
Optimized Methods for Analytical and Functional Comparison of Biosimilar mAb Drugs: A Case Study for Avastin, Mvasi, and Zirabev

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Intact Protein Analysis
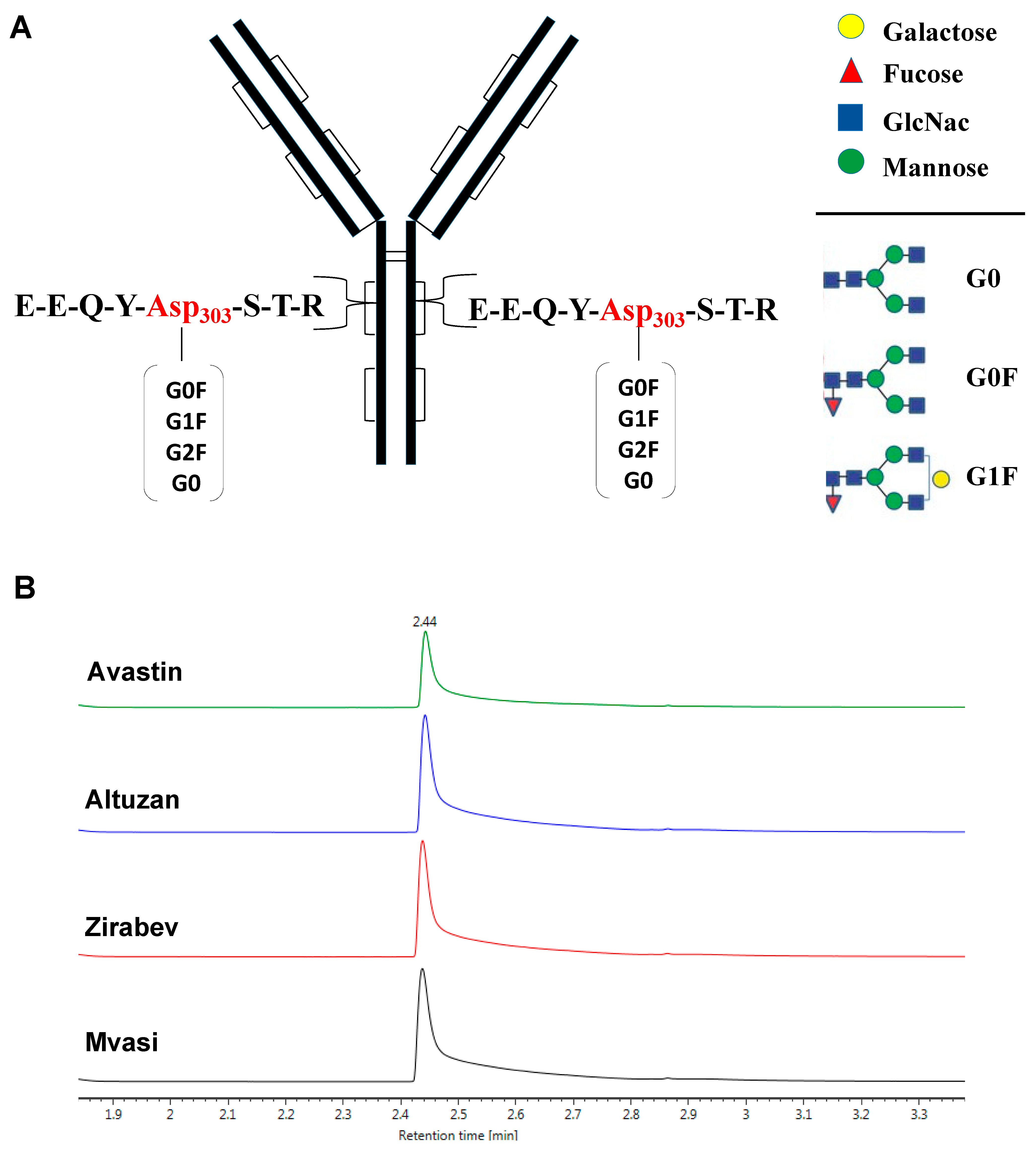
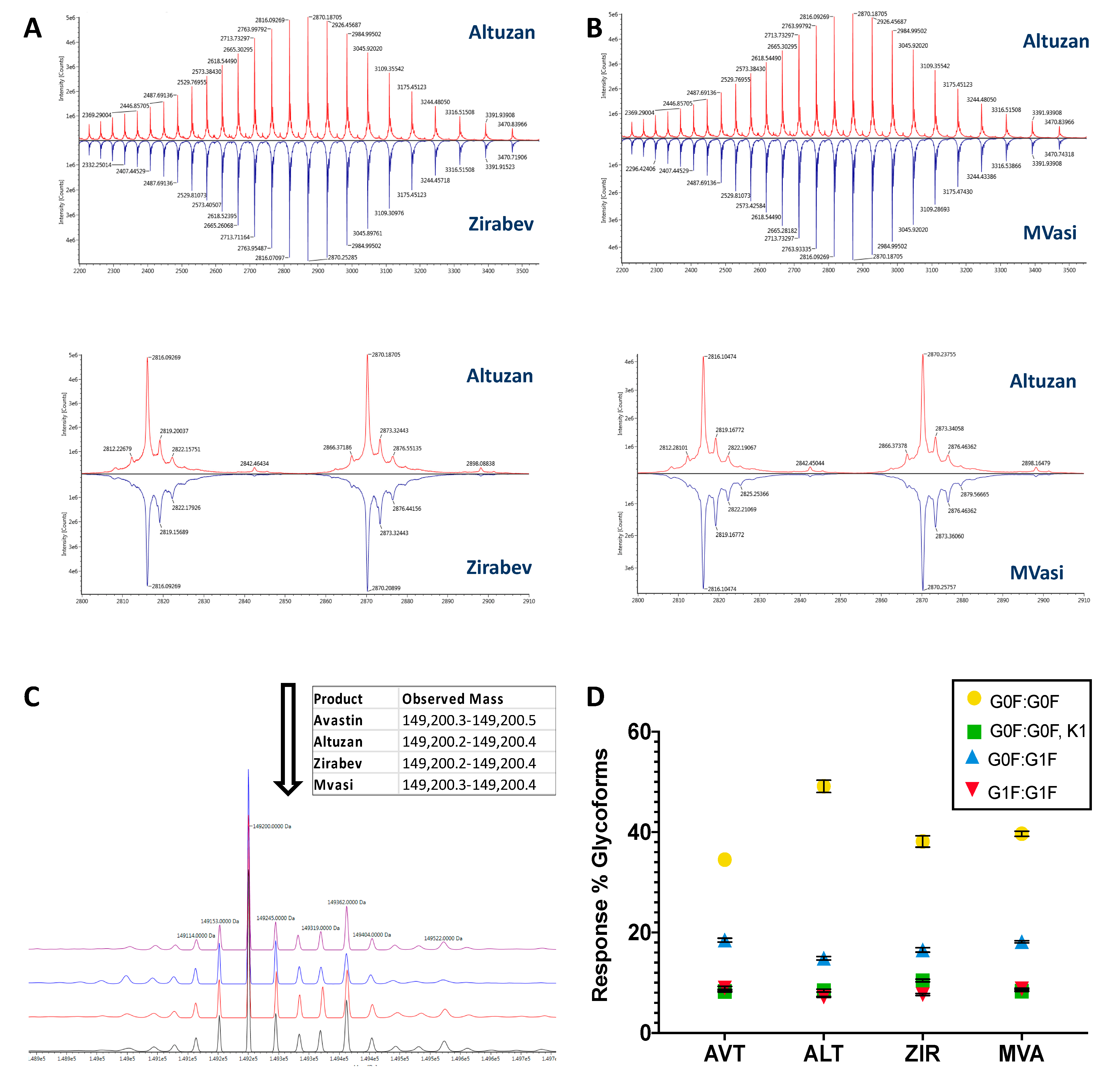
2.2. Peptide Mapping Analysis
2.3. VEGF Binding Assay
2.4. FcRn Binding Assay
2.5. FcγRIa Binding Assay
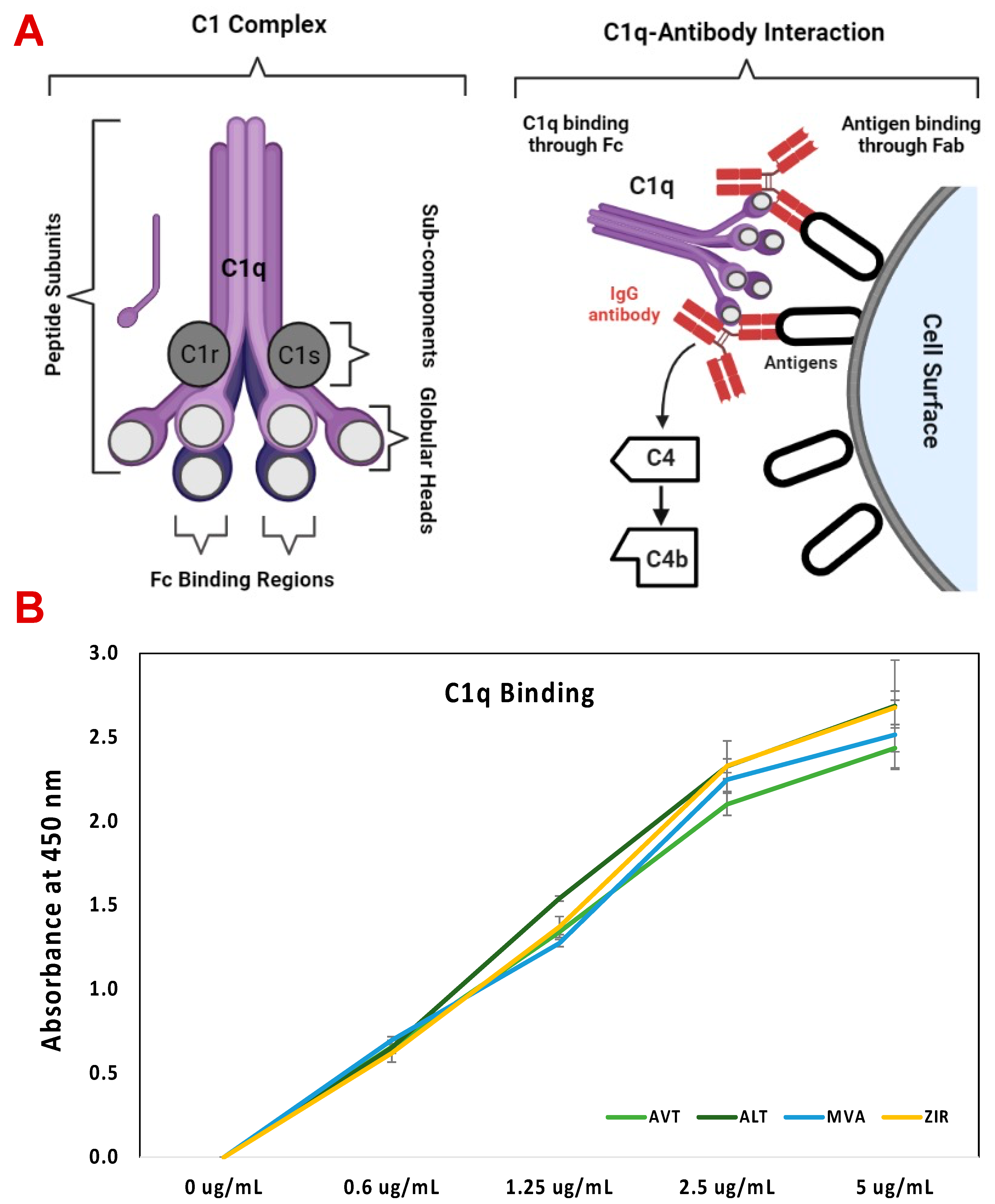
2.6. C1q ELISA Assay
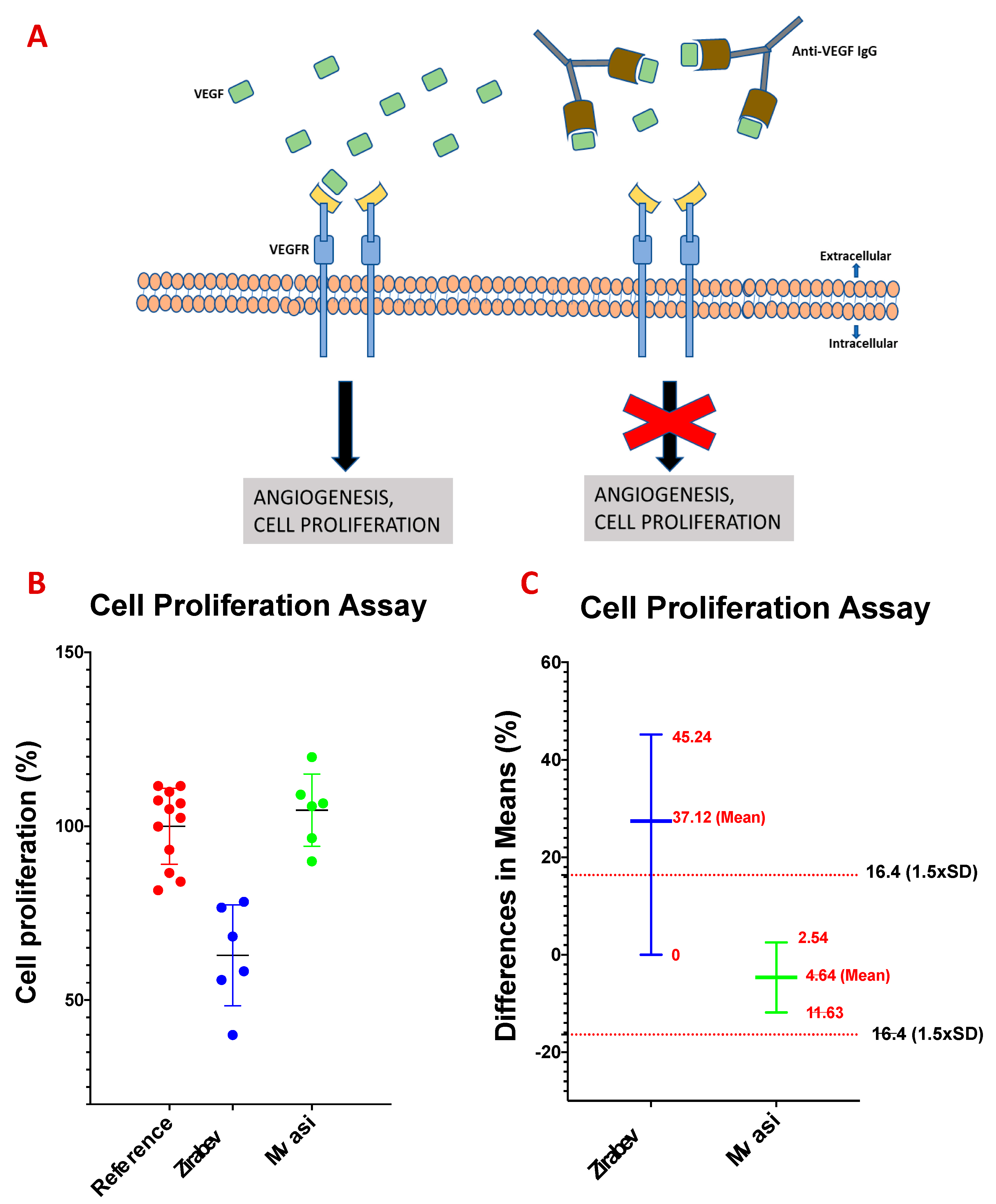
2.7. Cell Proliferation Assay
3. Results and Discussion
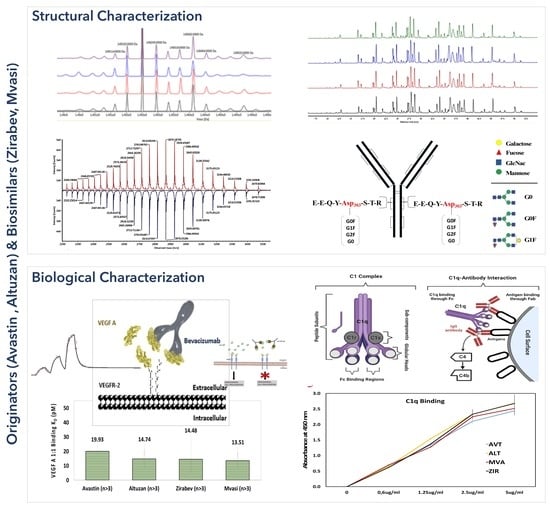
3.1. Intact Protein Analysis of Bevacizumab and Its Biosimilars
