
Olanzapine Loaded Nanostructured Lipid Carriers via High Shear Homogenization and Ultrasonication


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Preparation of Blank and OLZ-Loaded NLCs
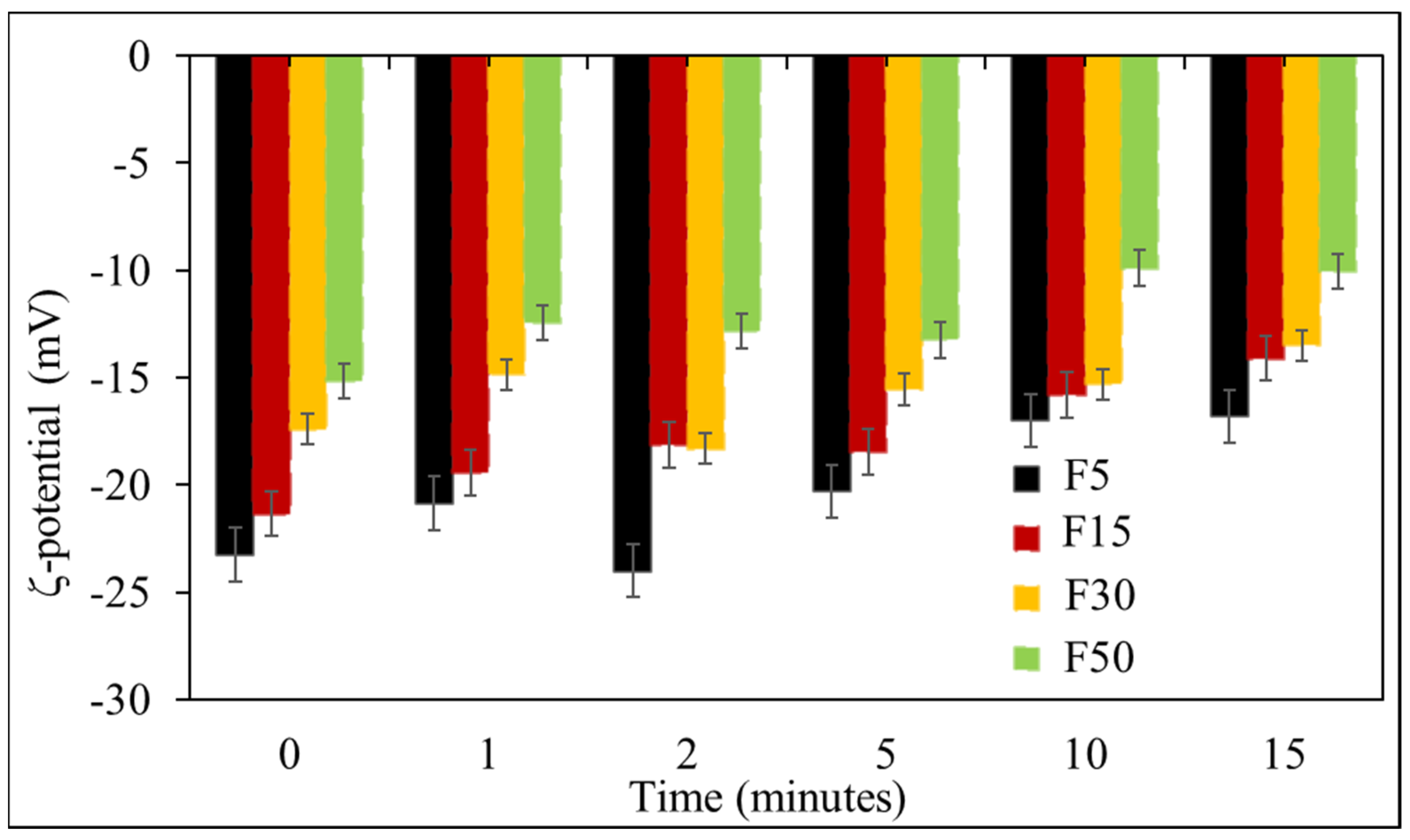
2.2.2. Determination of Particle Size, PDI and Zeta Potential
2.2.3. Bright Field Scanning Transmission Electron Microscopy (BF-STEM)
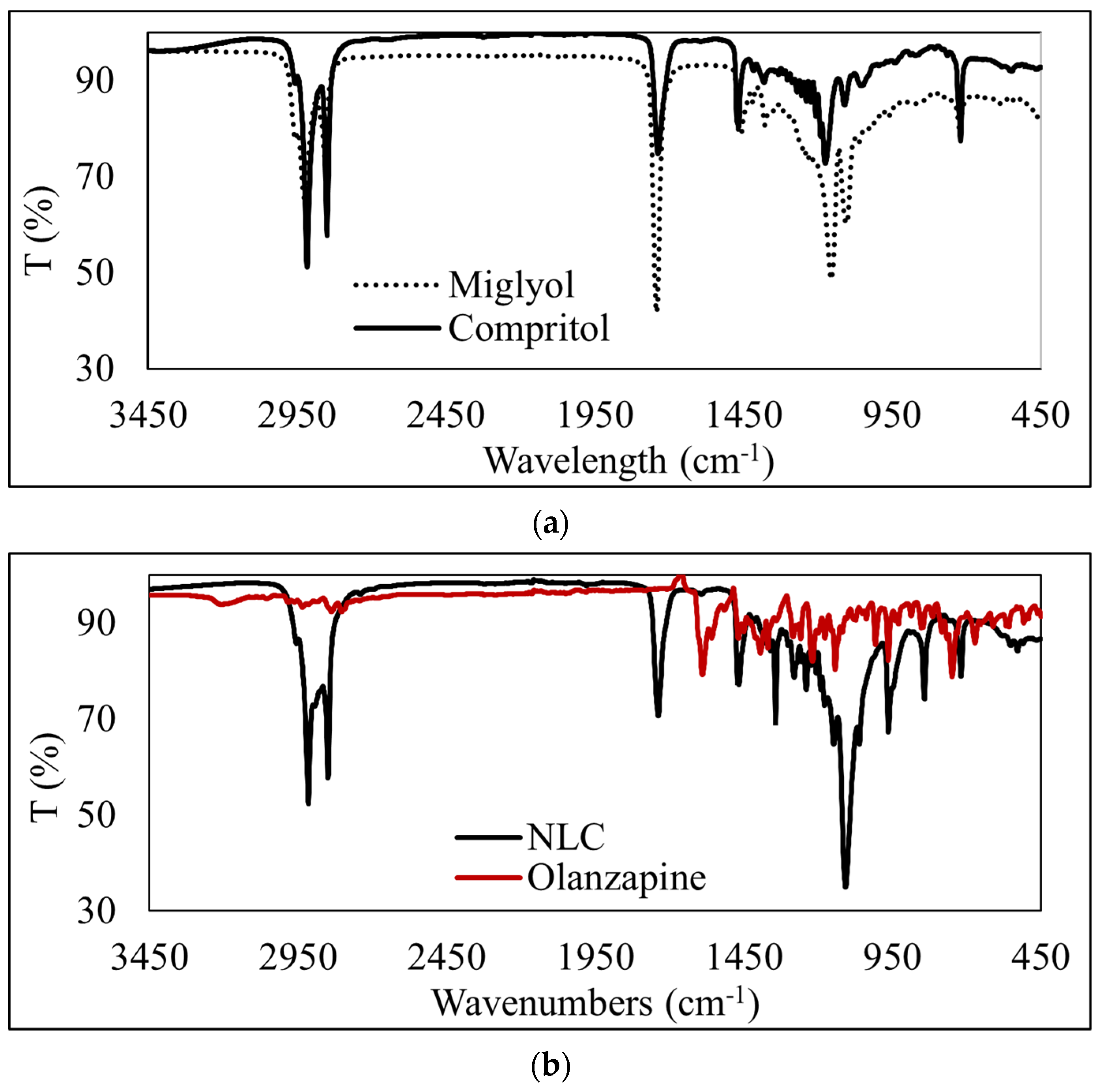
2.2.4. Attenuated Total Reflectance-Fourier Transform Infrared (ATR-FTIR) Spectroscopy
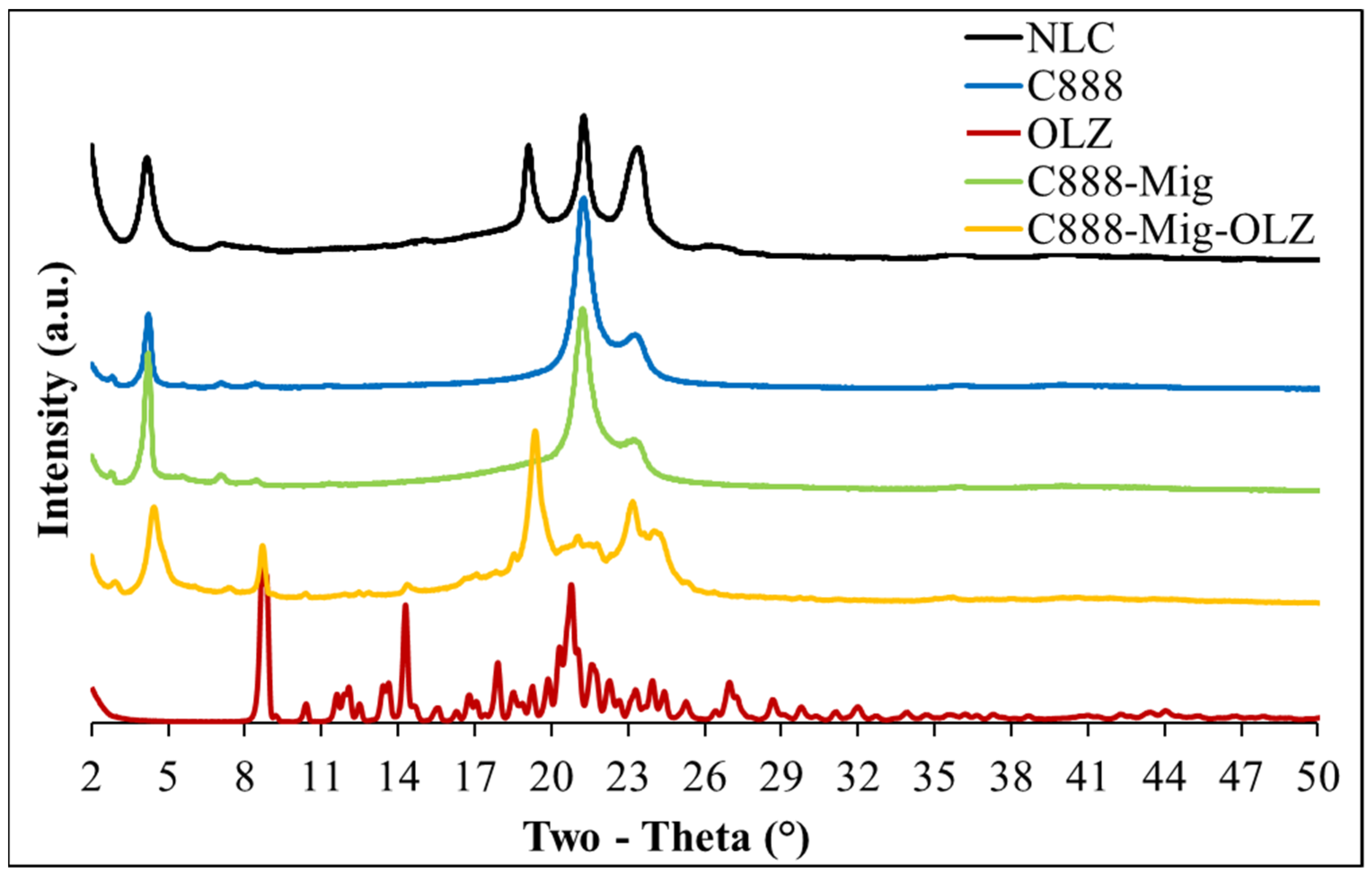
2.2.5. X-Ray Diffraction (XRD) Analysis
2.2.6. Drug Entrapment Efficiency
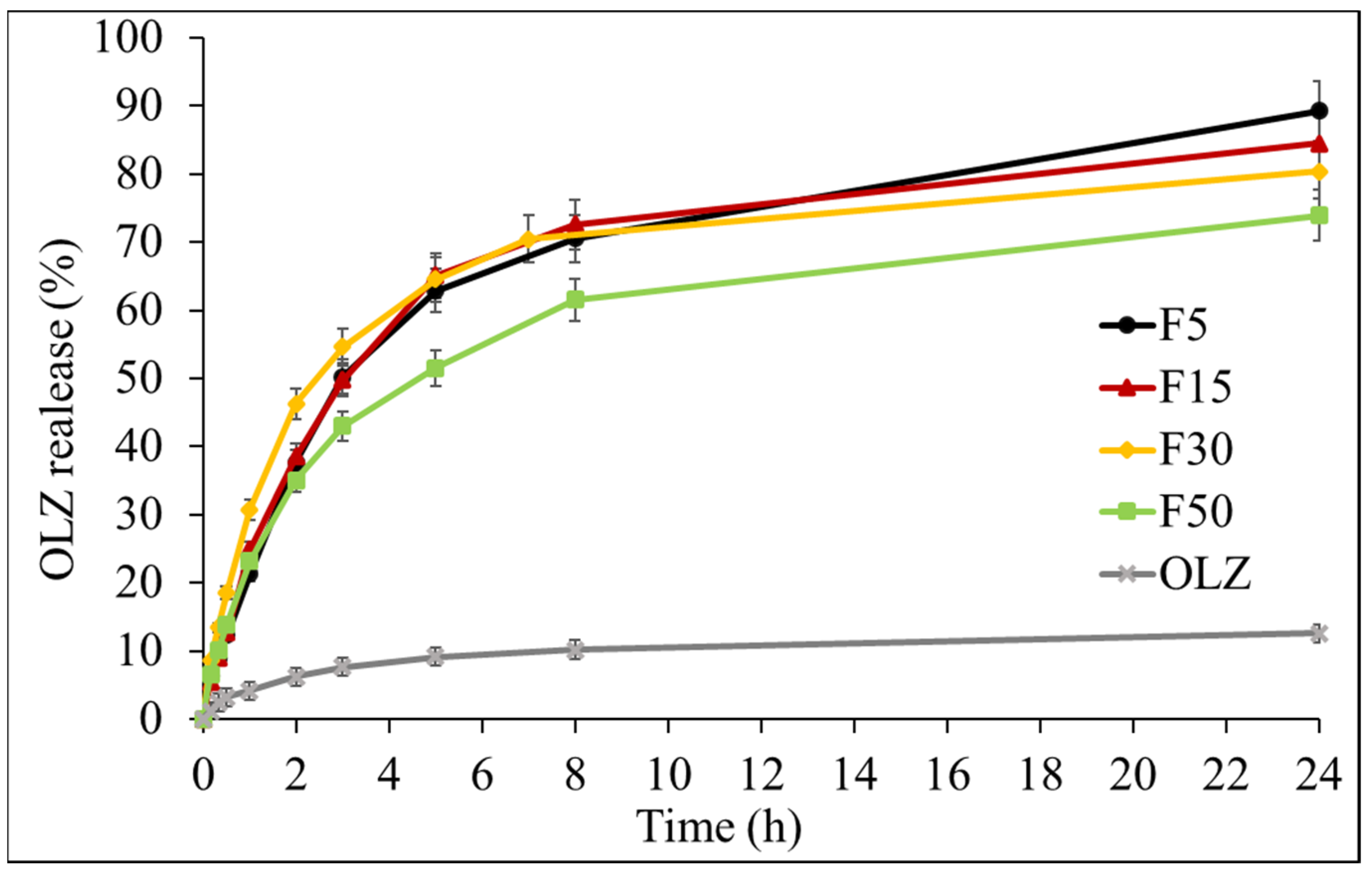
2.2.7. Drug Release Studies
2.2.8. Storage Stability Studies
3. Results and Discussion
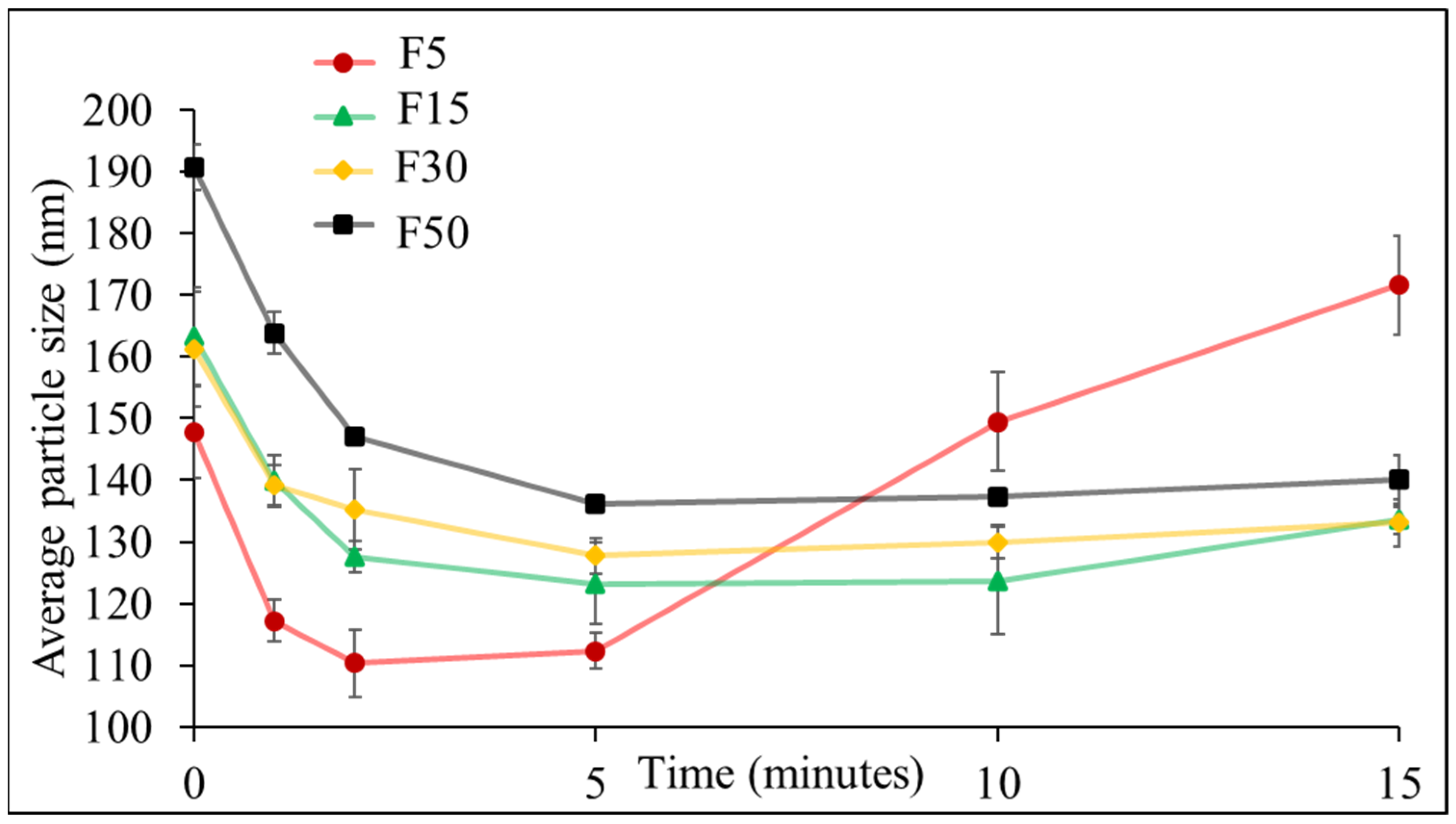
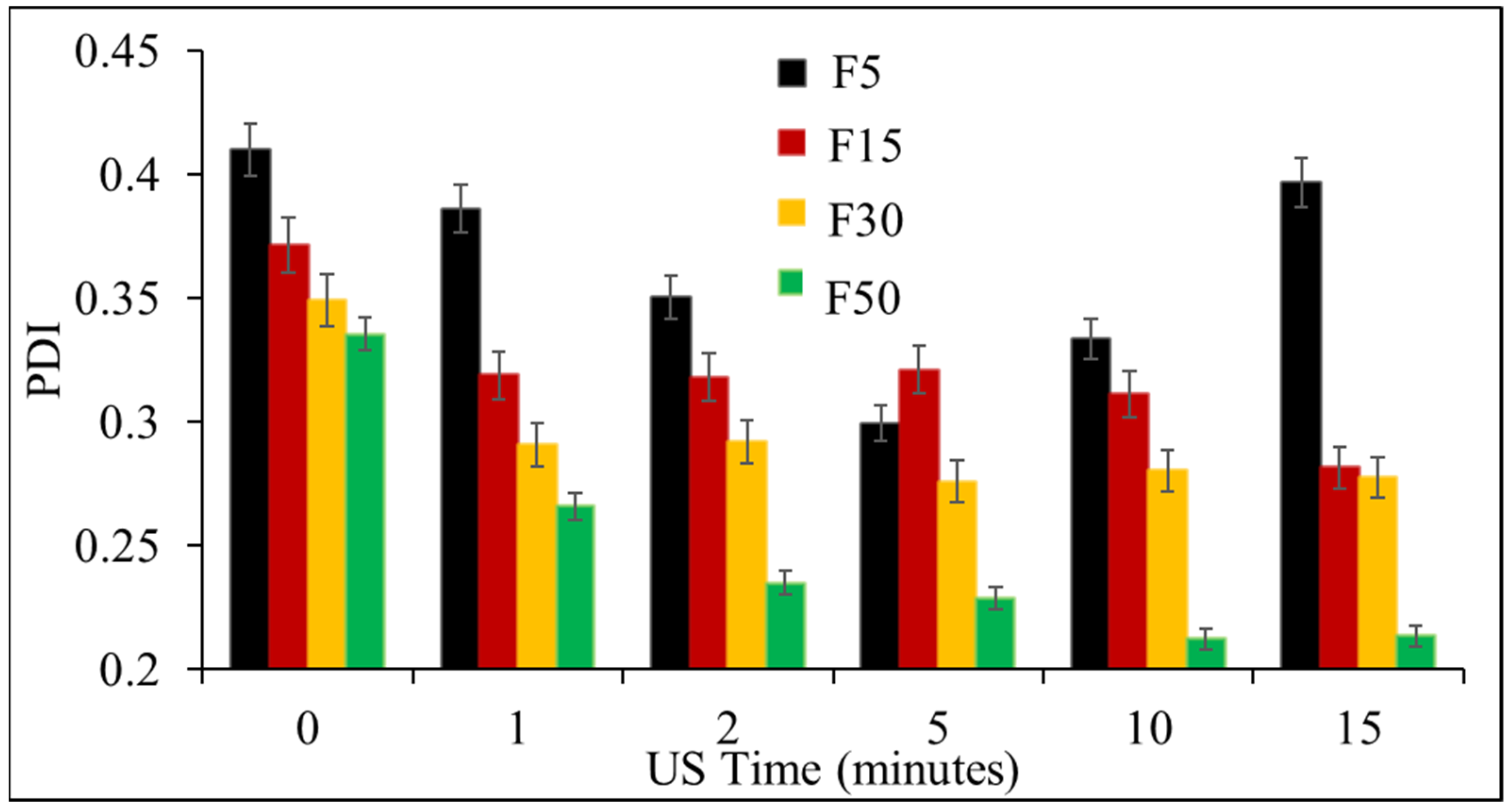
3.1. Effect of Ultrasonication on the Formulation of NLCs
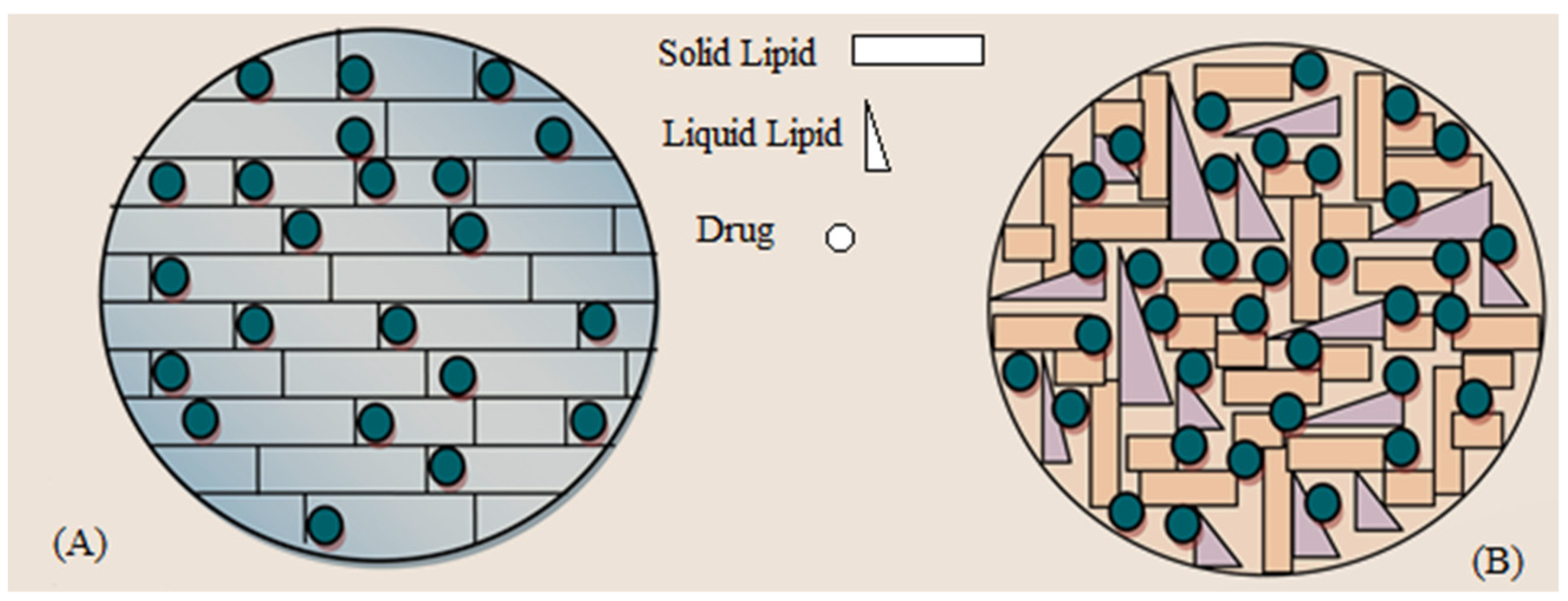
3.2. Effect of Solid to Liquid Lipid Ratio on OLZ Encapsulation
3.3. BF-STEM Analysis and Encapsulation Efficiency
3.4. ATR-FTIR Analysis of OLZ-Loaded NLCs
3.5. XRD Analysis of OLZ-Loaded NLCs
3.6. Drug Release Studies
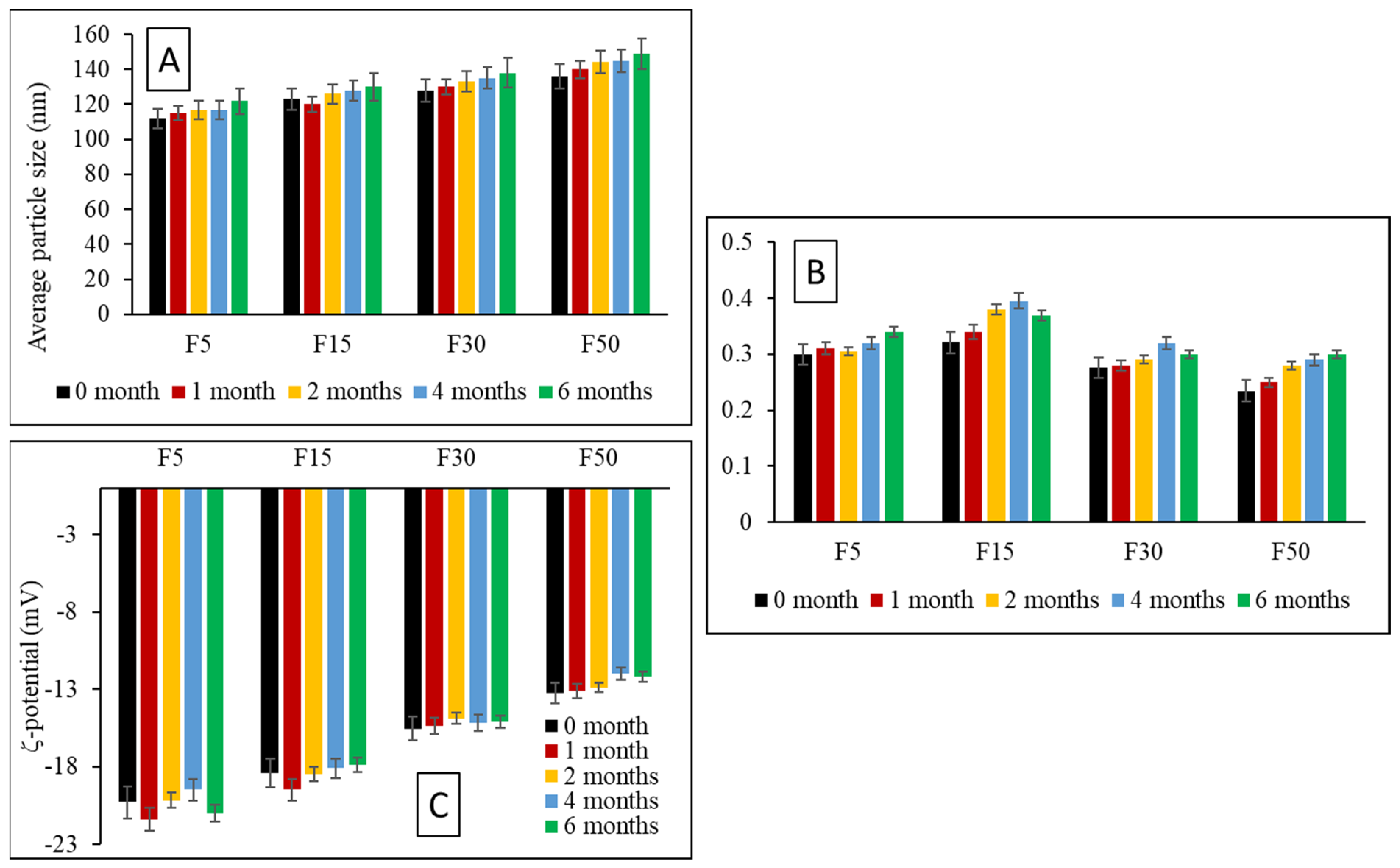
3.7. Storage Stability Studies
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Pinheiro, M.; Ribeiro, R.; Vieira, A.; Andrade, F.; Reis, S. Design of a nanostructured lipid carrier intended to improve the treatment of tuberculosis. Drug Des. Dev. Ther. 2016, 10, 2467–2475. [Google Scholar] [CrossRef] [PubMed]
- Nasirizadeh, S.; Malaekeh-Nikouei, B. Solid lipid nanoparticles and nanostructured lipid carriers in oral cancer drug delivery. J. Drug Deliv. Sci. Technol. 2020, 55. [Google Scholar] [CrossRef]
- Souto, E.B.; Müller, R.H. Lipid Nanoparticles: Effect on Bioavailability and Pharmacokinetic Changes. Organotypic Models Drug Dev. 2009, 197, 115–141. [Google Scholar] [CrossRef]
- Salvi, V.R.; Pawar, P. Nanostructured lipid carriers (NLC) system: A novel drug targeting carrier. J. Drug Deliv. Sci. Technol. 2019, 51, 255–267. [Google Scholar] [CrossRef]
