
Pharmacokinetic Study of Mucoadhesive Itopride Hydrochloride In Situ Nasal Gel Formulations in a Comparative In Vivo Study and Histopathological Safety Evaluation


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Preparation of In Situ Nasal Gels
2.2.2. In Vivo Study and Evaluation of ITO HCl in Rabbit Plasma
2.2.3. Animal Handling, Study Design, and Drug Administration
2.2.4. Sample Collection
2.2.5. Chromatographic Procedure
2.2.6. Determination of ITO HCl in Rabbit Plasma
2.2.7. Histopathological Study
3. Results and Discussion
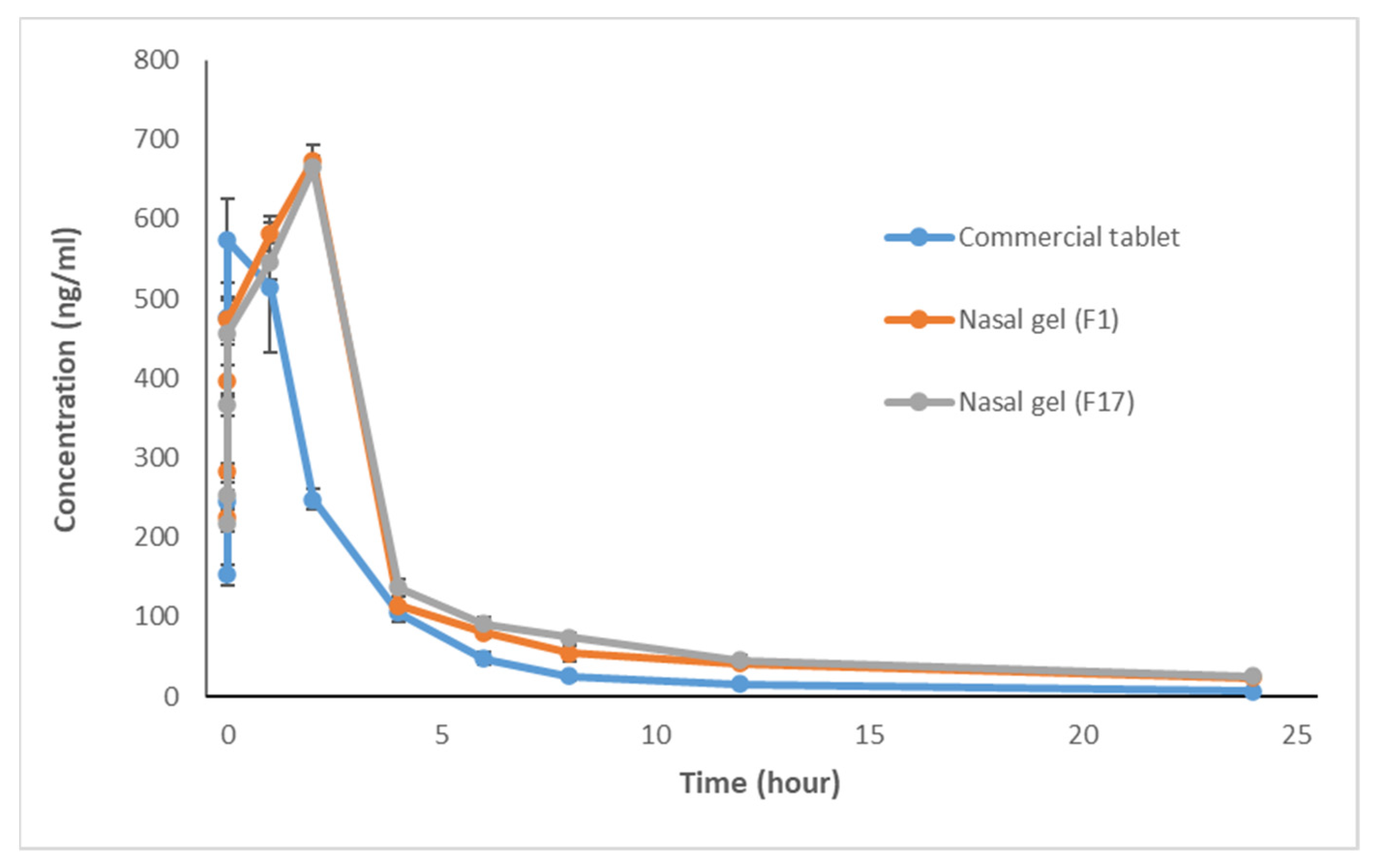
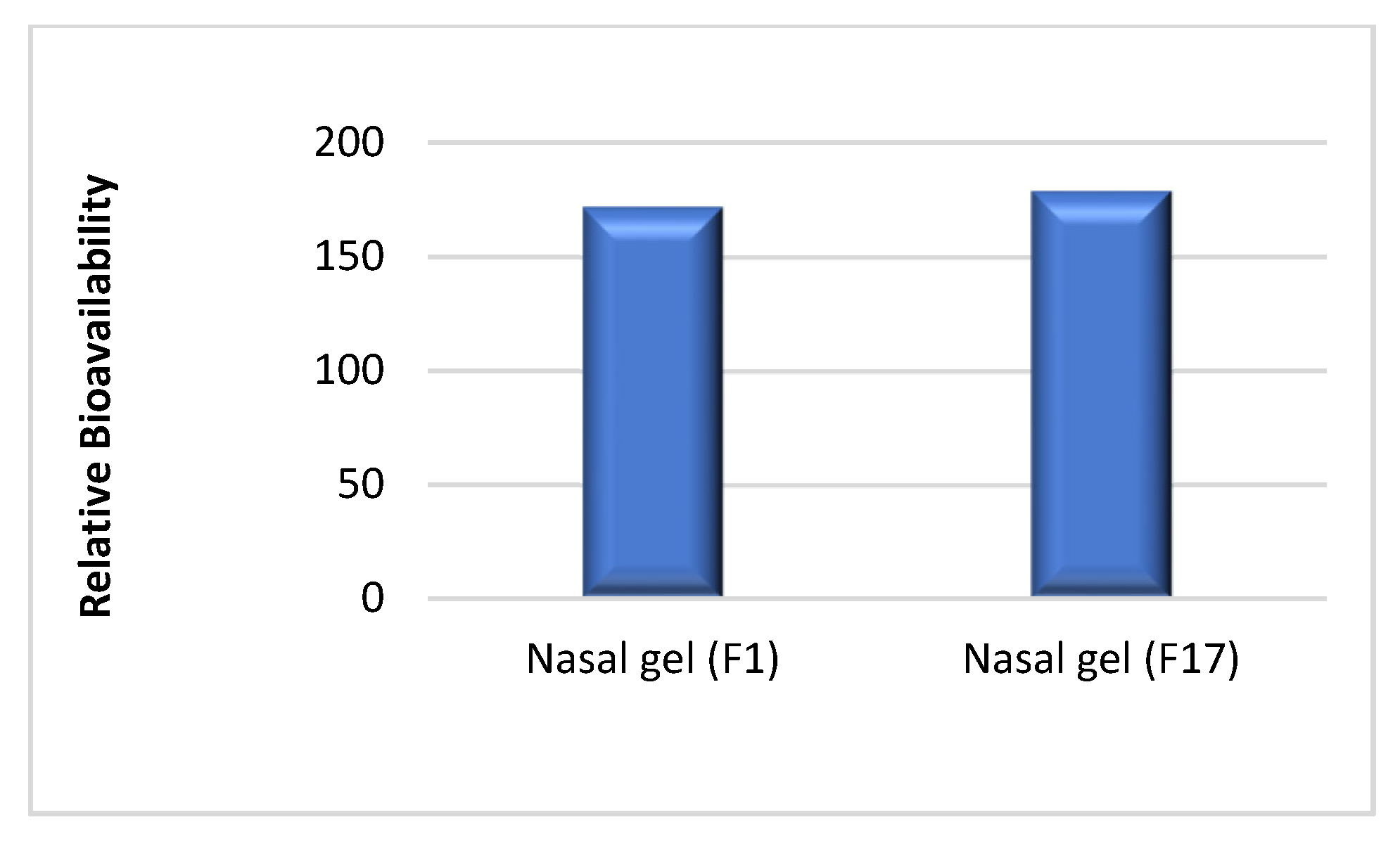
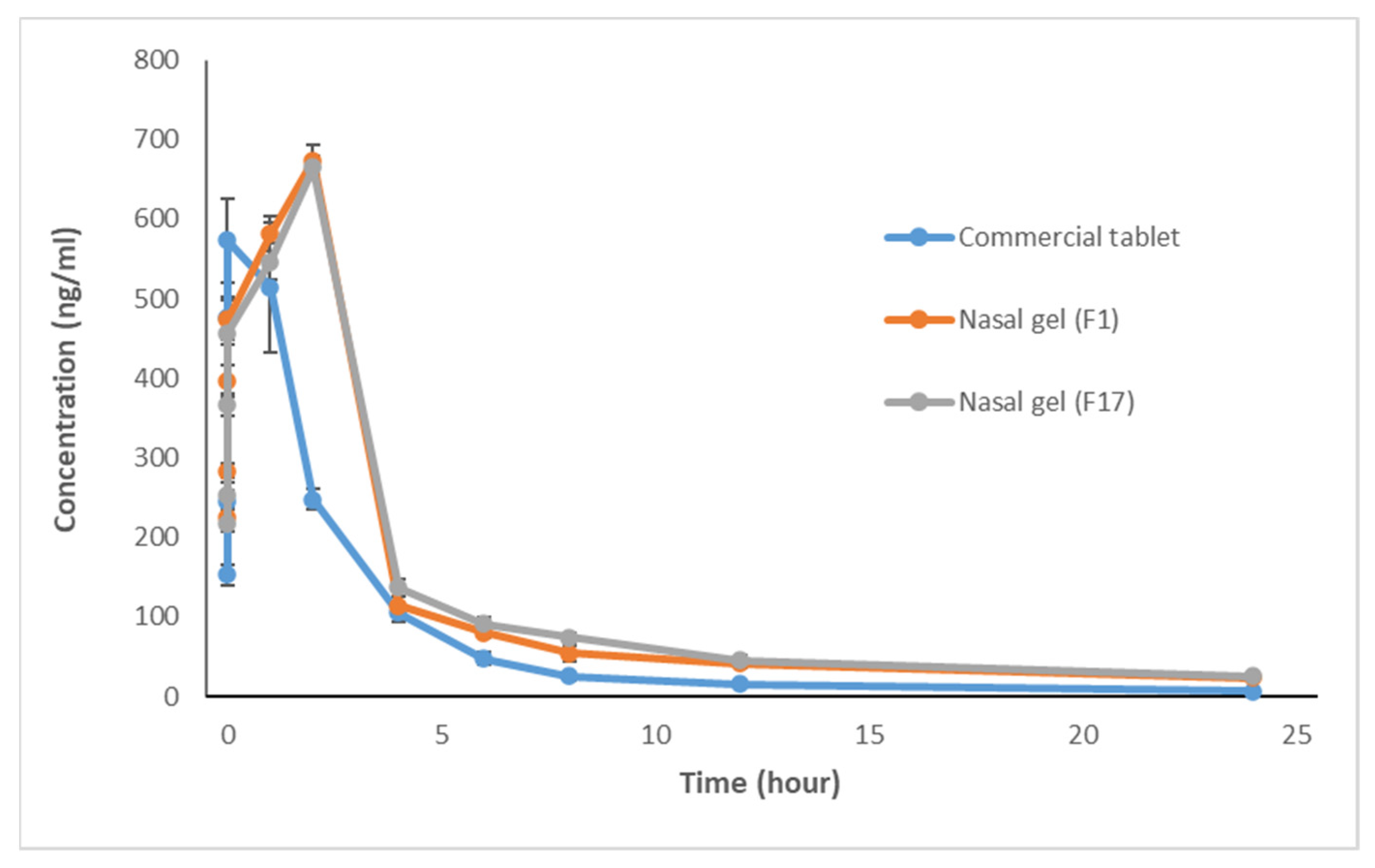
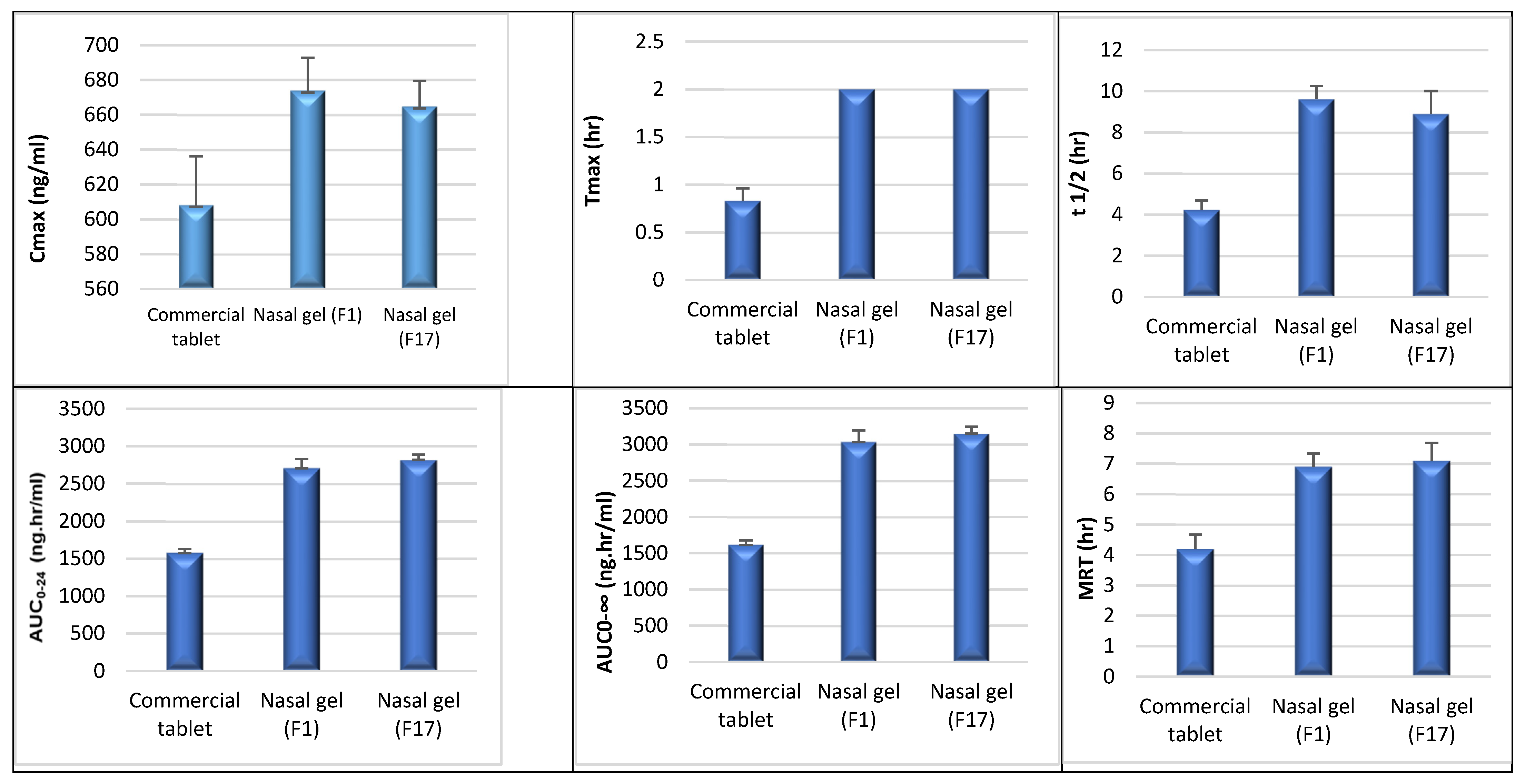
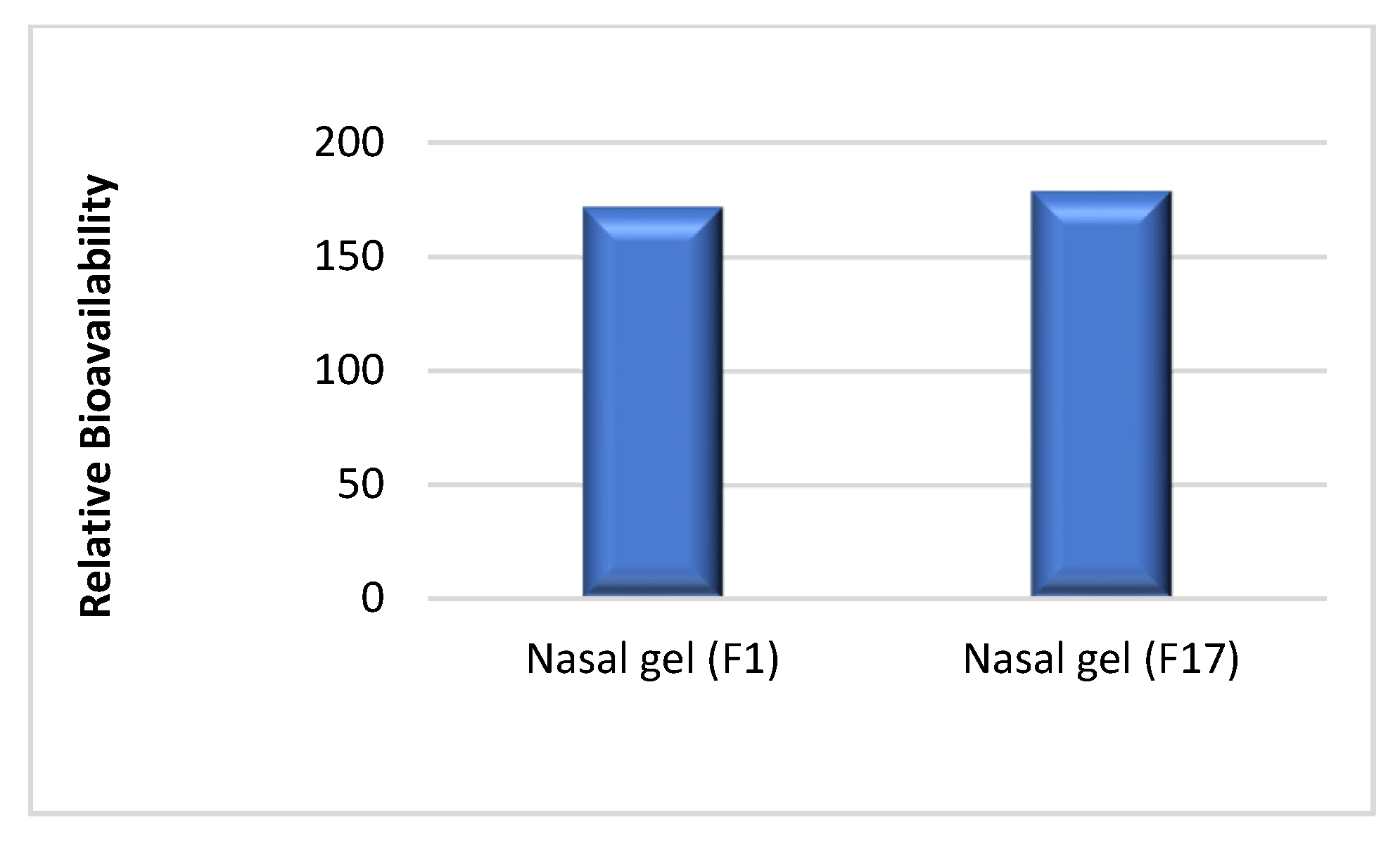
3.1. In Vivo Study and Evaluation of ITO HCl in Rabbit Plasma
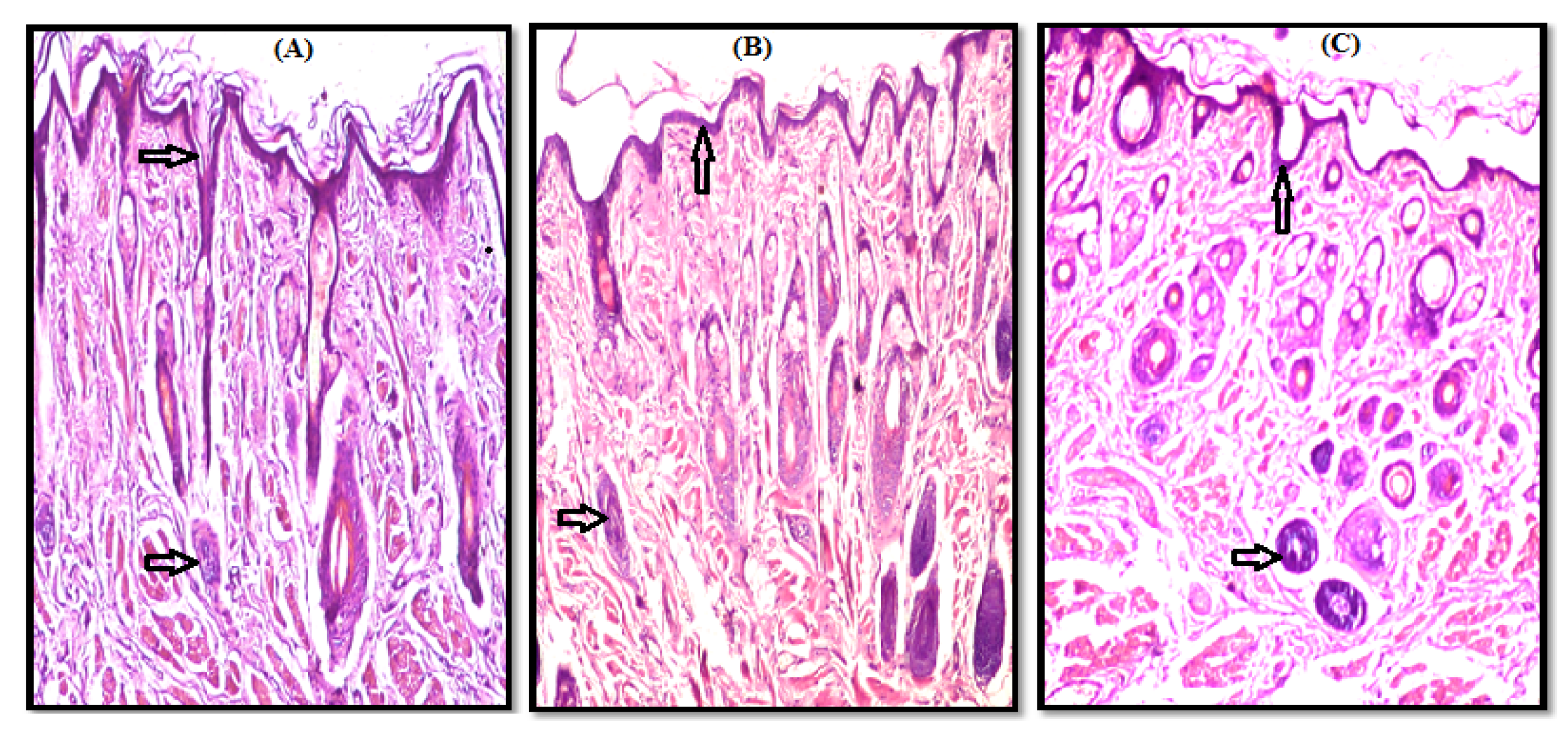
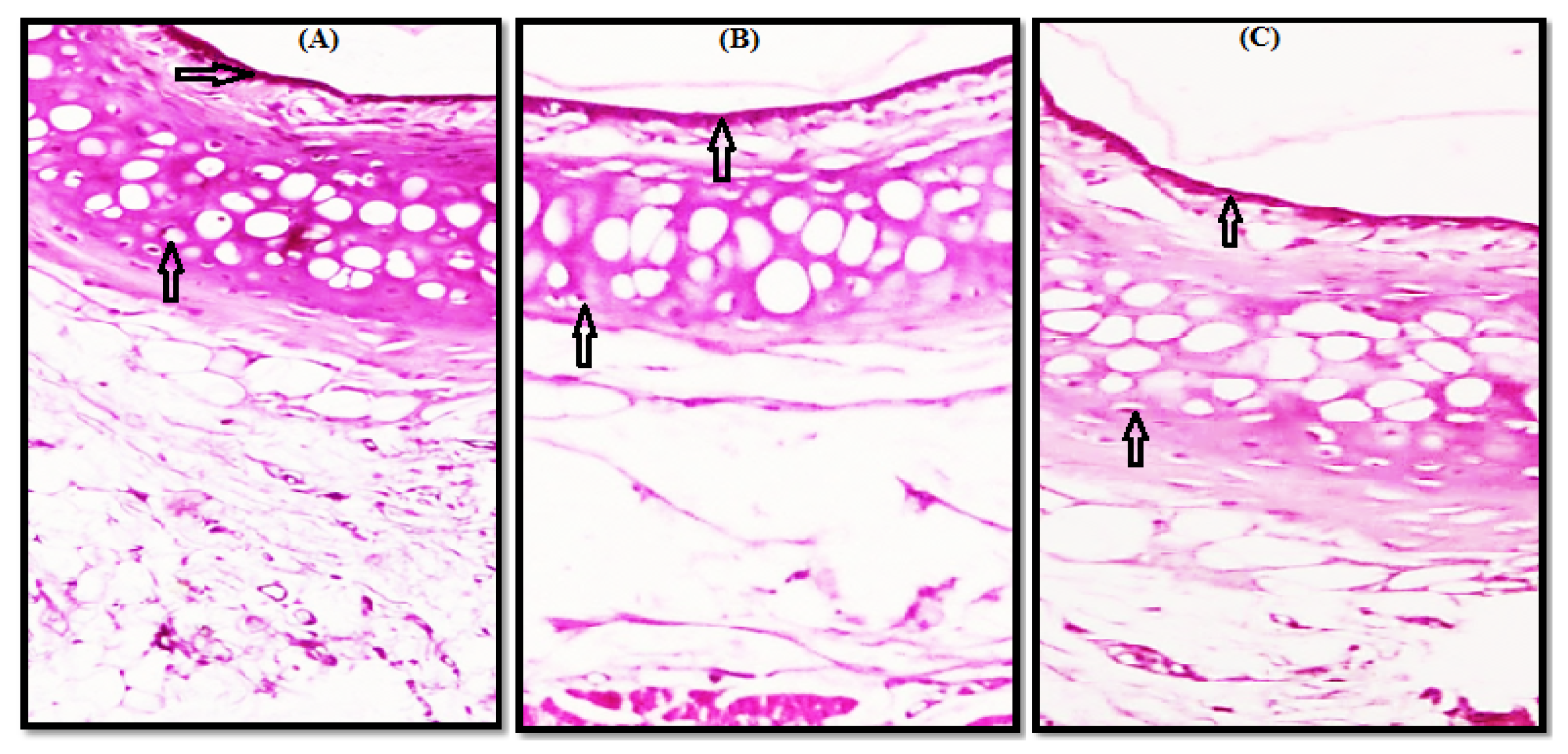
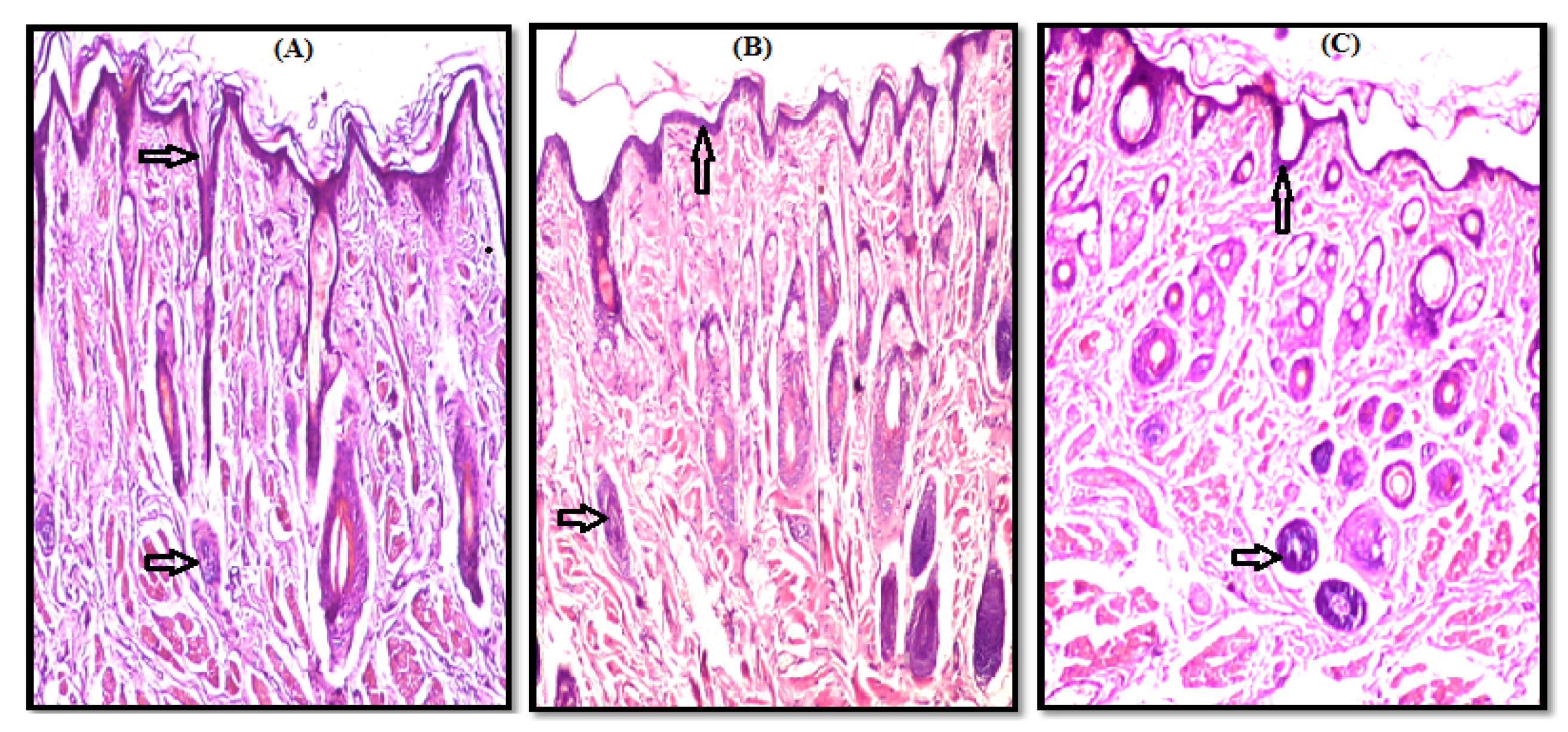
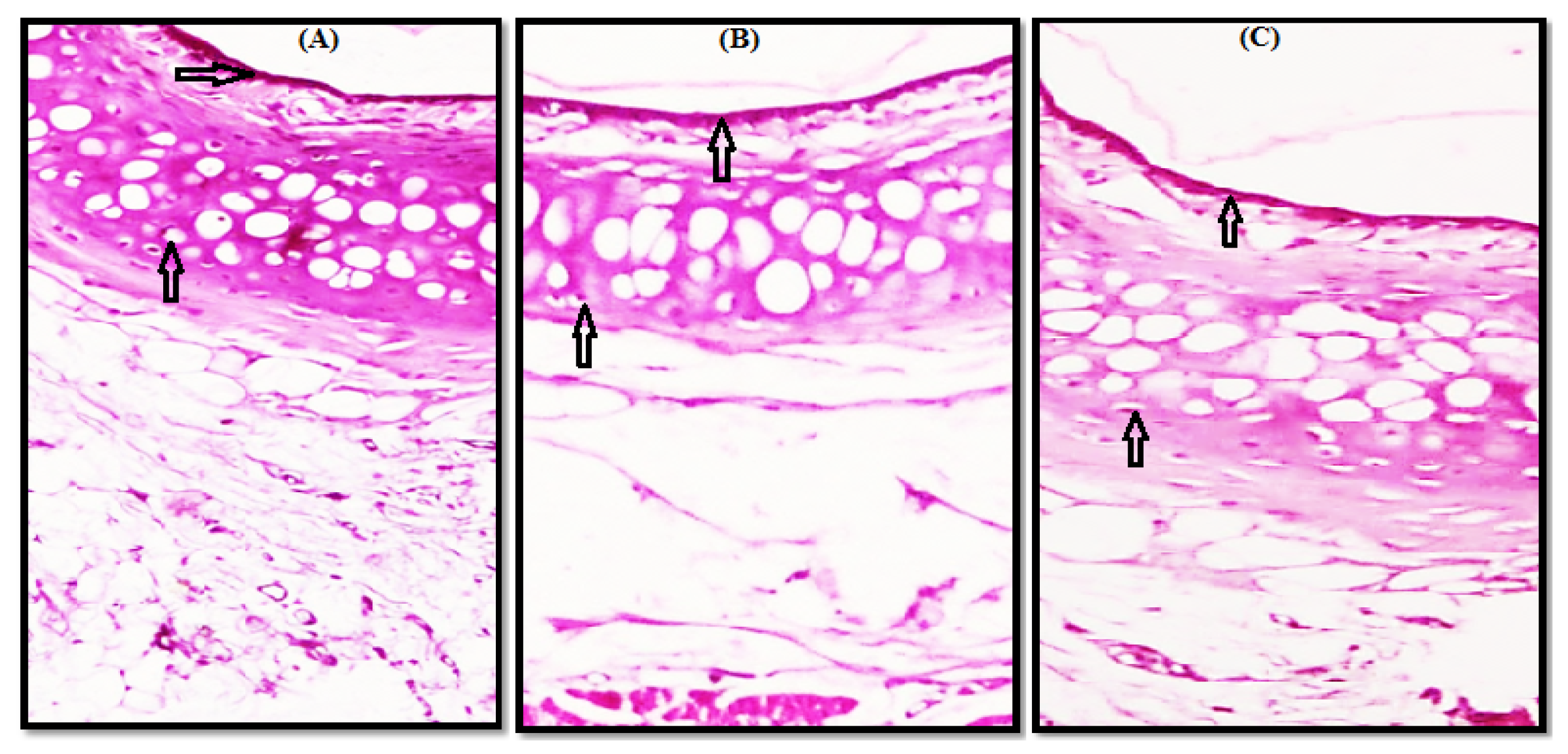
3.2. Histopathological Study
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Verma, P.; Prashar, N.; Kumar, D.; Chaudhary, H. Nasal (In-situ) Gel (Phenylepherine HCl) for Allergic Rhinitis Congestion treatment: Development and Characterization. Am. J. PharmTech Res. 2016, 6, 299–314. [Google Scholar]
- Ali, A.; Prajapati, S.K.; Devendra, S.; Kumar, B.; Kausar, S. Enhanced Bioavailability of Drugs Via Intranasal Drug Delivey System. Int. Res. J. Pharm. 2012, 3, 68–74. [Google Scholar]