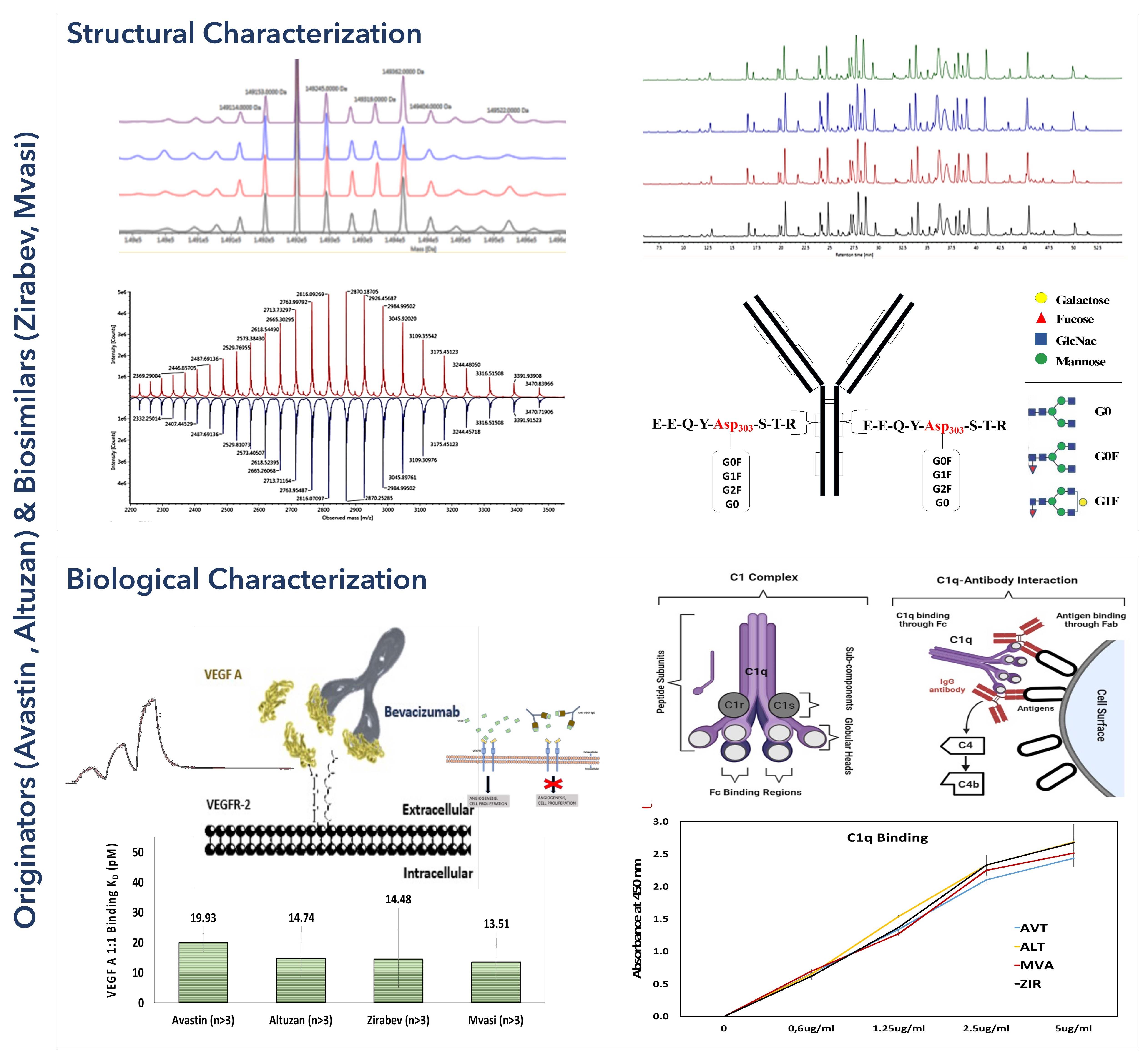
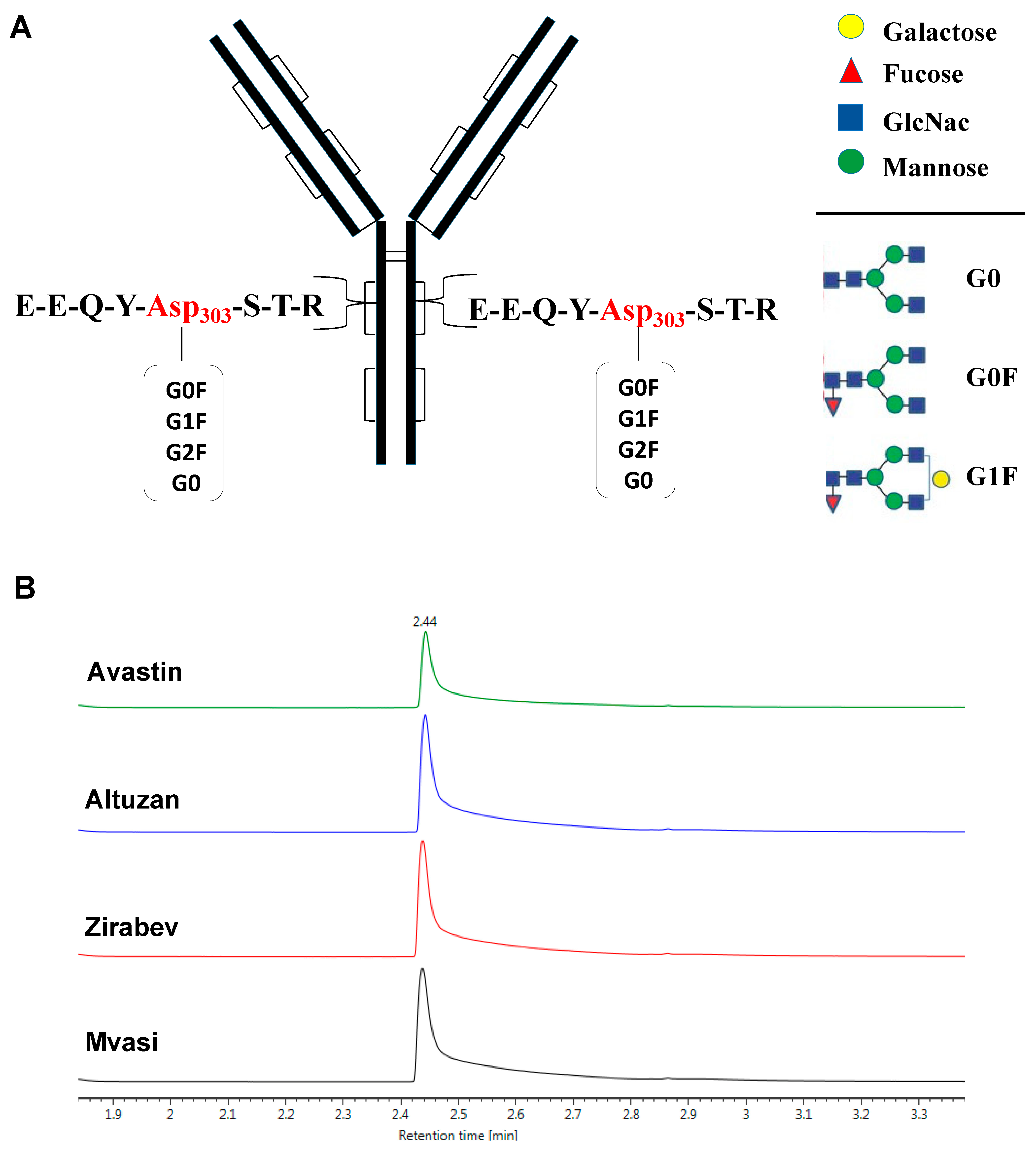
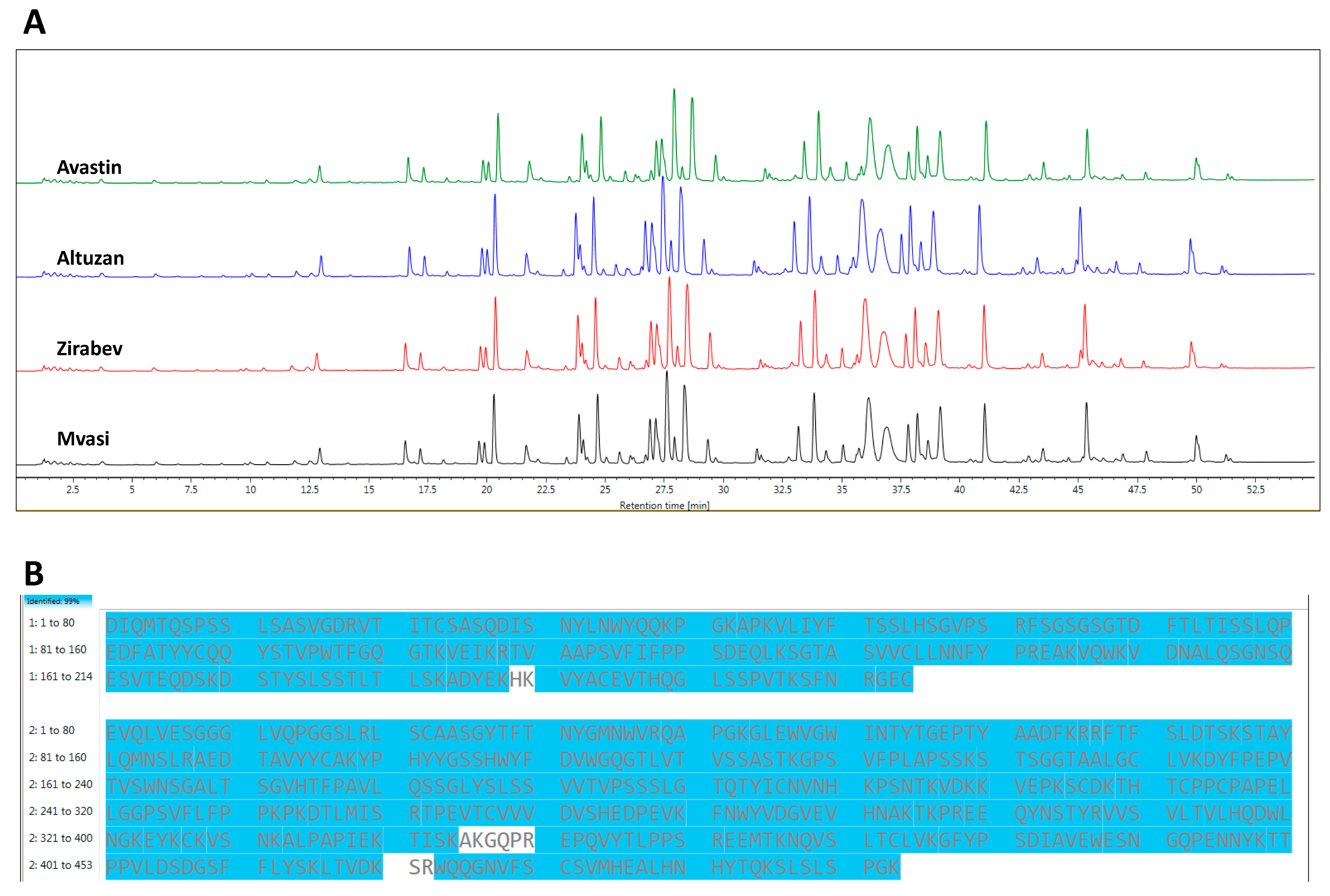
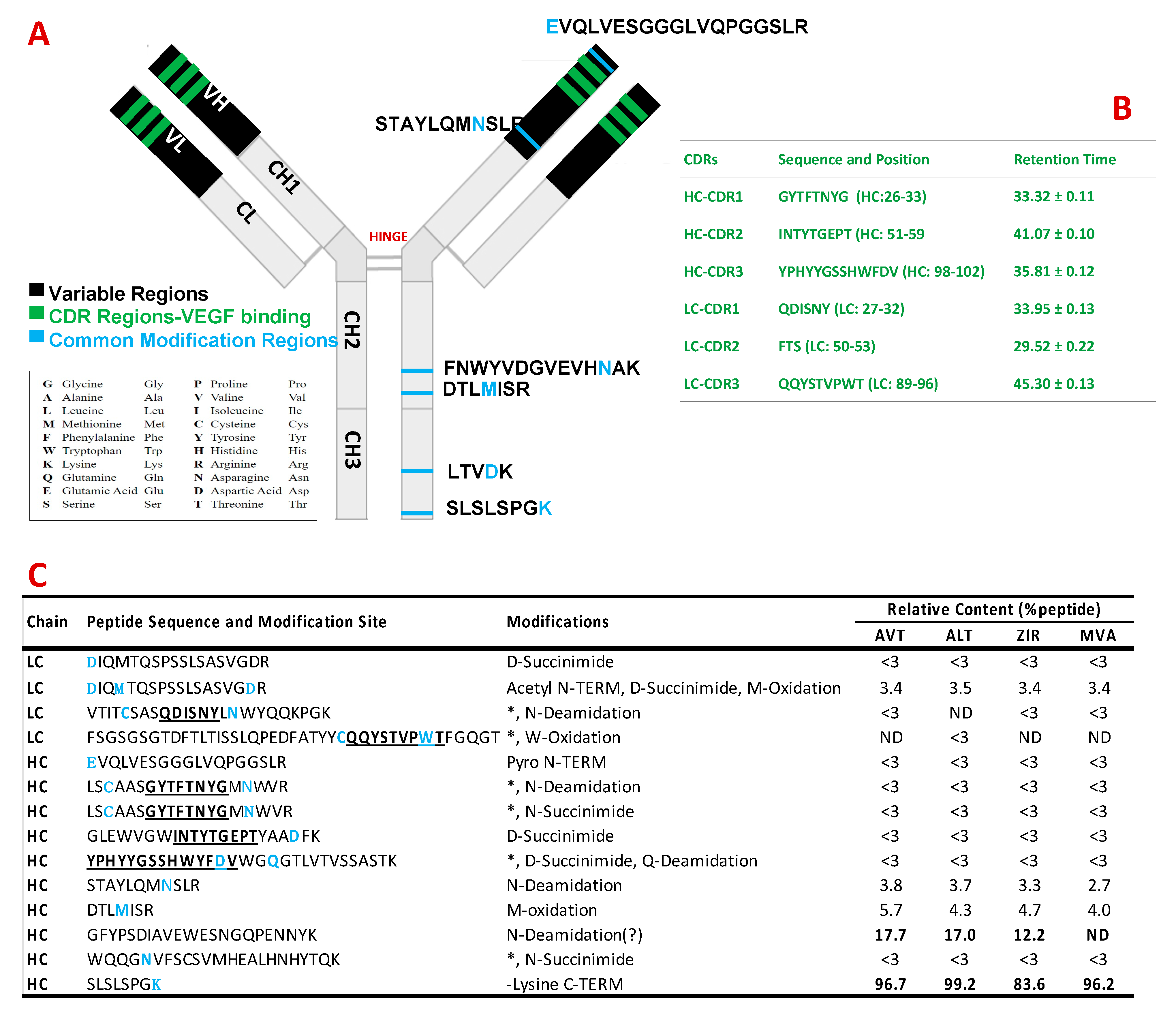
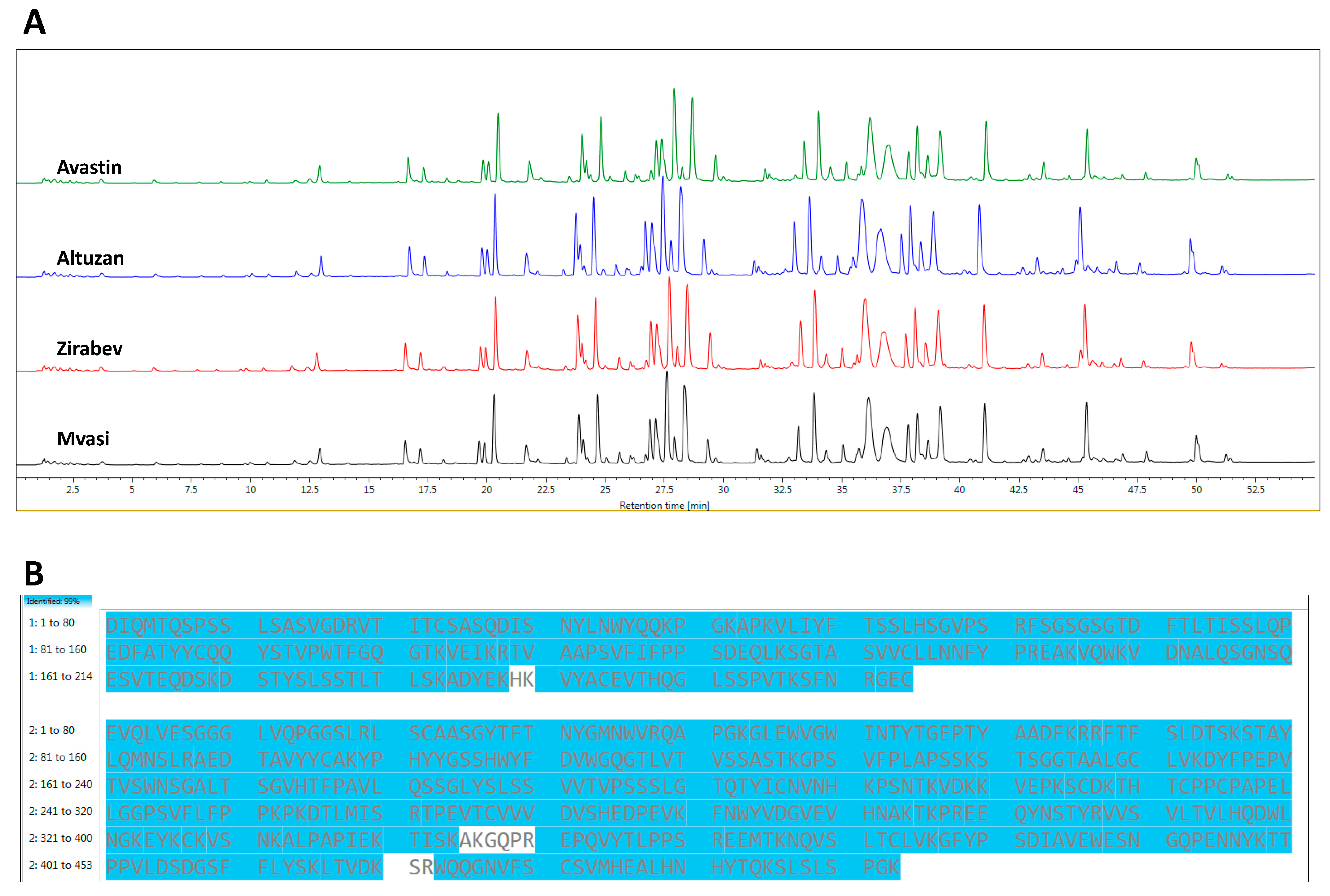
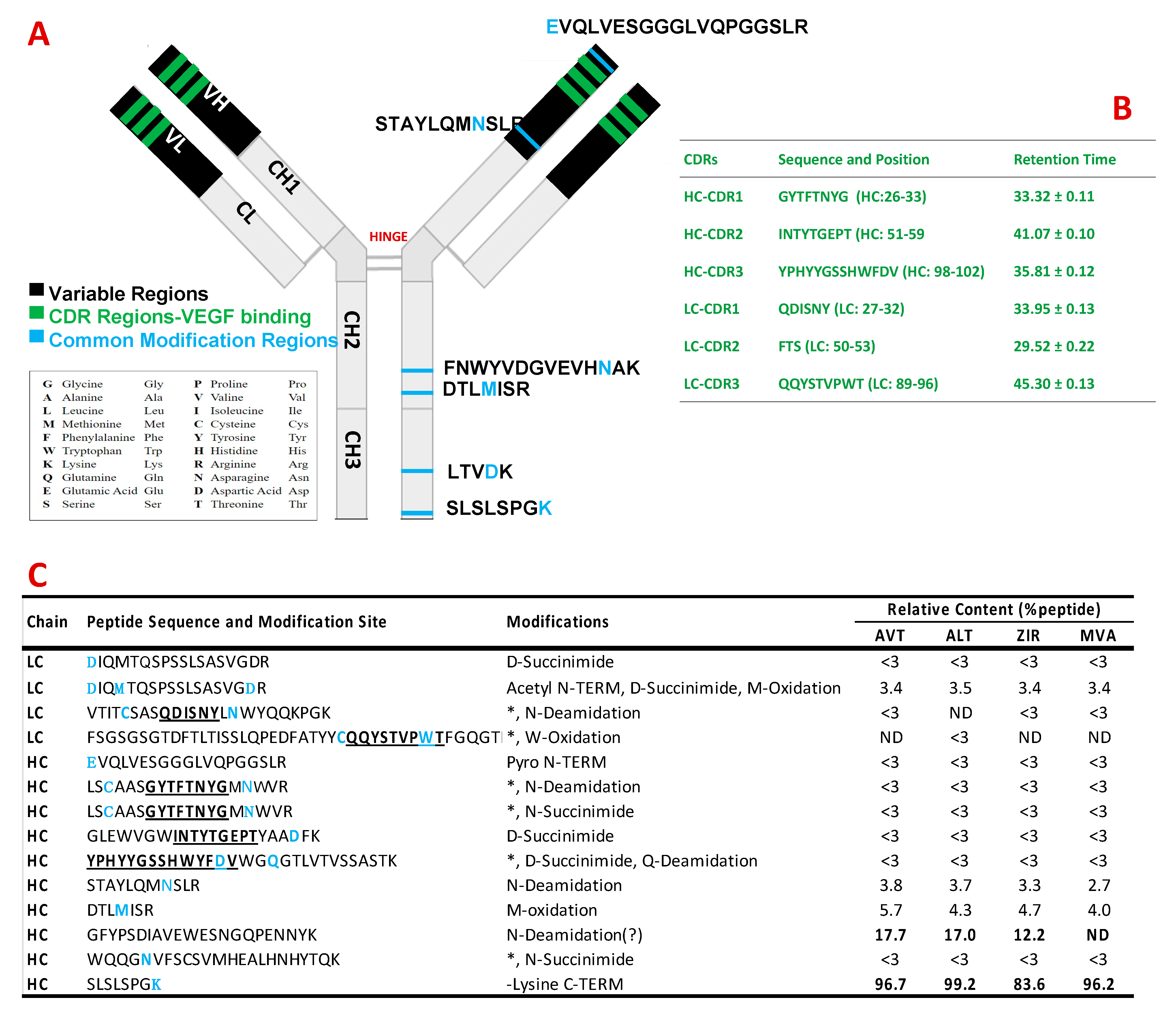
3.2. Peptide Mapping Analysis of Bevacizumab and Its Biosimilars
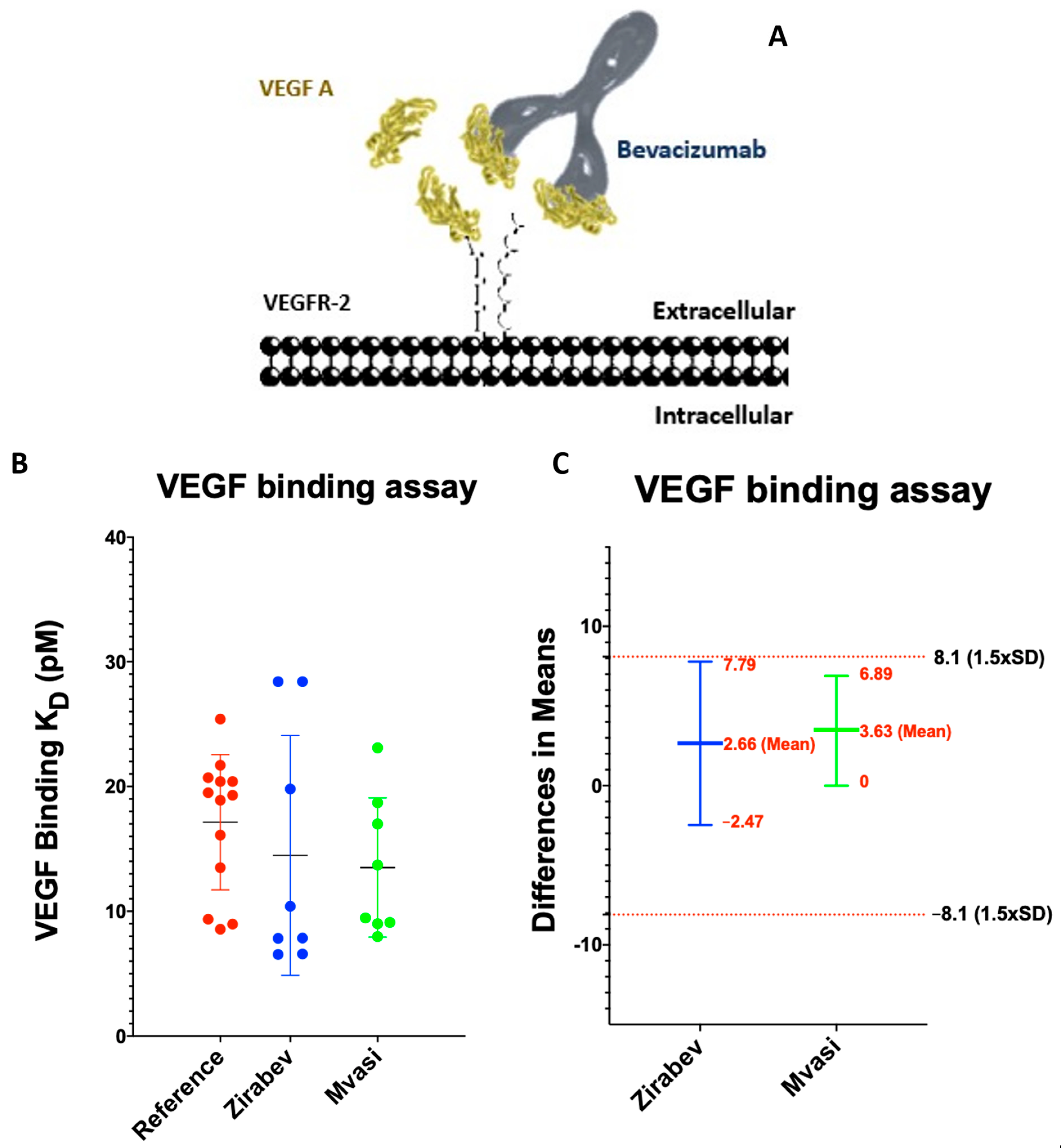
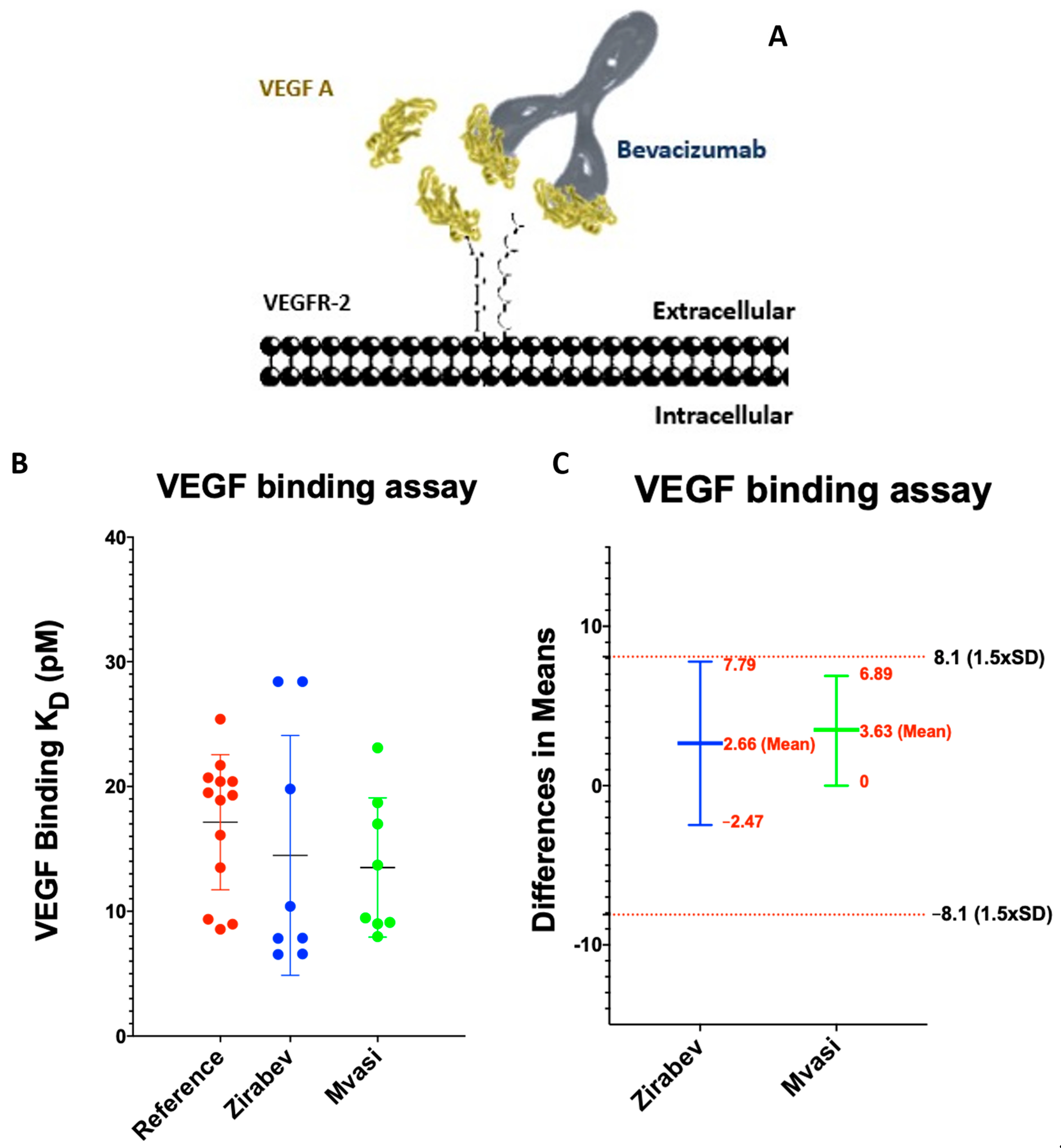
3.3. VEGF Binding Assays of Bevacizumab and Its Biosimilars
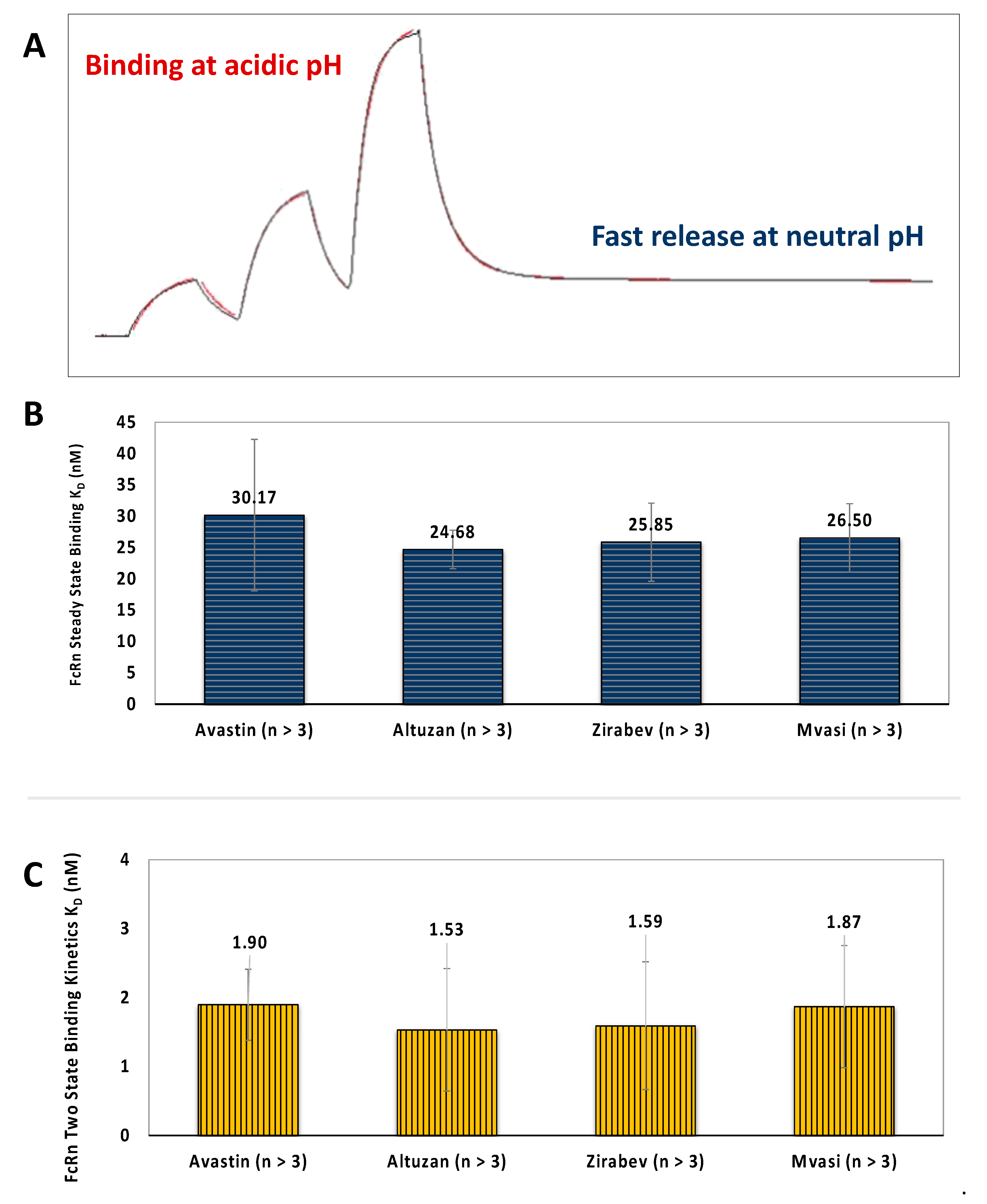
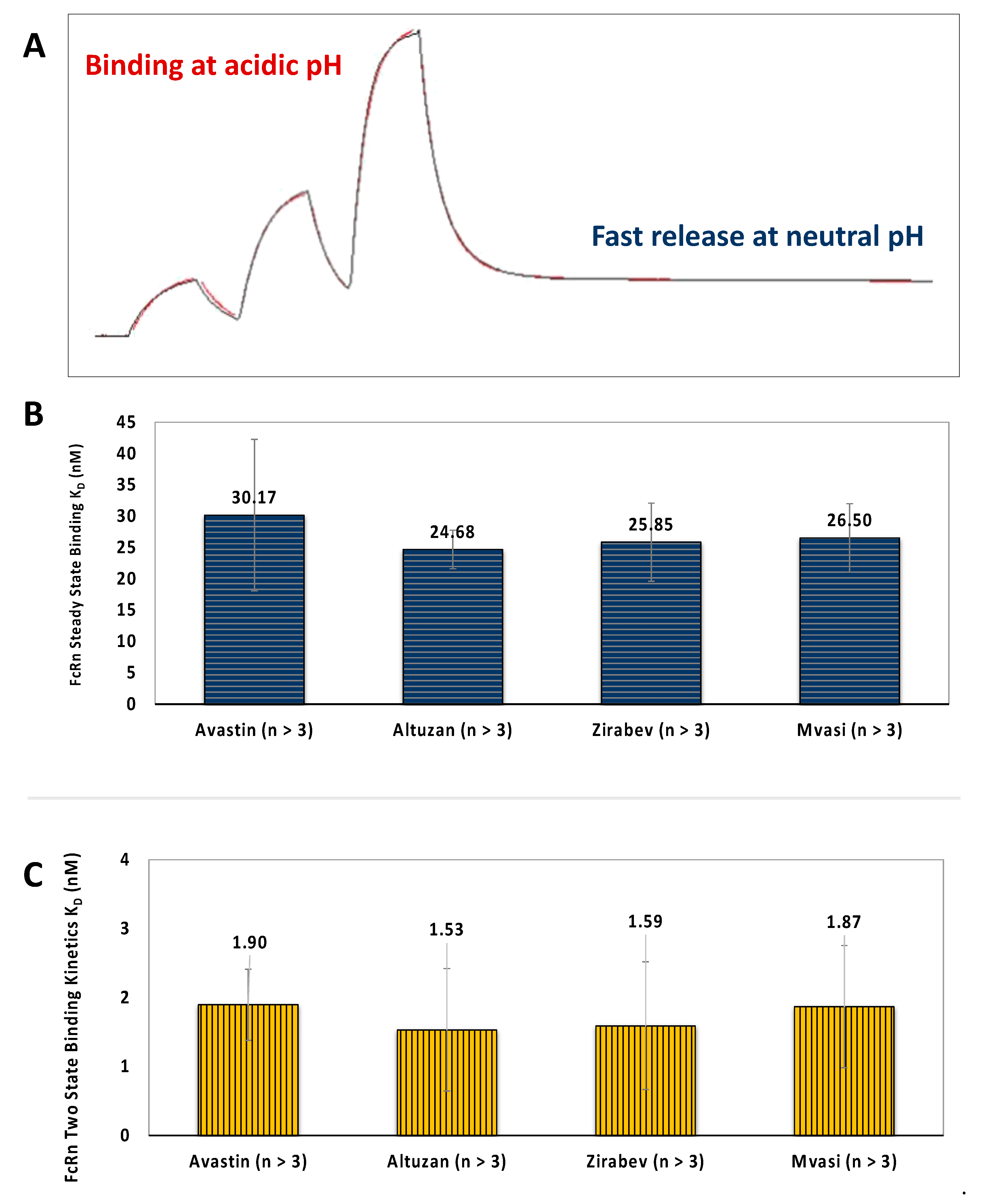
3.4. FcRn Binding Assays of Bevacizumab and Its Biosimilars
