents. Tetrahedron 2000, 56, 949–953. [Google Scholar] [CrossRef]
- Thengyai, S.; Maitarat, P.; Hannongbua, S.; Suwanborirux, K.; Plubrukarn, A. Probing the structural requirements for antitubercular activity of scalarane derivatives: 2D-QSAR and CoMFA approaches. Monatsh. Chem. 2009, 141, 621–629. [Google Scholar] [CrossRef]
- Changsen, C.; Franzblau, S.G.; Palittapongarnpim, P. Improved green fluorescent protein reporter gene-based microplate screening for antituberculosis compounds by utilizing and acetamidase promoter. Antimicrob. Agents Chemother. 2003, 47, 3682–3687. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.A.; Franzblau, S.G. Microplate Alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob. Agents Chemother. 1997, 41, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Gavagnin, M.; Mollo, E.; Docimo, T.; Guo, Y.-W.; Cimino, G. Scalarane metabolites of the nudibranch Glossodoris rufomarginata and its dietary sponge from the South China Sea. J. Nat. Prod. 2004, 67, 2104–2107. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Manzaneda, E.J.; Chahboun, R.; Cabrera Torres, E.; Alvarez, E.; Alvarez-Manzaneda, R.; Haidour, A.; Ramos, J. Triphenylphosphine-iodine: An effective reagent for the regioselective dehydration of tertiary alcohols. Tetrahedron Lett. 2004, 45, 4453–4455. [Google Scholar] [CrossRef]
- Alvarez-Manzaneda, E.J.; Chahboun, R.; Cabrera Torres, E.; Alvarez, E.; Alvarez-Manzaneda, R.; Haidour, A.; Ramos, J. Synthesis of alkenes from tertiary esters utilizing the triphenylphosphine-iodine system. Tetrahedron Lett. 2005, 46, 1075–1077. [Google Scholar] [CrossRef]
- Doi, Y.; Shigemori, H.; Ishibashi, M.; Mizobe, F.; Kawashia, A.; Nakaike, S.; Kobayashi, J. New sesterterpenes with nerve growth factor systhesis-stimulating activity from the Okinawan marine sponge Hyrtios sp. Chem. Pharm. Bull. 1993, 41, 2190–2191. [Google Scholar] [CrossRef]
- Tsukamoto, S.; Miura, S.; Van Soest, R.W.M.; Ohta, T. Three new cytotoxic sesterterpenes from a marine sponge Spongia sp. J. Nat. Prod. 2003, 66, 438–440. [Google Scholar] [CrossRef]
- Cui, J.-G.; Fan, L.; Huang, L.-L.; Liu, H.-L.; Zhou, A.-M. Synthesis and evaluation of some steroidal oximes as cytotoxic agents: Structure/activity study (I). Steroids 2009, 74, 62–72. [Google Scholar] [CrossRef]
- Hawkes, G.E.; Herwig, K.; Roberts, J.D. Nuclear magnetic resonance spectroscopy. Use of 13C spectra to establish configurations of oximes. J. Org. Chem. 1974, 39, 1017–1028. [Google Scholar] [CrossRef]
- Bunnel, C.A.; Fuchs, P.L. Rapid and unequivocal determination of syn-anti stereochemistry for toluenesulfonylhydrazones and other imine derivatives via carbon-13 nuclear magnetic resonance spectroscopy. A synthetic adjunct. J. Org. Chem. 1977, 42, 2614–2617. [Google Scholar] [CrossRef]
- Pettit, G.R.; Cichacz, Z.A.; Tan, R.; Herald, D.L.; Melody, N.; Hoard, M.S.; Doubek, D.L.; Hooper, J.N.A. Antineoplastic agents 385. The isolation and structure of a scalarane-type sesterterpene from the Indian Ocean porifera Hyrtios erecta. Collect. Czech. Chem. Commum. 1998, 63, 1671–1677. [Google Scholar] [CrossRef]
- Furuichi, N.; Hata, T.; Soetjipto, H.; Kato, M.; Katsumura, S. Common synthetic strategy for optically active cyclic terpenoids having a 1,1,5-trimethyl-trans-decalin nucleus: Syntheses of (+)-acuminolide, (−)-spongianolide A, and (+)-scalarenedial. Tetrahedron 2001, 57, 8425–8442. [Google Scholar] [CrossRef]
- Rosso, G.B.; Pilli, R.A. Diastereoselective addition of nitro compounds to α,β-unsaturated γ-butyrolactones. Tetrahedron Lett. 2006, 47, 185–188. [Google Scholar] [CrossRef]
- Ismail, K.A.; El-Tombary, A.A.; AboulWafa, O.M.; Omar, A.M.E.; El-Rewini, S.H. Novel steroidal 1,4-diketones and pyridazine derivatives as potential antiestrogens. Arch. Pharm. Pharm. Med. Chem. 1996, 329, 433–437. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | MIC (µM) a | Compounds | MIC (µM) a | ||||||