- Kim, D.; Kim, Y.H.; Kwon, S. Enhanced Nasal Drug Delivery Efficiency by Increasing Mechanical Loading Using Hypergravity. Sci. Rep. 2018, 8, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, J.; Pandey, V.P. Antiemetic Drugs as a Nasal Drug Delivery—A Review. Int. J. Pharm. Sci. Res. 2014, 5, 1624. [Google Scholar] [CrossRef]
- Ban, M.; Dhembre, G.; Joshi, D. In-Situ Gel for Nasal Drug Delivery. Int. J. Dev. Res. 2018, 8, 18763–18769. [Google Scholar]
- Chen, S.Y.; Feng, Z.; Yi, X. A General Introduction to Adjustment for Multiple Comparisons. J. Thorac. Dis. 2017, 9, 1725–1729. [Google Scholar] [CrossRef] [Green Version]
- Chaturvedi, M.; Kumar, M.; Pathak, K. A Review on Mucoadhesive Polymer Used in Nasal Drug Delivery System. J. Adv. Pharm. Technol. Res. 2011, 2, 215–222. [Google Scholar] [CrossRef]
- Yehia, S.; Elshafeey, A.; Elmeshad, A.; Al-Bialey, H. A Pharmaceutical Study on Different Approaches for Itopride Hydrochloride Sustainment: In-Vitro and In-Vivo Evaluation. J. Chem. Pharm. Res. 2012, 4, 1398–1412. [Google Scholar]
- Danish Khan, A.; Singh, L. Various Techniques of Bioavailibility Enhancement: A Review. J. Drug Deliv. Ther. 2016, 6, 34–41. [Google Scholar] [CrossRef]
- Marzouk, M.A.; Osman, D.A.; Abd El-Fattah, A.I. Formulation and In Vitro Evaluation of a Thermoreversible Mucoadhesive Nasal Gel of Itopride Hydrochloride. Drug Dev. Ind. Pharm. 2018, 44, 1857–1867. [Google Scholar] [CrossRef]