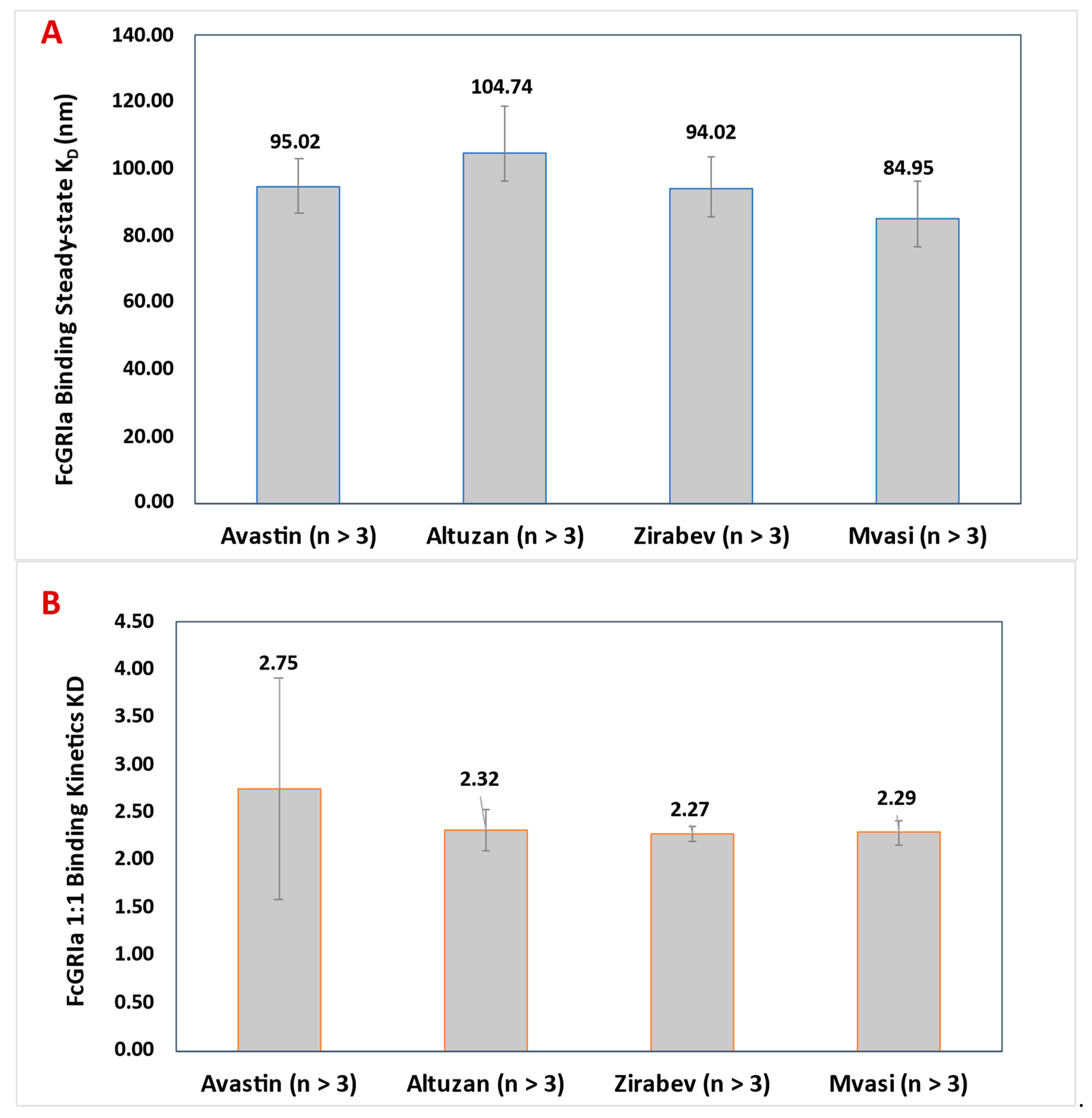
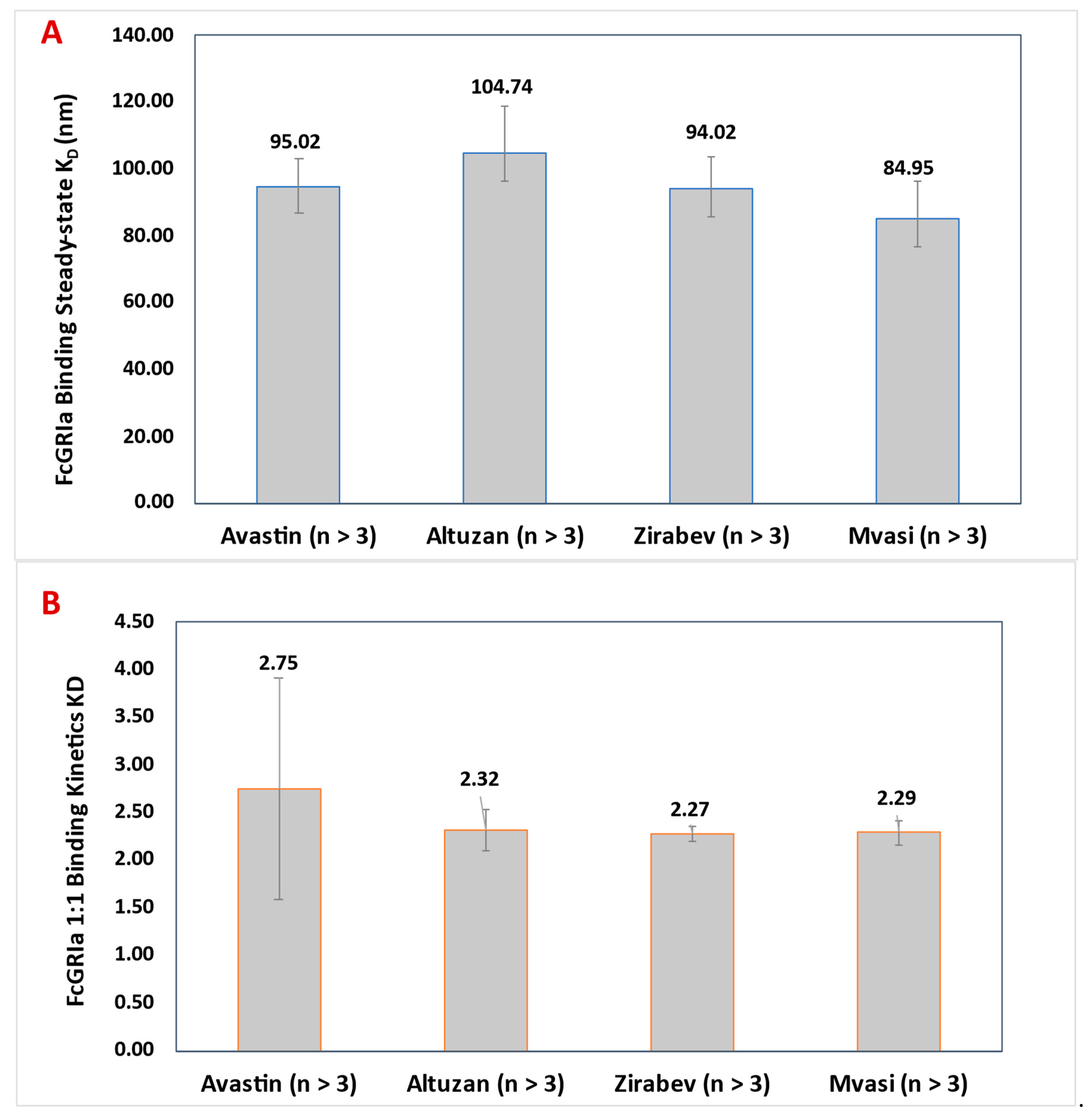
3.5. FcγRIa Binding Assay of Bevacizumab and Its Biosimilars
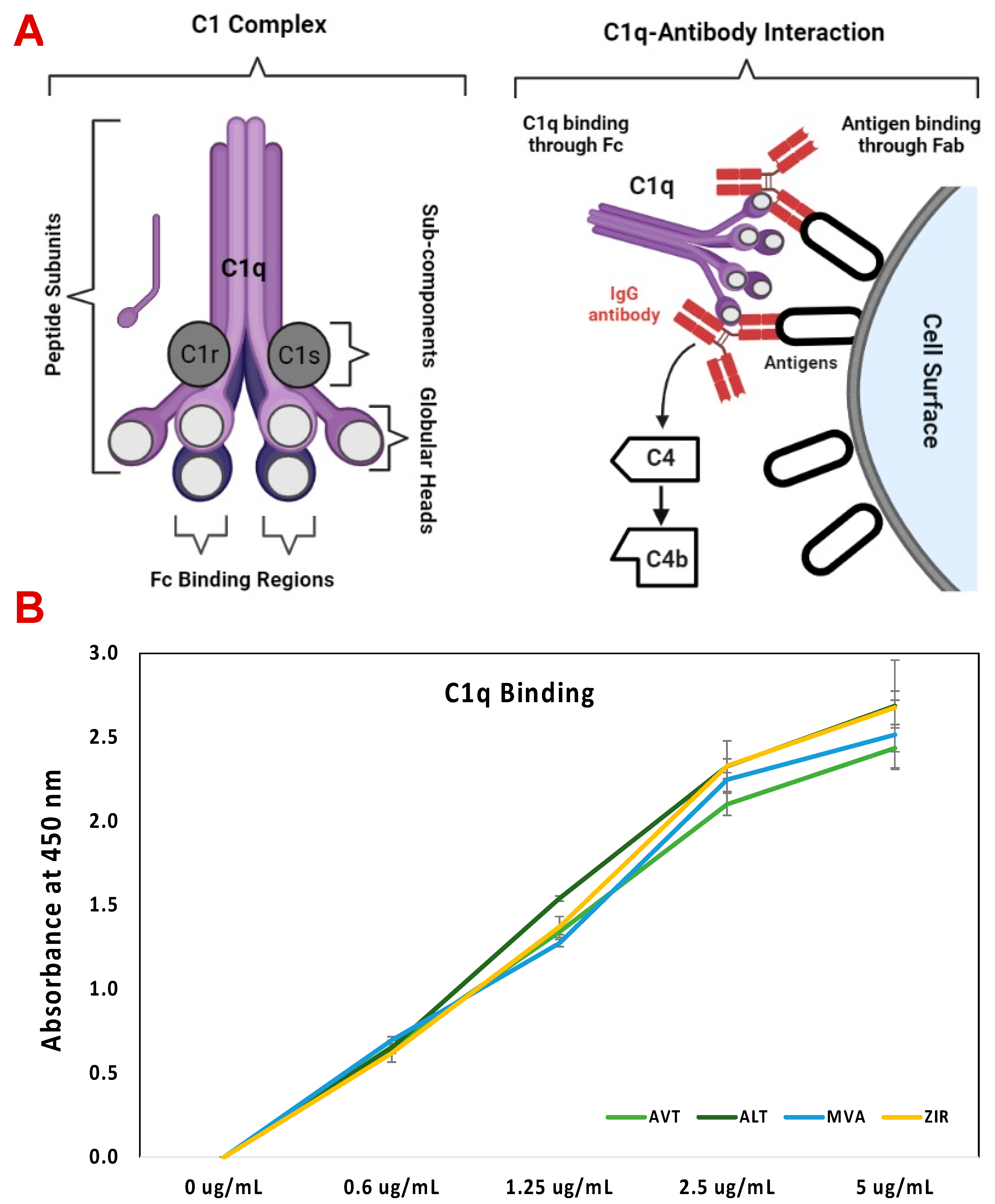
3.6. C1q ELISA Assay of Bevacizumab and Its Biosimilars
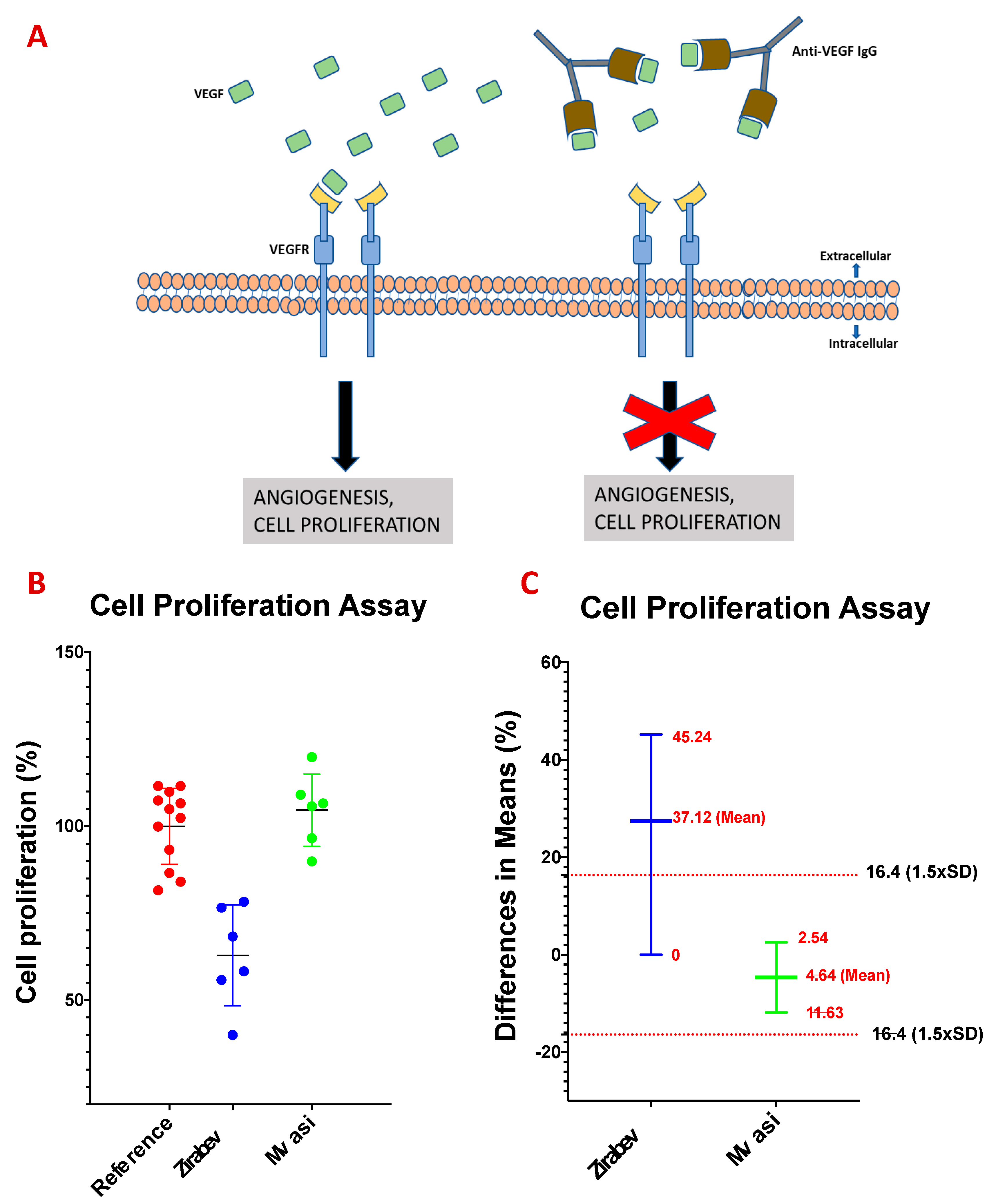
3.7. Cell Proliferation Assay of Bevacizumab and Its Biosimilars
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Wang, X.; An, Z.; Luo, W.; Xia, N.; Zhao, Q. Molecular and functional analysis of monoclonal antibodies in support of biologics development. Protein Cell 2018, 9, 74–85. [Google Scholar] [CrossRef]