- Müller, R.H.; Radtke, M.; Wissing, S.A. Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) in cosmetic and dermatological preparations. Adv. Drug. Deliv. Rev. 2002, 54, S131–S155. [Google Scholar] [CrossRef]
- Liu, D.; Liu, Z.; Wang, L.; Zhang, C.; Zhang, N. Nanostructured lipid carriers as novel carrier for parenteral delivery of docetaxel. Colloids Surfaces B Biointerfaces 2011, 85, 262–269. [Google Scholar] [CrossRef]
- Lin, Y.-K.; Huang, Z.-R.; Zhuo, R.-Z.; Fang, J.-Y. Combination of calcipotriol and methotrexate in nanostructured lipid carriers for topical delivery. Int. J. Nanomed. 2010, 5, 117. [Google Scholar]
- Desai, P.; Date, A.A.; Patravale, V.B. Overcoming poor oral bioavailability using nanoparticle formulations—opportunities and limitations. Drug Discov. Today Technol. 2012, 9, e87–e95. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.-Y.; Li, N.; Wang, M.; Zhang, X.-N.; Pan, W.-S.; Peng, J.-J.; Pan, Y.-S.; Tang, X. Preparation and characterization of vinpocetine loaded nanostructured lipid carriers (NLC) for improved oral bioavailability. Int. J. Pharm. 2010, 394, 179–185. [Google Scholar] [CrossRef]
- Medha, J.; Sulabha, P.; Shobhona, S. Design and in vivo pharmacodynamic evaluation of nanostructured lipid carriers for parenteral delivery of artemether: Nanoproject. Int. J. Pharm. 2008, 364, 119–126. [Google Scholar]
- Shen, J.; Deng, Y.; Jin, X.; Ping, Q.; Su, Z.; Li, L. Thiolated nanostructured lipid carriers as a potential ocular drug delivery system for cyclosporine A: Improving in vivo ocular distribution. Int. J. Pharm. 2010, 402, 248–253. [Google Scholar] [CrossRef]
- Patlolla, R.R.; Chougule, M.; Patel, A.R.; Jackson, T.; Tata, P.N.; Singh, M. Formulation, characterization and pulmonary deposition of nebulized celecoxib encapsulated nanostructured lipid carriers. J. Control. Release 2010, 144, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-Y.; Li, Y.-X.; Li, M.; Zhang, L.; Feng, L.-X.; Zhang, N. Hyaluronic acid-coated nanostructured lipid carriers for targeting paclitaxel to cancer. Cancer Lett. 2013, 334, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Mousa, S.A.; Bharali, D.J.; Khalil, M.; Gurbuz, M.; Simone, T.M. Nanoparticles and cancer therapy: A concise review with emphasis on dendrimers. Int. J. Nanomed. 2009, 4, 1–7. [Google Scholar] [CrossRef]
- Zhu, Q.; Feng, C.; Liao, W.; Zhang, Y.; Tang, S. Target delivery of MYCN siRNA by folate-nanoliposomes delivery system in a metastatic neuroblastoma model. Cancer Cell Int. 2013, 13, 65. [Google Scholar] [CrossRef]
- Taratula, O.; Kuzmov, A.; Shah, M.; Garbuzenko, O.B.; Minko, T. Nanostructured lipid carriers as multifunctional nanomedicine platform for pulmonary co-delivery of anticancer drugs and siRNA. J. Control. Release 2013, 171, 349–357. [Google Scholar] [CrossRef]
- Brugè, F.; Damiani, E.; Marcheggiani, F.; Offerta, A.; Puglia, C.; Tiano, L. A comparative study on the possible cytotoxic effects of different nanostructured lipid carrier (NLC) compositions in human dermal fibroblasts. Int. J. Pharm. 2015, 495, 879–885. [Google Scholar] [CrossRef]
- Gokce, E.; Korkmaz, E.; Dellera, E.; Sandri, G.; Bonferoni, M.C.; Ozer, O. Resveratrol-loaded solid lipid nanoparticles versus nanostructured lipid carriers: Evaluation of antioxidant potential for dermal applications. Int. J. Nanomed. 2012, 7, 1841–1850. [Google Scholar] [CrossRef] [PubMed]
- Gadhave, D.G.; Tagalpallewar, A.A.; Kokare, C.R. Agranulocytosis-Protective Olanzapine-Loaded Nanostructured Lipid Carriers Engineered for CNS Delivery: Optimization and Hematological Toxicity Studies. AAPS PharmSciTech 2019, 20, 22. [Google Scholar] [CrossRef]
- Gonçalves, M.L.C.M.; Lyra, M.A.M.; Oliveira, F.J.V.E.; Rolim, L.; Nadvorny, D.; Vilarinho, A.C.S.G.; Nunes, L.C.C.; Soares, M.F.D.L.R.; Silva-Filho, E.C.; Soares-Sobrinho, J.L. Use of phyllosilicate clay mineral to increase solubility olanzapine. J. Therm. Anal. Calorim. 2016, 127, 1743–1750. [Google Scholar] [CrossRef]
- Natarajan, J.; Baskaran, M.; Humtsoe, L.C.; Vadivelan, R.; Justin, A. Enhanced brain targeting efficacy of Olanzapine through solid lipid nanoparticles. Artif. Cells Nanomed. Biotechnol. 2017, 45, 364–371. [Google Scholar] [CrossRef]
- Salama, H.A.; Mahmoud, A.A.; Kamel, A.O.; Hady, M.A.; Awad, G.A. Brain delivery of olanzapine by intranasal administration of transfersomal vesicles. J. Liposome Res. 2012, 22, 336–345. [Google Scholar] [CrossRef]
- Joseph, E.; Reddi, S.; Rinwa, V.; Balwani, G.; Saha, R. Design and in vivo evaluation of solid lipid nanoparticulate systems of Olanzapine for acute phase schizophrenia treatment: Investigations on antipsychotic potential and adverse effects. Eur. J. Pharm. Sci. 2017, 104, 315–325. [Google Scholar] [CrossRef]
- Naseri, N.; Valizadeh, H.; Zakeri-Milani, P. Solid Lipid Nanoparticles and Nanostructured Lipid Carriers: Structure, Preparation and Application. Adv. Pharm. Bull. 2015, 5, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Puglia, C.; Offerta, A.; Rizza, L.; Zingale, G.; Bonina, F.; Ronsisvalle, S. Optimization of Curcumin Loaded Lipid Nanoparticles Formulated Using High Shear Homogenization (HSH) and Ultrasonication (US) Methods. J. Nanosci. Nanotechnol. 2013, 13, 6888–6893. [Google Scholar] [CrossRef] [PubMed]
- Cirri, M.; Bragagni, M.; Mennini, N.; Mura, P. Development of a new delivery system consisting in “drug–in cyclodextrin–in nanostructured lipid carriers” for ketoprofen topical delivery. Eur. J. Pharm. Biopharm. 2012, 80, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.V.; Seth, A.K.; Balaraman, R.; Aundhia, C.J.; Maheshwari, R.A.; Parmar, G.R. Nanostructured lipid carriers for oral bioavailability enhancement of raloxifene: Design and in vivo study. J. Adv. Res. 2016, 7, 423–434. [Google Scholar] [CrossRef]
- Singh, A.; Neupane, Y.R.; Mangla, B.; Kohli, K. Nanostructured Lipid Carriers for Oral Bioavailability Enhancement of Exemestane: Formulation Design, In Vitro, Ex Vivo, and In Vivo Studies. J. Pharm. Sci. 2019, 108, 3382–3395. [Google Scholar] [CrossRef]
- Soleimanian, Y.; Goli, S.A.H.; Varshosaz, J.; Sahafi, S.M. Formulation and characterization of novel nanostructured lipid carriers made from beeswax, propolis wax and pomegranate seed oil. Food Chem. 2018, 244, 83–92. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, D.; Duan, C.; Jia, L.; Wang, Y.; Feng, F.; Zhang, Q. Preparation and characteristics of oridonin-loaded nanostructured lipid carriers as a controlled-release delivery system. J. Microencapsul. 2010, 27, 234–241. [Google Scholar] [CrossRef]
- Gardouh, A.R.; Faheim, S.H.; Noah, A.T.; Ghorab, M.M. Influence Of Formulation Factors On The Size Of Nanostructured Lipid Carriers And Nanoemulsions Prepared By High Shear Homogenization. Int. J. Pharm. Pharm. Sci. 2018, 10, 61–75. [Google Scholar] [CrossRef]
- de Souza, I.D.L.; Saez, V.; de Campos, V.E.B.; Mansur, C.R.E. Size and Vitamin E Release of Nanostructured Lipid Carriers with Different Liquid Lipids, Surfactants and Preparation Methods. Macromol. Symp. 2019, 383, 1800011. [Google Scholar] [CrossRef]
- Gambhire, M.S.; Bhalekar, M.R.; Gambhire, V.M. Statistical optimization of dithranol-loaded solid lipid nanoparticles using factorial design. Braz. J. Pharm. Sci. 2011, 47, 503–511. [Google Scholar] [CrossRef]
- Gupta, B.; Poudel, B.K.; Pathak, S.; Tak, J.W.; Lee, H.H.; Jeong, J.-H.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Effects of Formulation Variables on the Particle Size and Drug Encapsulation of Imatinib-Loaded Solid Lipid Nanoparticles. AAPS PharmSciTech 2015, 17, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Emami, J.; Mohiti, H.; Hamishehkar, H.; Varshosaz, J. Formulation and optimization of solid lipid nanoparticle formulation for pulmonary delivery of budesonide using Taguchi and Box-Behnken design. Res. Pharm. Sci. 2015, 10, 17–33. [Google Scholar] [PubMed]
- Sheikh, F.A.; Barakat, N.A.M.; Kanjwal, M.A.; Aryal, S.; Khil, M.S.; Kim, H.-Y. Novel self-assembled amphiphilic poly(ε-caprolactone)-grafted-poly(vinyl alcohol) nanoparticles: Hydrophobic and hydrophilic drugs carrier nanoparticles. J. Mater. Sci. Mater. Electron. 2009, 20, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, S.; Garg, T.; Murthy, R.; Rath, G.; Goyal, A.K. Development, optimization and evaluation of long chain nanolipid carrier for hepatic delivery of silymarin through lymphatic transport pathway. Int. J. Pharm. 2015, 485, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Sanad, R.A.; Abdelmalak, N.S.; Elbayoomy, T.S.; Badawi, A.A. Formulation of a Novel Oxybenzone-Loaded Nanostructured Lipid Carriers (NLCs). AAPS PharmSciTech 2010, 11, 1684–1694. [Google Scholar] [CrossRef]
- Teeranachaideekul, V.; Souto, E.B.; Junyaprasert, V.B.; Müller, R.H. Cetyl palmitate-based NLC for topical delivery of Coenzyme Q10–Development, physicochemical characterization and in vitro release studies. Eur. J. Pharm. Biopharm. 2007, 67, 141–148. [Google Scholar] [CrossRef]
- Seyfoddin, A.; Al-Kassas, R. Development of solid lipid nanoparticles and nanostructured lipid carriers for improving ocular delivery of acyclovir. Drug Dev. Ind. Pharm. 2012, 39, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Teeranachaideekul, V.; Müller, R.H.; Junyaprasert, V.B. Encapsulation of ascorbyl palmitate in nanostructured lipid carriers (NLC)—Effects of formulation parameters on physicochemical stability. Int. J. Pharm. 2007, 340, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Teeranachaideekul, V.; Boonme, P.; Souto, E.B.; Müller, R.H.; Junyaprasert, V.B. Influence of oil content on physicochemical properties and skin distribution of Nile red-loaded NLC. J. Control. Release 2008, 128, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Vitorino, C.; Almeida, A.; Sousa, J.; Lamarche, I.; Gobin, P.; Marchand, S.; Couet, W.; Olivier, J.-C.; Pais, A. Passive and active strategies for transdermal delivery using co-encapsulating nanostructured lipid carriers: In vitro vs. in vivo studies. Eur. J. Pharm. Biopharm. 2014, 86, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.H.; Ramasamy, T.; Truong, D.H.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Preparation and Characterization of Fenofibrate-Loaded Nanostructured Lipid Carriers for Oral Bioavailability Enhancement. AAPS PharmSciTech 2014, 15, 1509–1515. [Google Scholar] [CrossRef] [PubMed]
- Tatke, A.; Dudhipala, N.; Janga, K.Y.; Balguri, S.P.; Avula, B.; Jablonski, M.M.; Majumdar, S. In Situ Gel of Triamcinolone Acetonide-Loaded Solid Lipid Nanoparticles for Improved Topical Ocular Delivery: Tear Kinetics and Ocular Disposition Studies. Nanomateries 2018, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Fini, A.; Cavallari, C.; Ospitali, F.; Gonzalez-Rodriguez, M.L. Theophylline-Loaded Compritol Microspheres Prepared by Ultrasound-Assisted Atomization. J. Pharm. Sci. 2011, 100, 743–757. [Google Scholar] [CrossRef]
- Bright, A.; Devi, T.S.R.; Gunasekaran, S. Qualitative and quantitative analysis of antipsychotic drugs-a spectroscopic study. Asian J. Chem. Asian J. Chem. 2010, 22, 5871. [Google Scholar]
- Shankarrao, K.A.; Mahadeo, G.D.; Balavantrao, K.P. Formulation and In-vitro Evaluation of Orally Disintegrating Tablets of Olanzapine-2-Hydroxypropyl-β-Cyclodextrin Inclusion Complex. Iran. J. Pharm. Res. IJPR 2010, 9, 335–347. [Google Scholar]
- Acevedo-Morantes, C.Y.; Acevedo-Morantes, M.T.; Suleiman-Rosado, D.; Ramirez-Vick, J.E. Evaluation of the cytotoxic effect of camptothecin solid lipid nanoparticles on MCF7 cells. Drug Deliv. 2013, 20, 338–348. [Google Scholar] [CrossRef]
- Dixit, M.; Kini, A.G.; Kulkarni, P.K. Enhancing the aqueous solubility and dissolution of olanzapine using freeze-drying. Braz. J. Pharm. Sci. 2011, 47, 743–749. [Google Scholar] [CrossRef]
- Hu, F.-Q.; Jiang, S.-P.; Du, Y.-Z.; Yuan, H.; Ye, Y.-Q.; Zeng, S. Preparation and characterization of stearic acid nanostructured lipid carriers by solvent diffusion method in an aqueous system. Colloids Surfaces B Biointerfaces 2005, 45, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Harisa, G.I.; Badran, M.M. Simvastatin nanolipid carriers decreased hypercholesterolemia induced cholesterol inclusion and phosphatidylserine exposure on human erythrocytes. J. Mol. Liq. 2015, 208, 202–210. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | C888 (mg) | Mig (mg) | P188 (mg) | Tween 80 (mg) |
|---|---|---|---|---|
| F5 | 285 | 15 | 150 | 50 |
| F15 | 255 | 45 | 150 | 50 |
| F30 | 210 | 90 | 150 | 50 |
| F50 | 150 | 150 | 150 | 50 |
| Formulation | HSH Time (Minutes) | US Time (Minutes) | Particle Size (nm) | PDI | ζ-Potential (mV) |
|---|---|---|---|---|---|
| F30 | 15 | 0 | 161 (±5) | 0.35 | −15 (±3) |
| 5 | 5 | 120 (±4) | 0.28 | −16 (±4) | |
| 10 | 5 | 121 (±5) | 0.29 | −15 (±2) | |
| 15 | 5 | 121 (±6) | 0.31 | −15 (±3) |
| Formulation | Blank NLCs | PDI | OLZ-NLCs | PDI |
|---|---|---|---|---|
| F5 | 112 (±2.9) | 0.30 | 114 (±4.0) | 0.40 |
| F15 | 123 (±6.6) | 0.32 | 125 (±3.7) | 0.40 |
| F30 | 128 (±2.9) | 0.28 | 123 (±17.7) | 0.38 |
| F50 | 136 (± 0.5) | 0.23 | 123 (± 31.5) | 0.33 |
| ζ-Potential (mV) | ||
|---|---|---|
| Formulation | Blank NLCs | OLZ-Loaded NLCs |
| F5 | −20 | −40 |
| F15 | −18 | −39 |
| F30 | −16 | −38 |
| F50 | −13 | −35 |
| Kinetic Model | F5 | F15 | F30 | F50 |
|---|---|---|---|---|
| Zero order | 0.7972 | 0.6742 | 0.5988 | 0.7219 |
| First order | 0.9191 | 0.8959 | 0.8759 | 0.8386 |
| Higuchi | 0.9054 | 0.8357 | 0.7735 | 0.879 |
| Korsmeyer-Peppas | 0.9751 | 0.9624 | 0.9701 | 0.9861 |
| Korsmeyer-Peppas (n) | 0.403 | 0.355 | 0.324 | 0.369 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ajiboye, A.L.; Nandi, U.; Galli, M.; Trivedi, V. Olanzapine Loaded Nanostructured Lipid Carriers via High Shear Homogenization and Ultrasonication. Sci. Pharm. 2021, 89, 25. https://doi.org/10.3390/scipharm89020025
Ajiboye AL, Nandi U, Galli M, Trivedi V. Olanzapine Loaded Nanostructured Lipid Carriers via High Shear Homogenization and Ultrasonication. Scientia Pharmaceutica. 2021; 89(2):25. https://doi.org/10.3390/scipharm89020025
Chicago/Turabian StyleAjiboye, Adejumoke Lara, Uttom Nandi, Martin Galli, and Vivek Trivedi. 2021. "Olanzapine Loaded Nanostructured Lipid Carriers via High Shear Homogenization and Ultrasonication" Scientia Pharmaceutica 89, no. 2: 25. https://doi.org/10.3390/scipharm89020025
APA StyleAjiboye, A. L., Nandi, U., Galli, M., & Trivedi, V. (2021). Olanzapine Loaded Nanostructured Lipid Carriers via High Shear Homogenization and Ultrasonication. Scientia Pharmaceutica, 89(2), 25. https://doi.org/10.3390/scipharm89020025