- Council, N.R. Guide for the Care and Use of Laboratory Animals: Eighth Edition; The National Academies Press: Washington, DC, USA, 2011; p. 246. [Google Scholar]
- Zaki, N.M.; Awad, G.A.; Mortada, N.D.; Abd Elhady, S.S. Enhanced Bioavailability of Metoclopramide Hcl by Intranasal Administration of a Mucoadhesive In Situ Gel with Modulated Rheological and Mucociliary Transport Properties. Eur. J. Pharm. Sci. 2007, 32, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, A.A.; Abdel-Rahman, S.I.; Abdel-Aleem, J.A. Formulation of Sustained Release Itopride Hydrochloride Matrix Tablets Using Direct Compression Technique. Unique J. Pharm. Biol. Sci. 2013, 1, 72–81. [Google Scholar]
- Shah, S.; Madan, S.; Agrawal, S. Formulation and Evaluation of Microsphere Based Oro Dispersible Tablets of Itopride Hcl. Daru 2012, 20, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aboud, H.M.; El Komy, M.H.; Ali, A.A.; El Menshawe, S.F.; Abd Elbary, A. Development, Optimization, and Evaluation of Carvedilol-Loaded Solid Lipid Nanoparticles for Intranasal Drug Delivery. AAPS PharmSciTech 2016, 17, 1353–1365. [Google Scholar] [CrossRef] [PubMed]
- Patro, S. Determination and Validation of A HPLC Method with UV Detectionof Itopride Hydrochloride in Human Serum. Int. J. Pharm. Pharm. Sci. 2015, 7, 246–249. [Google Scholar]
