Design, Synthesis and Antibacterial Studies of Novel Cationic Amphipathic Cyclic Undecapeptides and Their Linear Counterparts against Virulent Bacterial Strains

Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
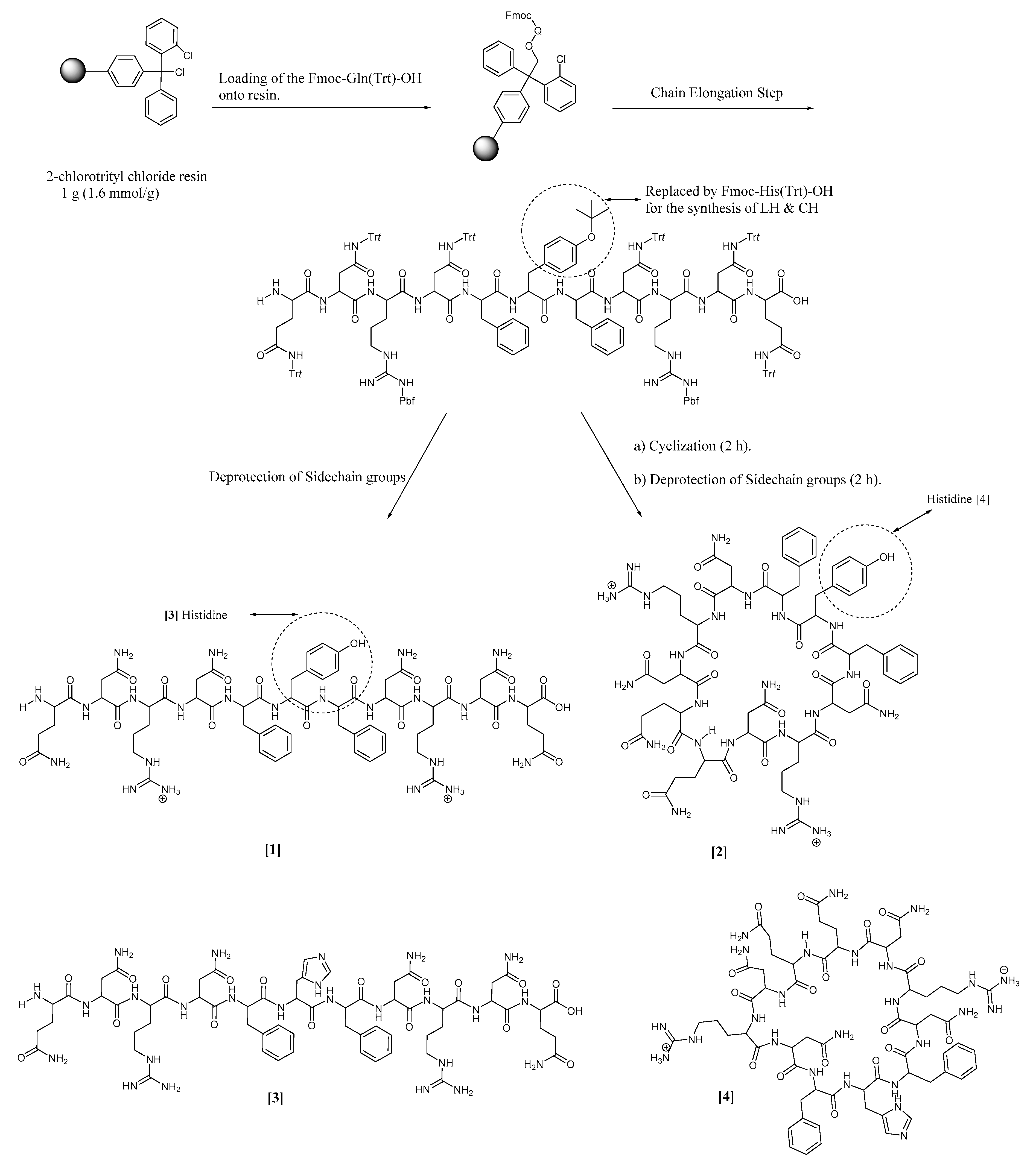
2.2. Synthetic Protocol
2.3. MIC Evaluation
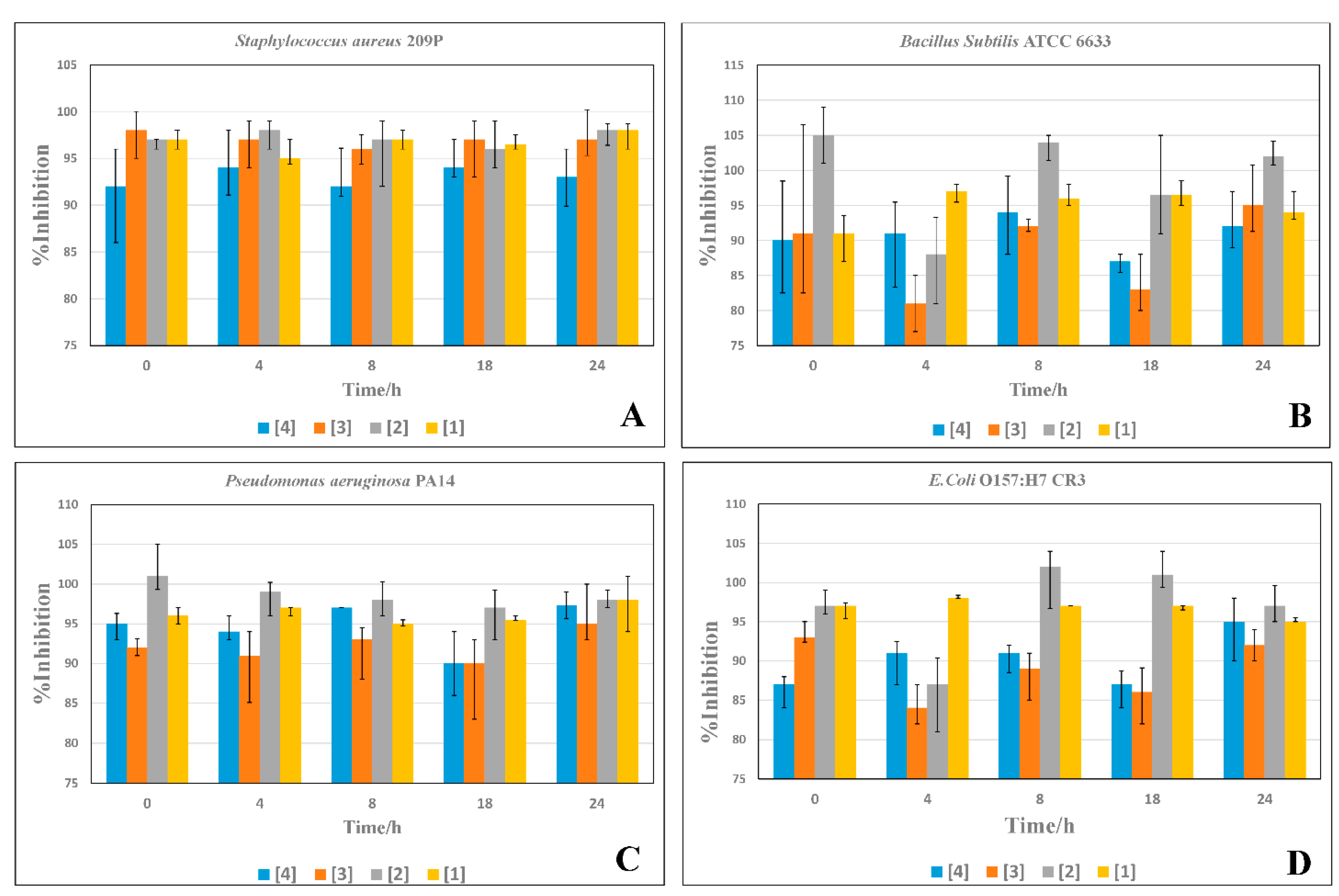
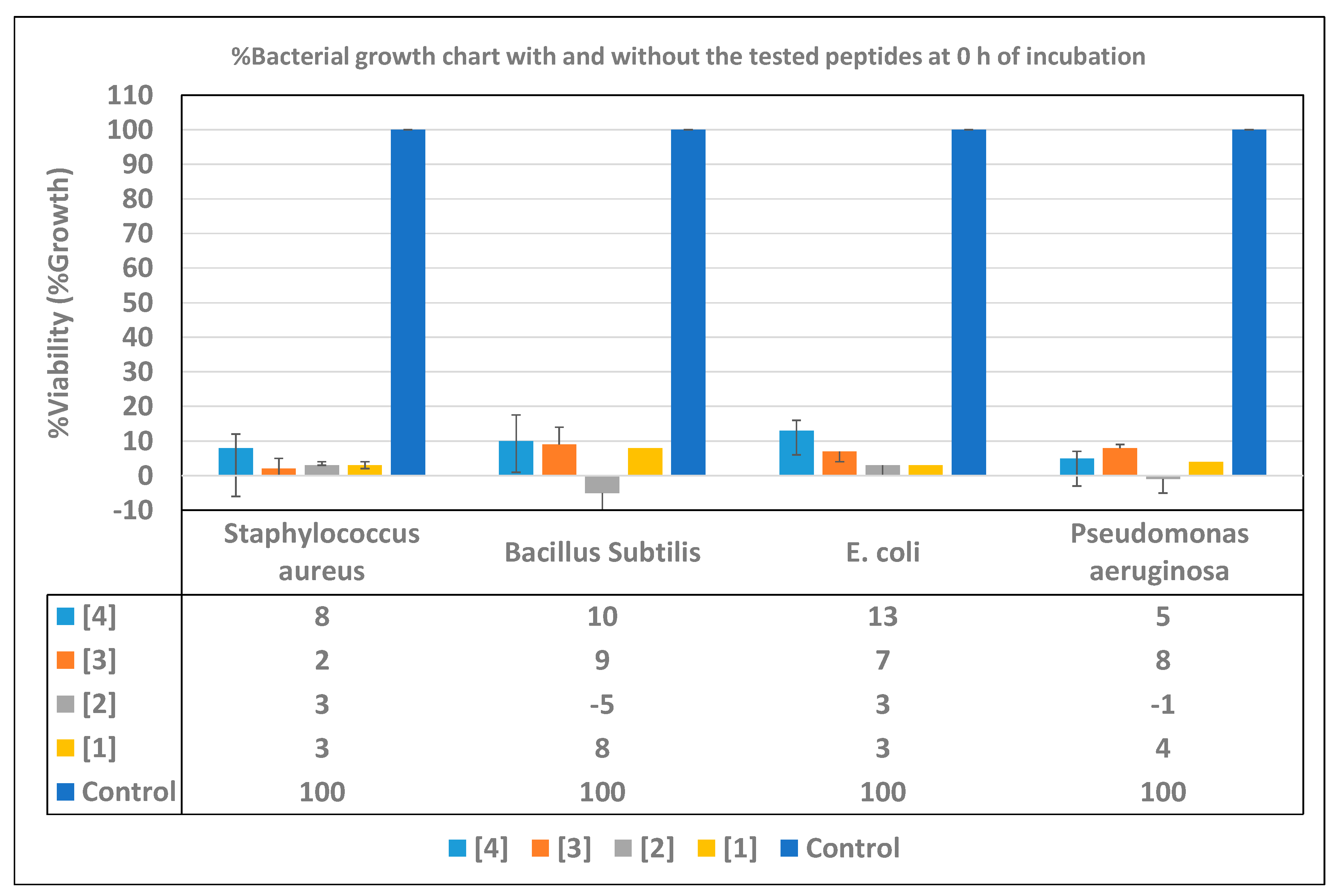
2.4. Time-Inhibition Studies
3. Results and Discussion
3.1. Design of Peptides
3.2. Synthesis and Characterization
3.3. MIC Evaluations
3.4. Time Inhibition Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| (1) | Linear tyrosine peptide |
| (2) | Cyclic tyrosine peptide |
| (3) | Linear histidine peptide |
| (4) | