
Development of a Novel, Fast, Simple HPLC Method for Determination of Atorvastatin and its Impurities in Tablets


 , , ,
, , ,
Abstract
1. Introduction
2. Results
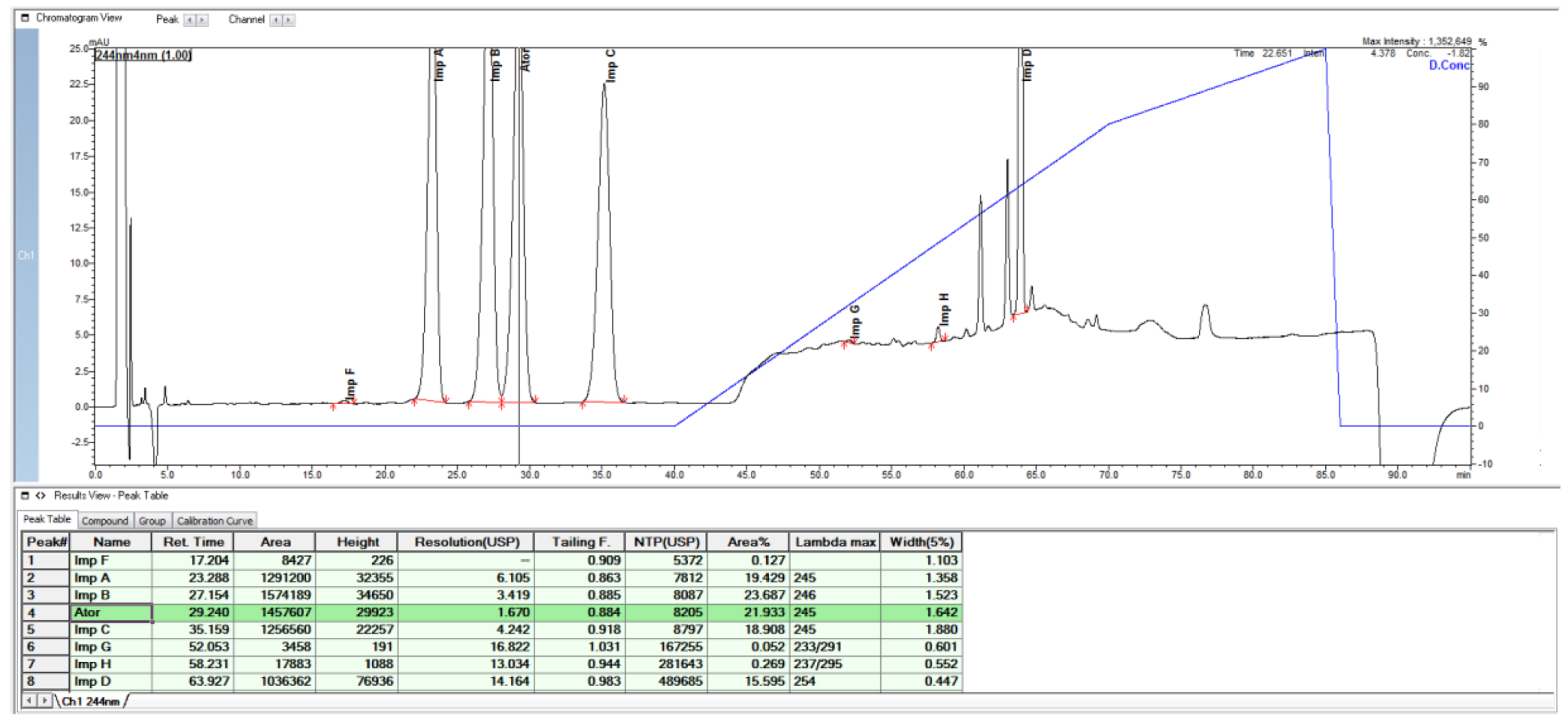