- Jensen, K. Theory and Practice of Histological Techniques, 6th Edition. J. Neuropathol. Exp. Neurol. 2008, 67, 633. [Google Scholar] [CrossRef] [Green Version]
- Omar, M.M.; Eleraky, N.E.; El Sisi, A.M.; Ali Hasan, O. Development and Evaluation of in-situ Nasal Gel Formulations of Nanosized Transferosomal Sumatriptan: Design, Optimization, in vitro and in vivo Evaluation. Drug Des. Devel. 2019, 13, 4413–4430. [Google Scholar] [CrossRef] [Green Version]
- Ravi, P.R.; Aditya, N.; Patil, S.; Cherian, L. Nasal In-Situ Gels for Delivery of Rasagiline Mesylate: Improvement in Bioavailability and Brain Localization. Drug Deliv. 2015, 22, 903–910. [Google Scholar] [CrossRef]
- Ahmad, W.M.A.; Shafiq, M.; Abdul Halim, N.; Aleng, N. Statistical Analysis Using SPSS Version 24; Penerbit USM: Gelugor, Malaysia, 2019; ISBN 978-967-461-339-6. [Google Scholar]
- Ahmed, S.M.; Ahmed Ali, A.; Ali, A.M.; Hassan, O.A. Design and In Vitro/In Vivo Evaluation of Sustained-Release Floating Tablets of Itopride Hydrochloride. Drug Des. Devel. 2016, 10, 4061–4071. [Google Scholar] [CrossRef] [Green Version]
- Bary, G.A. Preparation and Characterization of Thermosensitive Mucoadhesive In_Situ Gels for Nasal Delivery of Ondansetron Hydrochloride. Al-Azhar J. Pharm. Sci. 2014, 50, 191–207. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Shentu, J.-z.; Wu, L.-h.; Dou, J.; Xu, Q.-y.; Zhou, H.