- Chan, K.K.; Bass, A.R. Autoimmune complications of immunotherapy: Pathophysiology and management. BMJ 2020, 369, m736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milne, R. The rare and the common: Scale and the genetic imaginary in Alzheimer’s disease drug development. New Genet. Soc. 2020, 39, 101–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, O.; Joung, J.; Park, Y.; Kim, C.W.; Hong, S.H. Considerations of critical quality attributes in the analytical comparability assessment of biosimilar products. Biologicals 2017, 48, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A.; Cohen, A.D.; Puig, L.; Vender, R.; Van Der Walt, J.; Wu, J.J. Biosimilars for psoriasis: Preclinical analytical assessment to determine similarity. Br. J. Dermatol. 2016, 174, 282–286. [Google Scholar] [CrossRef]
- Torkashvand, F.; Vaziri, B. Main quality attributes of monoclonal antibodies and effect of cell culture components. Iran. Biomed. J. 2017, 21, 131–141. [Google Scholar] [CrossRef]
- Ren, D.; Zhang, J.; Pritchett, R.; Liu, H.; Kyauk, J.; Luo, J.; Amanullah, A. Detection and identification of a serine to arginine sequence variant in a therapeutic monoclonal antibody. J. Chromatogr. B 2011, 879, 2877–2884. [Google Scholar] [CrossRef]
- Joshi, S.; Rathore, A.S. Assessment of Structural and Functional Comparability of Biosimilar Products: Trastuzumab as a Case Study. BioDrugs 2020, 34, 209–223. [Google Scholar] [CrossRef]
- Kaur, H. Stability testing in monoclonal antibodies. Crit. Rev. Biotechnol. 2021, 41, 692–714. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, J.; Lu, H. Antibody glycosylation: Impact on antibody drug characteristics and quality control. Appl. Microbiol. Biotechnol. 2020, 104, 1905–1914. [Google Scholar] [CrossRef]
- FDA. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/scientific-considerations-demonstrating-biosimilarity-reference-product (accessed on 1 March 2022).
- EMA. Guideline on Similar Biological Medicinal Products. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-rev1_en.pdf (accessed on 1 March 2022).
- World Health Organization Guidelines on Evaluation of Similar Biotherapeutic Products (SBPs). Available online: https://cdn.who.int/media/docs/default-source/biologicals/who-guidelines-on-evaluation-of-biosimilars---4-nov-2021.pdf?sfvrsn=f17799ae_5 (accessed on 1 March 2022).
- Q6B Ich Harmonised Tripartite Guideline Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-6-b-test-procedures-acceptance-criteria-biotechnological/biological-products-step-5_en.pdf (accessed on 1 March 2022).
- Beyer, B.; Walch, N.; Jungbauer, A.; Lingg, N. How Similar Is Biosimilar? A Comparison of Infliximab Therapeutics in Regard to Charge Variant Profile and Antigen Binding Affinity. Biotechnol. J. 2019, 14, 1800340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Fei, D.; Vanderlaan, M.; Song, A. Biological activity of Bevacizumab, a humanized anti-VEGF antibody in vitro. Angiogenesis 2004, 7, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef] [PubMed]
- Assoun, S.; Brosseau, S.; Steinmetz, C.; Gounant, V.; Zalcman, G. Bevacizumab in advanced lung cancer: State of the art. Future Oncol. 2017, 13, 2515–2535. [Google Scholar] [CrossRef]
- Pfaendler, K.S.; Liu, M.C.; Tewari, K.S. Bevacizumab in cervical cancer: 5 years after. Cancer J. 2018, 24, 187–192. [Google Scholar] [CrossRef]
- Diaz, R.J.; Ali, S.; Qadir, M.G.; De La Fuente, M.I.; Ivan, M.E.; Komotar, R.J. The role of Bevacizumab in the treatment of glioblastoma. J. Neurooncol. 2017, 133, 455–467. [Google Scholar] [CrossRef]
- Prakash, A.; Mishra, N.N.; Utpreksha, V.; Sharma, S.; Anand, A.; Mahajan, R.V.; Prasad, J.P.; Chand, S. Comparative analytical profiling of bevacizumab biosimilars marketed in India: A national control laboratory study. 3 Biotech 2020, 10, 516. [Google Scholar] [CrossRef]
- Sokolowska, I.; Mo, J.; Dong, J.; Lewis, M.J.; Hu, P. Subunit mass analysis for monitoring antibody oxidation. MAbs 2017, 9, 498–505. [Google Scholar] [CrossRef] [Green Version]
- Kiris, I.; Basar, M.K.; Sahin, B.; Gurel, B.; Coskun, J.; Mroczek, T.; Baykal, A.T. Evaluation of the Therapeutic Effect of Lycoramine on Alzheimer’s Disease in Mouse Model. Curr. Med. Chem. 2021, 28, 3449–3473. [Google Scholar] [CrossRef]
- Rogstad, S.; Yan, H.; Wang, X.; Powers, D.; Brorson, K.; Damdinsuren, B.; Lee, S. Multi-Attribute Method for Quality Control of Therapeutic Proteins. Anal. Chem. 2019, 91, 14170–14177. [Google Scholar] [CrossRef]
- Miao, S.; Xie, P.; Zou, M.; Fan, L.; Liu, X.; Zhou, Y.; Zhao, L.; Ding, D.; Wang, H.; Tan, W.-S.S. Identification of multiple sources of the acidic charge variants in an IgG1 monoclonal antibody. Appl. Microbiol. Biotechnol. 2017, 101, 5627–5638. [Google Scholar] [CrossRef] [PubMed]
- Kurt, H.; Eyüpoğlu, A.E.; Sütlü, T.; Budak, H.; Yüce, M. Plasmonic Selection of ssDNA Aptamers against Fibroblast Growth Factor Receptor. ACS Comb. Sci. 2019, 21, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; McKay, P.; Yee, L.T.; Dutina, G.; Hass, P.E.; Nijem, I.; Allison, D.; Cowan, K.J.; Lin, K.; Quarmby, V.; et al. Impact of SPR biosensor assay configuration on antibody: Neonatal Fc receptor binding data. MAbs 2017, 9, 319–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuber, T.; Frese, K.; Jaehrling, J.; Jäger, S.; Daubert, D.; Felderer, K.; Linnemann, M.; Höhne, A.; Kaden, S.; Kölln, J.; et al. Characterization and screening of IgG binding to the neonatal Fc receptor. MAbs 2014, 6, 928–942. [Google Scholar] [CrossRef] [Green Version]
- Bertolotti-Ciarlet, A.; Wang, W.; Lownes, R.; Pristatsky, P.; Fang, Y.; McKelvey, T.; Li, Y.; Drummond, J.; Prueksaritanont, T.; Vlasak, J. Impact of methionine oxidation on the binding of human IgG1 to FcRn and Fcγ receptors. Mol. Immunol. 2009, 46, 1878–1882. [Google Scholar] [CrossRef]
- Abdiche, Y.N.; Yeung, Y.A.; Chaparro-Riggers, J.; Barman, I.; Strop, P.; Chin, S.M.; Pham, A.; Bolton, G.; McDonough, D.; Lindquist, K.; et al. The neonatal Fc receptor (FcRn) binds independently to both sites of the IgG homodimer with identical affinity. MAbs 2015, 7, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Biacore SPR Assay Application Guide, Fc Receptor Binding Assays Using Surface Plasmon Resonance. 2020. Available online: https://www.cytivalifesciences.com/en/us/solutions/protein-research/knowledge-center/surface-plasmon-resonance/fc-receptor-binding-assays-using-surface-plasmon-resonance (accessed on 1 March 2022).
- Shortreed, M.R.; Frey, B.L.; Scalf, M.; Knoener, R.A.; Cesnik, A.J.; Smith, L.M. Elucidating Proteoform Families from Proteoform Intact-Mass and Lysine-Count Measurements. J. Proteome Res. 2016, 15, 1213–1221. [Google Scholar] [CrossRef] [Green Version]
- Seo, N.; Polozova, A.; Zhang, M.; Yates, Z.; Cao, S.; Li, H.; Kuhns, S.; Maher, G.; McBride, H.J.; Liu, J. Analytical and functional similarity of Amgen biosimilar ABP 215 to Bevacizumab. MAbs 2018, 10, 678–691. [Google Scholar] [CrossRef] [Green Version]
- US FDA Zirabev Summary Review: Application Number 761099Orig1s000. In Cent Drug Eval. Res.; 2019. Available online: https://www.accessdata.fda.gov/drugsatfda%7B%5C_%7Ddocs/nda/2019/761099Orig1s000SumR.pdf (accessed on 1 March 2022).
- Brorson, K.; Jia, A.Y. Therapeutic monoclonal antibodies and consistent ends: Terminal heterogeneity, detection, and impact on quality. Curr. Opin. Biotechnol. 2014, 30, 140–146. [Google Scholar] [CrossRef]
- Zhao, Y.-Y.; Wang, N.; Liu, W.-H.; Tao, W.-J.; Liu, L.-L.; Shen, Z.-D. Charge Variants of an Avastin Biosimilar Isolation, Characterization, In Vitro Properties and Pharmacokinetics in Rat. PLoS ONE 2016, 11, e0151874. [Google Scholar] [CrossRef] [Green Version]
- Bansal, R.; Dash, R.; Rathore, A.S. Impact of mAb Aggregation on Its Biological Activity: Rituximab as a Case Study. J. Pharm. Sci. 2020, 109, 2684–2698. [Google Scholar] [CrossRef]
- Vlasak, J.; Bussat, M.C.; Wang, S.; Wagner-Rousset, E.; Schaefer, M.; Klinguer-Hamour, C.; Kirchmeier, M.; Corvaïa, N.; Ionescu, R.; Beck, A. Identification and characterization of asparagine deamidation in the light chain CDR1 of a humanized IgG1 antibody. Anal. Biochem. 2009, 392, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Haberger, M.; Bomans, K.; Diepold, K.; Hook, M.; Gassner, J.; Schlothauer, T.; Zwick, A.; Spick, C.; Kepert, J.F.; Hienz, B.; et al. Assessment of chemical modifications of sites in the CDRs of recombinant antibodies. MAbs 2014, 6, 327–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamanna, W.C.; Heller, K.; Schneider, D.; Guerrasio, R.; Hampl, V.; Fritsch, C.; Schiestl, M. The in-use stability of the rituximab biosimilar Rixathon®/Riximyo® upon preparation for intravenous infusion. J. Oncol. Pharm. Pract. 2019, 25, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ponniah, G.; Zhang, H.; Neill, A.; Gonzalez-lopez, N.; Patel, R.; Cheng, G.; Kita, A.Z.; Andrien, B.; Neill, A.; et al. In vitro and in vivo modifications of recombinant and human IgG antibodies In vitro and in vivo modi fi cations of recombinant and human IgG antibodies. mAbs 2014, 6, 1145–1154. [Google Scholar] [CrossRef] [Green Version]
- Dashivets, T.; Stracke, J.; Dengl, S.; Knaupp, A.; Pollmann, J.; Buchner, J.; Schlothauer, T. Oxidation in the complementarity-determining regions differentially influences the properties of therapeutic antibodies. MAbs 2016, 8, 1525–1535. [Google Scholar] [CrossRef] [Green Version]
- Chelius, D.; Rehder, D.S.; Bondarenko, P.V. Identification and Characterization of Deamidation Sites in the Conserved Regions of Human Immunoglobulin Gamma Antibodies. Anal. Chem. 2005, 77, 6004–6011. [Google Scholar] [CrossRef]
- Sinha, S.; Zhang, L.; Duan, S.; Williams, T.D.; Vlasak, J.; Ionescu, R.; Topp, E.M. Effect of protein structure on deamidation rate in the Fc fragment of an IgG1 monoclonal antibody. Protein Sci. 2009, 18, 1573–1584. [Google Scholar] [CrossRef] [Green Version]
- Panoilia, E.; Schindler, E.; Samantas, E.; Aravantinos, G.; Kalofonos, H.P.; Christodoulou, C.; Patrinos, G.P.; Friberg, L.E.; Sivolapenko, G. A pharmacokinetic binding model for Bevacizumab and VEGF165 in colorectal cancer patients. Cancer Chemother. Pharmacol. 2015, 75, 791–803. [Google Scholar] [CrossRef]
- Beck, A.; Liu, H. Macro- and Micro-Heterogeneity of Natural and Recombinant IgG Antibodies. Antibodies 2019, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Beyer, B.; Schuster, M.; Jungbauer, A.; Lingg, N. Microheterogeneity of Recombinant Antibodies: Analytics and Functional Impact. Biotechnol. J. 2018, 13, 1700476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, S.; Tian, J.; Tan, Z.; Chen, J.; Lee, J.; Borys, M.; Li, Z.J. Industrial bioprocessing perspectives on managing therapeutic protein charge variant profiles. Biotechnol. Bioeng. 2018, 115, 1646–1665. [Google Scholar] [CrossRef] [PubMed]
- Presta, L.G.; Chen, H.; O’Connor, S.J.; Chisholm, V.; Meng, Y.G.; Krummen, L.; Winkler, M.; Ferrara, N. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997, 57, 4593–4599. [Google Scholar] [PubMed]
- Sreenivasan, S.; Kumar, D.; Malani, H.; Rathore, A.S. Does interaction of monoclonal antibody charge variants with VEGF-A and ELISA reagents affect its quantification? Quantification of anti-VEGF mAb charge variants by ELISA? Anal. Biochem. 2020, 590, 113513. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.K.; Park, C.S.; Jang, J.; Kim, M.R.; Na, H.J.; Lee, K.; Kim, H.J.; Heo, K.; Yoo, B.C.; Kim, Y.M.; et al. Inhibition of VEGF-dependent angiogenesis and tumor angiogenesis by an optimized antibody targeting CLEC14a. Mol. Oncol. 2018, 12, 356–372. [Google Scholar] [CrossRef] [Green Version]