|---|---|---|---|---|---|---|---|---|---|
| obs. | calc. | obs. | calc. | ||||||
| 2D-QSAR | CoMFA | 2S-QSAR | CoMFA | ||||||
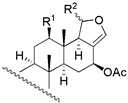 | 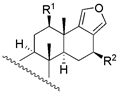 | ||||||||
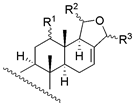
| 1 | R1 = −OH; R2 = α − OAC | 3 | 1.6 | 3.4 | 13 | R1 = −OH; R2 = −OAC | 14 | 16.2 | 11.2 |
| 2 | R1 = −OAc; R2 = α − OAC | 3 | 1.6 | 1.8 | 14 | R1 = −OH; Δ15 | 135 | 104.7 | 128.8 |
| 3 | R1 = =O; R2 = α − OAC | 0.2 | 0.6 | 0.4 | 15 c | R1 = −OH; R2 = −H | 270 | - | - |
| 29 b | R1 = −OAc; R2 = β − OAC | 12 | 5.5 | 13.1 | 16 c | R1 = −OAc; R2 = −OAC | NA | - | - |
 | 17 c | R1 = −OAc; R2 = −H | 970 | - | - | ||||
| 24 | R1 = −OH; R2 = −OH | 65 | 66.1 | 87.1 | |||||
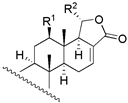
 | |||||||||
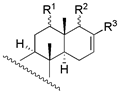
| 4 | R1 = −OH; R2 = −H | 16 | 34.7 | 22.4 | |||||
| 5 | R1 = −OAc; R2 = −H | 117 | 63.1 | 83.2 | |||||
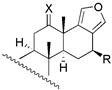
| 6 | R1 = −H; R2 = −OAC | 4 | 15.8 | 3.6 | 18 c | X = O; R = −OAc | 235 | - | - |
 | 19 c | X = O; R = −H | 272 | - | - | ||||
| 20 | X = E − NOH; R = −OAc | 28 | 30.9 | 33.9 | |||||
| 21 | X = E − NOH; R = −H | 65 | 57.5 | 53.7 | |||||
| 22 | X = E − NOCH3; R = −OAc | 110 | 151.4 | 97.7 | |||||
| 23 b | X = E − NOCH3; R = −H | 126 | 228.1 | 266.0 | |||||
| 7 | R1 = β − OH; R2 = α − OAC; R3 = β − OCH3 | 54 | 46.8 | 46.8 | 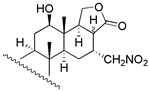 | ||||
| 8 | R1 = β − OH; R2 = α − OH; R3 = −H | 257 | 446.7 | 182.0 | |||||
| 30 | R1 = β − OH; R2 = β − OCH3; R3 = α − OCH3 | 56 | 72.4 | 64.6 | |||||
| 31 c | R1 = α − OAc; R2 = β − OCH3; R3 = α − OCH3 | NA | - | - | |||||
 | 26 | β − H − 17 | 28 | 13.8 | 38.9 | ||||
| 27 | α − H − 17 | 28 | 50.1 | 26.3 | |||||
| 28 | 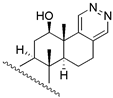 | 263 | 263.0 | 295.1 | |||||
| 9 | R1 = β − OH; R2 = β − CHO; R3 = −CHO | 65 | 41.7 | 52.5 | |||||
| 10 | R1 = β − OH; R2 = α − CHO; R3 = −CHO | 8 | 13.5 | 6.8 | |||||
| 11 | R1 = =O; R2 = β − CHO; R3 = −CHO | 130 | 45.7 | 120.2 | 32 c | 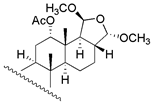 | 210 | - | - |
| 12 | R1 = β − OAc; R2 = β − CHO; R3 = −CHO | 58 | 57.7 | 104.7 | |||||
| 25 c | R1 = β − OH; R2 = β − CH2OH; R3 =−CH2OH | NA | - | - | |||||
| 33 c | R1 = α − OAc; R2 = β − CHO; R3 = −CHO | NA | - | - | |||||
| Enb | vsufID2 | vsurfCW3 | PEOEVSA+1 | |
|---|---|---|---|---|
| Enb | 1 | |||
| vsufID2 | −0.2354 | 1 | ||
| vsurfCW3 | −0.2885 | 0.6236 | 1 | |
| PEOEVSA+1 | 0.4329 | −0.6096 | −0.3434 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thengyai, S.; Guo, Y.; Suwanborirux, K.; Berner, H.; Spreitzer, H.; Wolschann, P.; Hannongbua, S.; Plubrukarn, A. 2D-QSAR and CoMFA Models for Antitubercular Activity of Scalarane-Type Sesterterpenes. Sci. Pharm. 2022, 90, 47. https://doi.org/10.3390/scipharm90030047
Thengyai S, Guo Y, Suwanborirux K, Berner H, Spreitzer H, Wolschann P, Hannongbua S, Plubrukarn A. 2D-QSAR and CoMFA Models for Antitubercular Activity of Scalarane-Type Sesterterpenes. Scientia Pharmaceutica. 2022; 90(3):47. https://doi.org/10.3390/scipharm90030047
Chicago/Turabian StyleThengyai, Suriyan, Yuewei Guo, Khanit Suwanborirux, Heinz Berner, Helmut Spreitzer, Peter Wolschann, Supa Hannongbua, and Anuchit Plubrukarn. 2022. "2D-QSAR and CoMFA Models for Antitubercular Activity of Scalarane-Type Sesterterpenes" Scientia Pharmaceutica 90, no. 3: 47. https://doi.org/10.3390/scipharm90030047
APA StyleThengyai, S., Guo, Y., Suwanborirux, K., Berner, H., Spreitzer, H., Wolschann, P., Hannongbua, S., & Plubrukarn, A. (2022). 2D-QSAR and CoMFA Models for Antitubercular Activity of Scalarane-Type Sesterterpenes. Scientia Pharmaceutica, 90(3), 47. https://doi.org/10.3390/scipharm90030047