-l.; Wu, G.-l.; Huang, M.-z.; Hu, X.-j.; Chen, J.-c. Relative Bioavailability and Pharmacokinetic Comparison of Two Different Enteric Formulations of Omeprazole. J. Zhejiang Univ. Sci. B 2012, 13, 348–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Screening Number = 6 | Treatment period (I) | Wash Out Period (7 days) | Treatment period (II) | Wash Out Period (7 days) | Treatment period (III) |
| Group (I) n = 2 A, B | Group (I) n = 2 E, F | Group (I) n = 2 C, D | |||
| tablets | tablets | tablets | |||
| Group (II) n = 2 C, D | Group (II) n = 2 A, B | Group (II) n = 2 E, F | |||
| F1 | F1 | F1 | |||
| Group (III) n = 2 E, F | Group (III) n = 2 C, D | Group (III) n = 2A, B | |||
| F17 | F17 | F17 |
| Time (h) | Gp I (Commercial Tablet) | Gp II (Nasal Gel F1) | Gp III (Nasal Gel F17) | ANOVA p Value | P1 | P2 | P3 |
|---|---|---|---|---|---|---|---|
| 0.08 | 152.71 ± 12.57 | 224.96 ± 11.75 | 217.09 ± 10.33 | 70.216, 0.001 * | 0.001 * | 0.001 * | 0.258 |
| 0.25 | 244.37 ± 15.02 | 284.10 ± 9.29 | 252.40 ± 16.89 | 13.309, 0.003 * | 0.002 * | 0.340 | 0.01 * |
| 0.5 | 475.87 ± 26.89 | 397.28 ± 19.62 | 366.42 ± 13.86 | 44.093, 0.001 * | 0.011 * | 0.001 * | 0.021 * |
| 0.75 | 573.18 ± 53.32 | 473.47 ± 24.83 | 457.07 ± 15.39 | 19.226, 0.002 * | 0.013 * | 0.012 * | 0.431 |
| 1 | 514.65 ± 81.54 | 581.68 ± 21.70 | 546.59 ± 23.00 | 2.646,0.104 | 0.063 | 0.290 | 0.247 |
| 2 | 247.60 ± 12.92 | 673.87 ± 18.86 | 664.79 ± 14.72 | 444.1, 0.001 * | 0.001 * | 0.001 * | 0.33 |
| 4 | 105.45 ± 10.94 | 113.94 ± 12.29 | 137.33 ± 10.63 | 12.790, 0.016 * | 0.213 | 0.001 * | 0.003 * |
| 6 | 47.46 ± 8.24 | 79.97 ± 6.29 | 91.02 ± 8.69 | 50.464, 0.001 * | 0.001 * | 0.002 * | 0.027 * |
| 8 | 25.54 ± 5.49 | 54.19 ± 9.73 | 74.35 ± 6.09 | 66.897, 0.001 * | 0.001 * | 0.0013 * | 0.0011 * |
| 12 | 15.82 ± 4.25 | 42.06 ± 5.20 | 45.11 ± 6.17 | 56.116, 0.001 * | 0.001 * | 0.001 * | 0.323 |
| 24 | 6.80 ± 2.03 | 23.44 ± 2.92 | 25.47 ± 4.02 | 65.560, 0.001 * | 0.001 * | 0.001 * | 0.275 |
| Parameter | Commercial Tablet | Nasal Gel F1 | Nasal Gel F17 |
|---|---|---|---|
| Cmax (ng/mL) | 608.16 ± 28.10 | 673.87 ± 18.86 | 664.79 ± 14.72 |
| Tmax (h) | 0.83 ± 0.13 | 2 ± 0 | 2 ± 0 |
| t1/2el (h) | 4.23 ± 0.47 | 9.61 ± 0.65 | 8.89 ± 1.12 |
| AUC0–24 (ng·h/mL) | 1575.10 ± 52.91 | 2706.81 ± 119.84 | 2817.83 ± 68.00 |
| AUC0–∞ (ng·h/mL) | 1617.56 ± 62.35 | 3033.50 ± 158.16 | 3149.27 ± 96.01 |
| MRT (h) | 4.19 ± 0.48 | 6.90 ± 0.43 | 7.10 ± 0.59 |
| Relative bioavailability | - | 171.85 | 178.90 |
| Parameter | Formulations | ANOVA p Value | P1 | P2 | P3 | ||
|---|---|---|---|---|---|---|---|
| Commercial Tablets | F1 | F17 | |||||
| Tmax (h) | 0.83 ± 0.13 | 2 ± 0 | 2 ± 0 | 490.00, 0.001 * | 0.001 * | 0.001 * | 0.275 |
| Cmax (ng/mL) | 608.16 ± 28.10 | 673.87 ± 18.86 | 664.79 ± 14.72 | 16.754, 0.013 * | 0.011 * | 0.035 * | 0.472 |
| Ke (h−1) | 0.1655 ± 0.020 | 0.0724 ± 0.0049 | 0.07906 ± 0.0103 | 91.042, 0.001 * | 0.001 * | 0.001 * | 0.398 |
| t1/2 (h) | 4.23 ± 0.47 | 9.61 ± 0.65 | 8.89 ± 1.12 | 80.640, 0.001 * | 0.001 * | 0.002 * | 0.134 |
| AUC0–24 (ng·h/mL) | 1575.10 ± 52.91 | 2706.81 ± 119.84 | 2817.83 ± 68.00 | 390.735, 0.001 * | 0.002 * | 0.004 * | 0.039 |
| AUC24–inf (ng·h/mL) | 42.46 ± 15.71 | 326.69 ± 57.25 | 331.43 ± 88.87 | 43.160, 0.001 * | 0.001 * | 0.001 * | 0.896 |
| AUC0–inf (ng·h/mL) | 1617.56 ± 62.35 | 3033.50 ± 158.16 | 3149.27 ± 96.01 | 341.756, 0.001 * | 0.002 * | 0.001 * | 0.096 |
| AUMC0–24 (ng·h2/mL) | 5769.78 ± 642.57 | 13,125.27 ± 1137.66 | 14,456.03 ± 801.87 | 167.637, 0.001 * | 0.003 * | 0.001 * | 0.02 * |
| AUMC24–inf (ng·h2/mL) | 1019.05 ± 376.95 | 7840.597 ± 1373.89 | 7954.39 ± 2132.86 | 43.159, 0.001 * | 0.0013 * | 0.005 * | 0.896 |
| AUMC0–inf (ng·h2/mL) | 6788.82 ± 948.72 | 20,965.87 ± 2269.20 | 22,410.42 ± 2460.27 | 110.832, 0.001 * | 0.001 * | 0.001 * | 0.232 |
| MRT (h) | 4.19 ± 0.48 | 6.90 ± 0.43 | 7.10 ± 0.59 | 62.322, 0.001 * | 0.002 * | 0.001 * | 0.486 |
| Vd (L) | 13.72 ± 4.00 | 34.26 ± 4.29 | 26.40 ± 5.06 | 32.234, 0.006 * | 0.001 * | 0.001 * | 0.018 |
| TCR (mL/min) | 36.9376 ± 7.387 | 41.1472 ± 4.232 | 34.12 ± 2.79 | 2.80, 0.092 | 0.360 | 0.179 | 0.13 |
| Cmax/AUC0–24 (h−1) | 0.38672 ± 0.0268 | 0.2492 ± 0.0080 | 0.2361 ± 0.0098 | 142.296, 0.001 * | 0.001 * | 0.001 * | 0.260 |