2.1. HPLC Method from European Pharmacopoeia Monograph
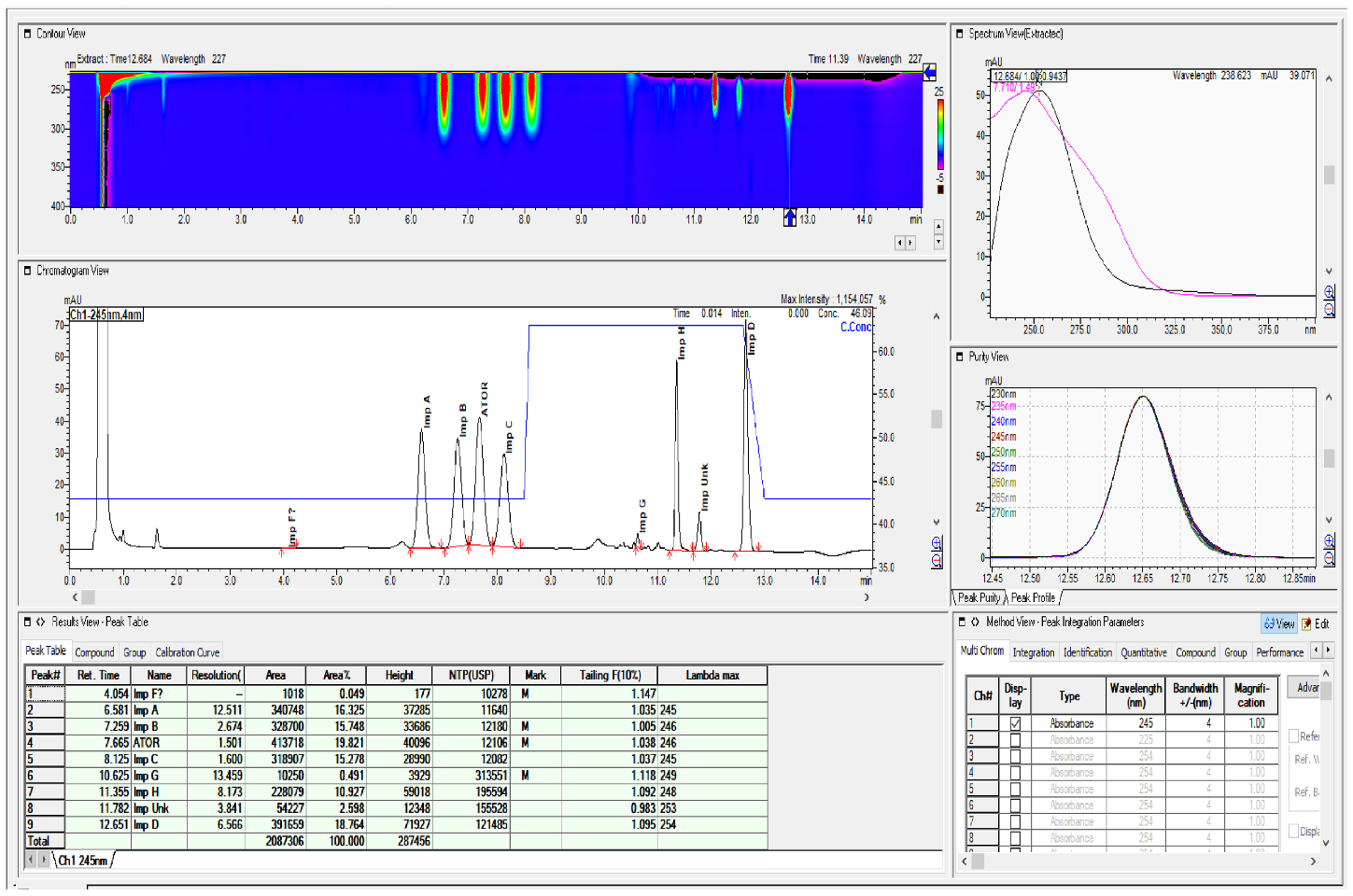
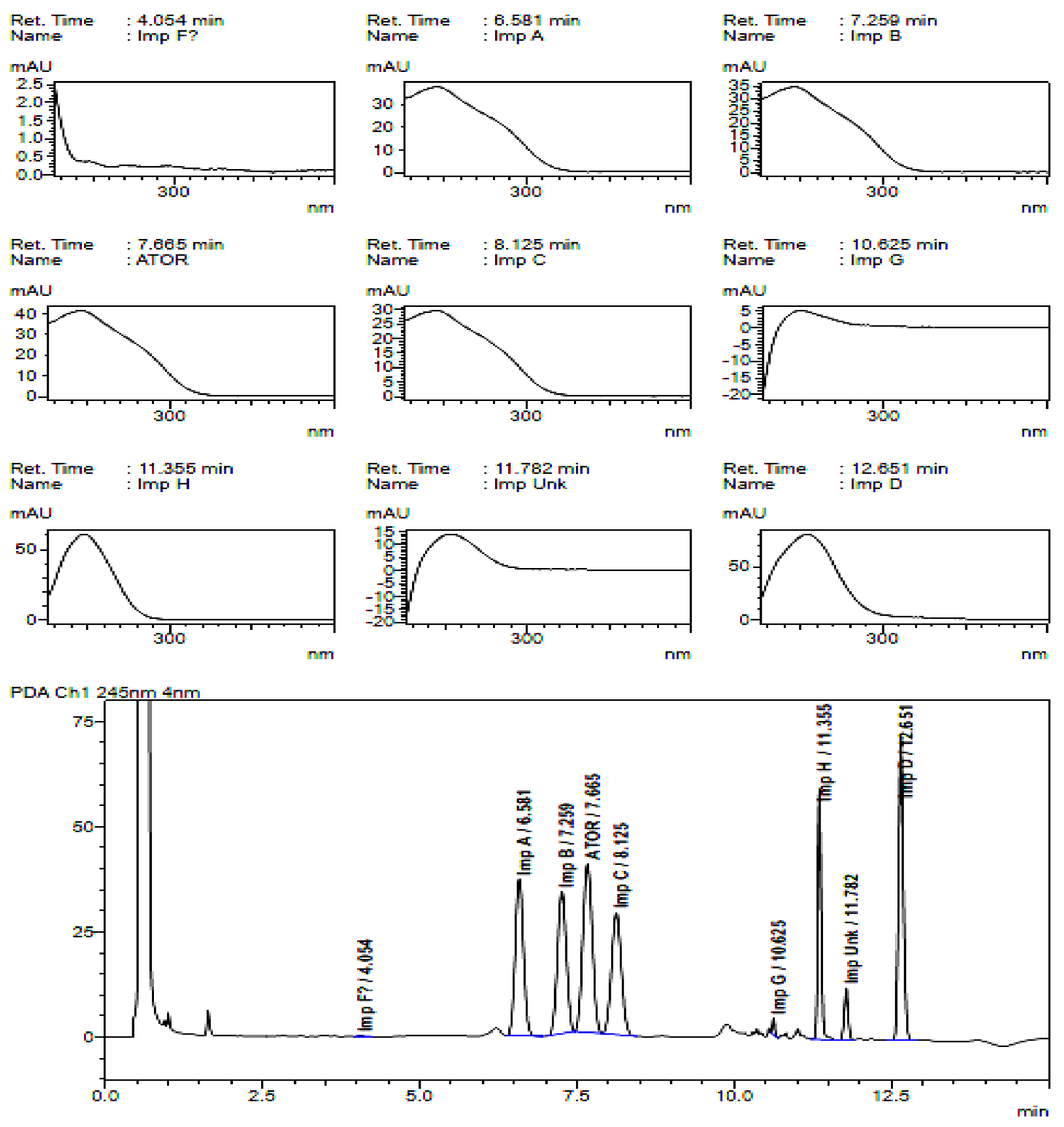
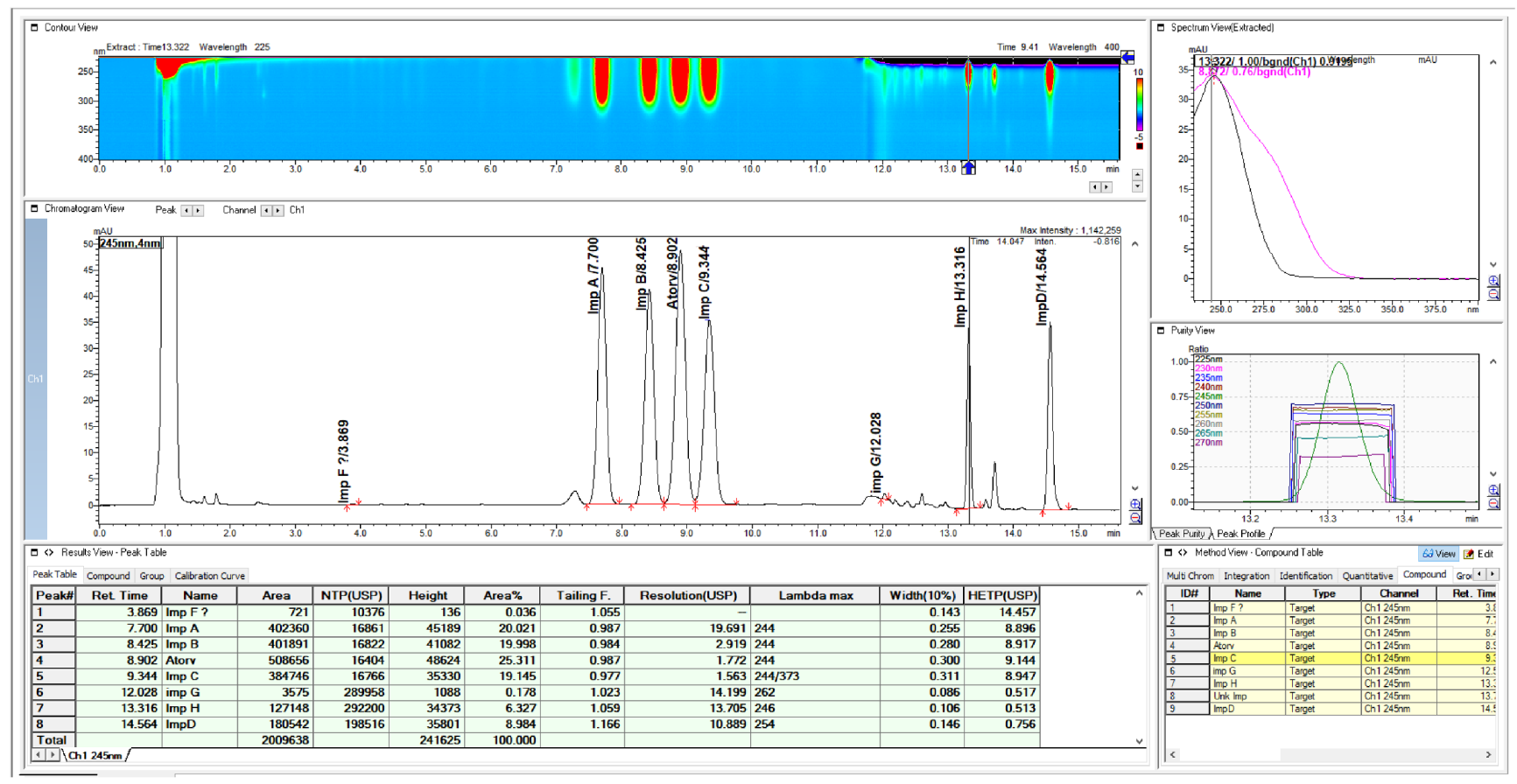
2.2. HPLC Method Development
2.3. Method Validation
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Instrumental and Conditions
4.3. Sample Preparation
4.4. Method Validation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- European Pharmacopoeia. European Pharmacopoeia, 10th ed.; 2020; Available online: https://www.edqm.eu/en/european_pharmacopoeia_10th_edition (accessed on 26 October 2020).
- Drugs.com [Internet]. Atorvastatin. Available online: http://www.drugs.com/atorvastatin.html (accessed on 5 November 2020).
- Lippi, G.; Mattiuzzi, C.; Cervellin, G. Letter to Editor—Statins popularity: A Global picture to statins. Br. J. Clin. Pharmacol. 2019, 85, 1614–1615. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, G.B.; Patel, M.N.; Shah, B.P. Stability indicating RP-HPLC method for simultaneous determination of atorvastatin and amlodipine from their combination drug products. Chem. Pharm. Bull. 2007, 55, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Hafez, H.M.; Elshanawany, A.A.; Abdelaziz, L.M.; Mohram, M.S. Development of a stability-indicating HPLC method for simultaneous determination of amlodipine besylate and atorvastatin calcium in tablets. Austin J. Anal. Pharm. Chem. 2014, 1, 1–11. [Google Scholar]
- Shah, A.D.; Bhatt, K.K.; Mehta, S.R.; Shankar, B.M.; Baldania, L.S. RP-HPLC method for the determination of atorvastatin calcium and nicotinic acid in combined tablet dosage form. Indian J. Pharm. Sci. 2007, 69, 700–703. [Google Scholar] [CrossRef]
- Patole, M.S.; Potale, V.L.; Khodke, S.A.; Damle, C.M. A validated HPLC method for analysis of atorvastatin calcium, ramipril and aspirin as the bulk drug and in combined capsule dosage form. Int. J. Pharm. Sci. Rev. Res. 2010, 4, 40–45. [Google Scholar]
- Londhe, V.S.; Deshmukh, S.R.; Mulgund, V.S.; Jain, S.K. Development and validation of a reversed-phase HPLC method for simultaneous determination of aspirin, atorvastatin calcium and clopidogrel bisulphate in capsules. Indian J. Pharm. Sci. 2011, 73, 23–29. [Google Scholar]
- Kumar, P.S.N.; Gowda, B.D.G.; Deepu, V.C.; Mantelingu, K.; Rangappa, K.S. Simultaneous estimation of statins like pravastatin, atorvastatin and simvastatin in bulk and in pharmaceutical dosage by means of High-Performance Liquid Chromatography. J. Chem. Pharm. 2013, 5, 359–364. [Google Scholar]
- Sathiyasundar, R.; Valliappan, K. An improved RP-HPLC method for the simultaneous estimation of aspirin, atorvastatin and clopidogrel in pharmaceutical formulation using experimental design methodology. Int. J. Pharm. Pharm. 2014, 6, 279–283. [Google Scholar]
- Ramesh, D.; Habibuddin, M. Stability indicating RP-HPLC method for the simultaneous determination of atorvastatin calcium, metformin hydrochloride, and glimepiride in bulk and combined tablet dosage form. Int. Sch. Res. Notices. 2014, 12, 33–46. [Google Scholar] [CrossRef]
- Lakshmi, R.V.; Sreedevi, B. Simultaneous estimation of atorvastatin and olmesartan in bulk and pharmaceutical formulations by RP-HPLC method. Int. J. Innov. Technol. Res. 2014, 2, 1559–1563. [Google Scholar]
- Gurram, C.S. RP-HPLC method development and validation for the simultaneous estimation of atorvastatin, fenofibrate and ezetimibe in a pharmaceutical dosage form. Biochem. Anal. Biochem. 2015, 4, 1–4. [Google Scholar]
- Simionato, L.D.; Ferello, L.; Stamer, S.G.; Repetto, M.F.; Zubata, P.D.; Segall, A.I. A validated reversed-phase HPLC method for the determination of atorvastatin calcium in tablets. Austin Chromatogr. 2014, 1, 1–4. [Google Scholar]
- Ertürk, S.; Aktas, S.E.; Ersoy, L.; Ficiciȯǧlu, S. An HPLC method for the determination of atorvastatin and its impurities in bulk drug and tablets. J. Pharm. Biomed. Anal. 2003, 33, 1017–1023. [Google Scholar] [CrossRef]
- Stanisz, B.; Kania, L. Validation of HPLC method for determination of atorvastatin in tablets and for monitoring stability in solid phase. Acta Pol. Pharm. 2006, 63, 471–476. [Google Scholar]
- Zaheer, Z.; Farooqui, M.; Mangle, A.A.; Nikalje, G.P.O. Stability-indicating high performance liquid chromatographic determination of atorvastatin calcium in pharmaceutical dosage form. Afr. J. Pharm. Pharmacol. 2008, 2, 204–210. [Google Scholar]
- Petkovska, R.; Cornett, C.; Dimitrovska, A. Development and validation of rapid resolution RP-HPLC method for simultaneous determination of atorvastatin and related compounds by use of chemometrics. Anal. Lett. 2008, 41, 992–1009. [Google Scholar] [CrossRef]
- Kumar, K.K.; Rao, K.C.; Lakshmi, V.M.; Mukkanti, K. A validated stability indicating RP-HPLC method for atorvastatin calcium. Am. J. Analyt. Chem. 2012, 3, 392–399. [Google Scholar]
- Vakkum, P.; Babu, M.J.; Muralikrishna, R. Stress degradation behavior of atorvastatin calcium and development of a suitable stability-indicating LC method for the determination of atorvastatin, its related impurities and its degradation products. Sci. Pharm. 2013, 81, 93–114. [Google Scholar] [CrossRef]
- Piponski, M.; Bakovska Stoimenova, T.; Piponska, M.; Trendovska Serafimovska, G. Concepts in development of fast, simple, stability indicating HPLC method for analysis of atorvastatin related compounds in tablets. J. Anal. Pharm. Res. 2018, 7, 450–457. [Google Scholar]
- Kumar Talluri, M.V.N.; Kalyankar, A.; Ragampeta, S. Synchronized separation of atorvastatin—An antihyperlipidemic drug with antihypertensive, antidiabetic, antithrombotic drugs by RP-LC for determination in combined formulations. J. Pharm. Anal. 2012, 2, 285–292. [Google Scholar] [CrossRef]
- Piponski, M.; Stoimenova, T.B.; Stefov, S.; Balkanov, T.; Serafimovska, G.T.; Logoyda, L. Development of a novel, fast, simple, nonderivative HPLC method with direct UV measurment for quantification of memantine hydrochloride in tablets. J. Sep. Sci. 2020, 43, 3482–3490. [Google Scholar] [CrossRef] [PubMed]
- Snyder, R.L.; Kirckland, J.; Dolan, W.J. Introduction to Modern Liquid Chromatography, 3rd ed.; John Willey & Sons: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- International Conference on Harmonization (ICH) of technical requirements for registration of pharmaceuticals for human use. In Validation of Analytical Procedures: Text and Methodology Q2(R1); ICH: Geneva, Switzerland, 2005.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Specified Impurities of Atorvastatin | RRT (EP Method) | RRT (New Developed Method) |
|---|---|---|
| Impurity A | 0.8 | 0.86 |
| Impurity B | 0.9 | 0.95 |
| Impurity C | 1.2 | 1.06 |
| Impurity D | 2.1 | 1.65 |
| Linearity Parameters | Impurities | Assay |
|---|---|---|
| Atorvastatin concentration range (% of working conc. of test sol.) | 0.05%–0.3% | 70%–130% |
| Linear regression equation | y = 3014.8108x − 70.2973 | y = 2832.6906x − 19,216.7744 |
| RSD of response factors, % | 2.47 | 0.93 |
| Correlation coefficient, R2 | 0.9998 | 0.9993 |
| Approx. conc. in % of the Working conc. in the Test sol. (imp.) | Recovery (%) for Impurities Testing (n = 3) | Approx. conc. in % of the Working conc. in the Test sol. (Assay) | Recovery (%) for Assay/Uniformity of Dosage Units Testing (n = 3) |
|---|---|---|---|
| 0.1 | 98.12 | 70 | 100.04 |
| 0.2 | 98.37 | 100 | 101.38 |
| 0.3 | 99.03 | 130 | 101.12 |
| RSD = 0.60 | RSD = 0.61 | ||
| R2 = 0.9999 | R2 = 0.9999 | ||
| Slope = 0.9948 | Slope = 1.0238 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shulyak, N.; Piponski, M.; Kovalenko, S.; Bakovska Stoimenova, T.; Balkanov, T.; El-Subbagh, H.I.; Drapak, I.; Omotosho, J.O.; Logoyda, L. Development of a Novel, Fast, Simple HPLC Method for Determination of Atorvastatin and its Impurities in Tablets. Sci. Pharm. 2021, 89, 16. https://doi.org/10.3390/scipharm89020016
Shulyak N, Piponski M, Kovalenko S, Bakovska Stoimenova T, Balkanov T, El-Subbagh HI, Drapak I, Omotosho JO, Logoyda L. Development of a Novel, Fast, Simple HPLC Method for Determination of Atorvastatin and its Impurities in Tablets. Scientia Pharmaceutica. 2021; 89(2):16. https://doi.org/10.3390/scipharm89020016
Chicago/Turabian StyleShulyak, Nataliia, Marjan Piponski, Sergiy Kovalenko, Tanja Bakovska Stoimenova, Trajan Balkanov, Hussein I. El-Subbagh, Iryna Drapak, Joy Oluwatobiloba Omotosho, and Liliya Logoyda. 2021. "Development of a Novel, Fast, Simple HPLC Method for Determination of Atorvastatin and its Impurities in Tablets" Scientia Pharmaceutica 89, no. 2: 16. https://doi.org/10.3390/scipharm89020016
APA StyleShulyak, N., Piponski, M., Kovalenko, S., Bakovska Stoimenova, T., Balkanov, T., El-Subbagh, H. I., Drapak, I., Omotosho, J. O., & Logoyda, L. (2021). Development of a Novel, Fast, Simple HPLC Method for Determination of Atorvastatin and its Impurities in Tablets. Scientia Pharmaceutica, 89(2), 16. https://doi.org/10.3390/scipharm89020016