- Drake, A.W.; Myszka, D.G.; Klakamp, S.L. Characterizing high-affinity antigen/antibody complexes by kinetic- and equilibrium-based methods. Anal. Biochem. 2004, 328, 35–43. [Google Scholar] [CrossRef]
- Yang, J.; Wang, X.; Fuh, G.; Yu, L.; Wakshull, E.; Khosraviani, M.; Day, E.S.; Demeule, B.; Liu, J.; Shire, S.J.; et al. Comparison of binding characteristics and in vitro activities of three inhibitors of vascular endothelial growth factor A. Mol. Pharm. 2014, 11, 3421–3430. [Google Scholar] [CrossRef]
- Wang, X.; Phan, M.M.; Li, J.; Gill, H.; Williams, S.; Gupta, N.; Quarmby, V.; Yang, J. Molecular Interaction Characterization Strategies for the Development of New Biotherapeutic Antibody Modalities. Antibodies 2020, 9, 7. [Google Scholar] [CrossRef] [Green Version]
- Patel, R.; Neill, A.; Liu, H.; Andrien, B. IgG subclass specificity to C1q determined by surface plasmon resonance using Protein L capture technique. Anal. Biochem. 2015, 479, 15–17. [Google Scholar] [CrossRef]
- Papadopoulos, N.; Martin, J.; Ruan, Q.; Rafique, A.; Rosconi, M.P.; Shi, E.; Pyles, E.A.; Yancopoulos, G.D.; Stahl, N.; Wiegand, S.J. Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF Trap, ranibizumab and Bevacizumab. Angiogenesis 2012, 15, 171–185. [Google Scholar] [CrossRef] [Green Version]
- Hintersteiner, B.; Lingg, N.; Zhang, P.; Woen, S.; Hoi, K.M.; Stranner, S.; Wiederkum, S.; Mutschlechner, O.; Schuster, M.; Loibner, H.; et al. Charge heterogeneity: Basic antibody charge variants with increased binding to Fc receptors. MAbs 2016, 8, 1548–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, B.; Steen, S.; Hambly, D.; Valliere-Douglass, J.; Vanden Bos, T.; Smallwood, S.; Yates, Z.; Arroll, T.; Han, Y.; Gadgil, H.; et al. Succinimide formation at Asn 55 in the complementarity determining region of a recombinant monoclonal antibody IgG1 heavy chain. J. Pharm. Sci. 2009, 98, 3509–3521. [Google Scholar] [CrossRef]
- Schmid, I.; Bonnington, L.; Gerl, M.; Bomans, K.; Thaller, A.L.; Wagner, K.; Schlothauer, T.; Falkenstein, R.; Zimmermann, B.; Kopitz, J.; et al. Assessment of susceptible chemical modification sites of trastuzumab and endogenous human immunoglobulins at physiological conditions. Commun. Biol. 2018, 1, 28. [Google Scholar] [CrossRef] [PubMed]
- Szikora, B.; Hiripi, L.; Bender, B.; Kacskovics, I.; Iliás, A. Characterization of the interactions of rabbit neonatal Fc receptor (FcRn) with rabbit and human IgG isotypes. PLoS ONE 2017, 12, e0185662. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Vlasak, J.; Li, Y.; Pristatsky, P.; Fang, Y.; Pittman, T.; Roman, J.; Wang, Y.; Prueksaritanont, T.; Ionescu, R. Impact of methionine oxidation in human IgG1 Fc on serum half-life of monoclonal antibodies. Mol. Immunol. 2011, 48, 860–866. [Google Scholar] [CrossRef]
- Pan, H.; Chen, K.; Chu, L.; Kinderman, F.; Apostol, I.; Huang, G. Methionine oxidation in human IgG2 Fc decreases binding affinities to protein A and FcRn. Protein Sci. 2009, 18, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khawli, L.A.; Goswami, S.; Hutchinson, R.; Kwong, Z.W.; Yang, J.; Wang, X.; Yao, Z.; Sreedhara, A.; Cano, T.; Tesar, D.B.; et al. Charge variants in IgG1. MAbs 2010, 2, 613–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subedi, G.P.; Barb, A.W. The immunoglobulin G1 N-glycan composition affects binding to each low affinity Fc γ receptor. MAbs 2016, 8, 1512–1524. [Google Scholar] [CrossRef] [Green Version]
- Bournazos, S.; Gupta, A.; Ravetch, J.V. The role of IgG Fc receptors in antibody-dependent enhancement. Nat. Rev. Immunol. 2020, 20, 633–643. [Google Scholar] [CrossRef]
- Bruhns, P.; Iannascoli, B.; England, P.; Mancardi, D.A.; Fernandez, N.; Jorieux, S.; Daëron, M. Specificity and affinity of human Fcγ receptors and their polymorphic variants for human IgG subclasses. Blood 2009, 113, 3716–3725. [Google Scholar] [CrossRef]
- Avastin: EPAR—European Medicines Agency. Avastin, INN-Bevacizumab—European Medicines Agency. Available online: https://www.ema.europa.eu/en/documents/product-information/avastin-epar-product-information_en.pdf (accessed on 1 March 2022).
- Zirabev: EPAR—European Medicines Agency. Zirabev: EPAR—Public Assessment Report. Available online: https://www.ema.europa.eu/en/documents/assessment-report/zirabev-epar-public-assessment-report_en.pdf (accessed on 1 March 2022).
- Mvasi: EPAR—European Medicines Agency. Mvasi: EPAR—Public Asssessment Report. Available online: https://www.ema.europa.eu/en/documents/assessment-report/mvasi-epar-public-assessment-report_en.pdf (accessed on 1 March 2022).
- Forest-Nault, C.; Gaudreault, J.; Henry, O.; Durocher, Y.; De Crescenzo, G. On the Use of Surface Plasmon Resonance Biosensing to Understand IgG-FcγR Interactions. Int. J. Mol. Sci. 2021, 22, 6616. [Google Scholar] [CrossRef] [PubMed]
- Dorion-Thibaudeau, J.; Raymond, C.; Lattová, E.; Perreault, H.; Durocher, Y.; De Crescenzo, G. Towards the development of a surface plasmon resonance assay to evaluate the glycosylation pattern of monoclonal antibodies using the extracellular domains of CD16a and CD64. J. Immunol. Methods 2014, 408, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement system part I—Molecular mechanisms of activation and regulation. Front. Immunol. 2015, 6, 1–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merle, N.S.; Noe, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement system part II: Role in immunity. Front. Immunol. 2015, 6, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Meyer, S.; Leusen, J.H.W.; Boross, P. Regulation of complement and modulation of its activity in monoclonal antibody therapy of cancer. MAbs 2014, 6, 1133–1144. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Yang, C.; Jin, X.; Du, Q.; Wu, H.; Dall’acqua, W.; Mazor, Y. Regulation of antibody-mediated complement-dependent cytotoxicity by modulating the intrinsic affinity and binding valency of IgG for target antigen. mAbs 2020, 12, 1690959. [Google Scholar] [CrossRef] [Green Version]
- Zeitlin, L.; Pettitt, J.; Scully, C.; Bohorova, N.; Kim, D.; Pauly, M.; Hiatt, A.; Ngo, L.; Steinkellner, H.; Whaley, K.J.; et al. Enhanced potency of a fucose-free monoclonal antibody being developed as an Ebola virus immunoprotectant. Proc. Natl. Acad. Sci. USA 2011, 108, 20690–20694. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Lai, H.; Engle, M.; Gorlatov, S.; Gruber, C.; Steinkellner, H.; Diamond, M.S.; Chen, Q. Generation and Analysis of Novel Plant-Derived Antibody-Based Therapeutic Molecules against West Nile Virus. PLoS ONE 2014, 9, e93541. [Google Scholar] [CrossRef]
- Jovic, M.; Cymer, F. Qualification of a surface plasmon resonance assay to determine binding of IgG-type antibodies to complement component C1q. Biologicals 2019, 61, 76–79. [Google Scholar] [CrossRef]
- Park, H.-J.; Zhang, Y.; Georgescu, S.P.; Johnson, K.L.; Kong, D.; Galper, J.B. Human Umbilical Vein Endothelial Cells and Human Dermal Microvascular Endothelial Cells Offer New Insights Into the Relationship Between Lipid Metabolism and Angiogenesis. Stem Cell Rev. 2006, 2, 93–102. [Google Scholar] [CrossRef]
- Jiménez, N.; Krouwer, V.J.D.; Post, J.A. A new, rapid and reproducible method to obtain high quality endothelium in vitro. Cytotechnology 2013, 65, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Htay, A.; Dos Santos, W.; Gillies, G.T.; Fillmore, H.L.; Sholley, M.M.; Broaddus, W.C. In vitro angiogenesis by human umbilical vein endothelial cells (HUVEC) induced by three-dimensional co-culture with glioblastoma cells. J. Neurooncol. 2009, 92, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Zhang, F.; Xu, G.; Wu, G.; Wang, W.; Liu, C.; Fu, Z.; Li, M.; Guo, S.; Yu, X.; et al. Analytical Similarity of a Proposed Biosimilar BVZ-BC to Bevacizumab. Anal. Chem. 2020, 92, 3161–3170. [Google Scholar] [CrossRef]
- Aslantürk, Ö.S. In Vitro Cytotoxicity and Cell Viability Assays: Principles, Advantages, and Disadvantages. In Genotoxicity—A Predictable Risk to Our Actual World; IntechOpen: London, UK, 2018; pp. 1–18. [Google Scholar] [CrossRef] [Green Version]
- Stone, V.; Johnston, H.; Schins, R.P.F. Development of in vitro systems for nanotoxicology: Methodological considerations. Crit. Rev. Toxicol. 2009, 39, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gürel, B.; Çapkın, E.; Parlar, A.; Özkan, A.; Çorbacıoğlu, M.; Dağlikoca, D.E.; Yüce, M. Optimized Methods for Analytical and Functional Comparison of Biosimilar mAb Drugs: A Case Study for Avastin, Mvasi, and Zirabev. Sci. Pharm. 2022, 90, 36. https://doi.org/10.3390/scipharm90020036
Gürel B, Çapkın E, Parlar A, Özkan A, Çorbacıoğlu M, Dağlikoca DE, Yüce M. Optimized Methods for Analytical and Functional Comparison of Biosimilar mAb Drugs: A Case Study for Avastin, Mvasi, and Zirabev. Scientia Pharmaceutica. 2022; 90(2):36. https://doi.org/10.3390/scipharm90020036
Chicago/Turabian StyleGürel, Büşra, Eda Çapkın, Ayhan Parlar, Aylin Özkan, Meltem Çorbacıoğlu, Duygu Emine Dağlikoca, and Meral Yüce. 2022. "Optimized Methods for Analytical and Functional Comparison of Biosimilar mAb Drugs: A Case Study for Avastin, Mvasi, and Zirabev" Scientia Pharmaceutica 90, no. 2: 36. https://doi.org/10.3390/scipharm90020036
APA StyleGürel, B., Çapkın, E., Parlar, A., Özkan, A., Çorbacıoğlu, M., Dağlikoca, D. E., & Yüce, M. (2022). Optimized Methods for Analytical and Functional Comparison of Biosimilar mAb Drugs: A Case Study for Avastin, Mvasi, and Zirabev. Scientia Pharmaceutica, 90(2), 36. https://doi.org/10.3390/scipharm90020036