| Relative Bioavailability (%) | - | 171.85 | 178.90 | T = 1.01, 0.211 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marzouk, M.A.; Osman, D.A.; Abd El-Fattah, A.I.; Aldeeb, R.A. Pharmacokinetic Study of Mucoadhesive Itopride Hydrochloride In Situ Nasal Gel Formulations in a Comparative In Vivo Study and Histopathological Safety Evaluation. Sci. Pharm. 2022, 90, 8. https://doi.org/10.3390/scipharm90010008
Marzouk MA, Osman DA, Abd El-Fattah AI, Aldeeb RA. Pharmacokinetic Study of Mucoadhesive Itopride Hydrochloride In Situ Nasal Gel Formulations in a Comparative In Vivo Study and Histopathological Safety Evaluation. Scientia Pharmaceutica. 2022; 90(1):8. https://doi.org/10.3390/scipharm90010008
Chicago/Turabian StyleMarzouk, Maha A., Dina A. Osman, Amany I. Abd El-Fattah, and Reem A. Aldeeb. 2022. "Pharmacokinetic Study of Mucoadhesive Itopride Hydrochloride In Situ Nasal Gel Formulations in a Comparative In Vivo Study and Histopathological Safety Evaluation" Scientia Pharmaceutica 90, no. 1: 8. https://doi.org/10.3390/scipharm90010008
APA StyleMarzouk, M. A., Osman, D. A., Abd El-Fattah, A. I., & Aldeeb, R. A. (2022). Pharmacokinetic Study of Mucoadhesive Itopride Hydrochloride In Situ Nasal Gel Formulations in a Comparative In Vivo Study and Histopathological Safety Evaluation. Scientia Pharmaceutica, 90(1), 8. https://doi.org/10.3390/scipharm90010008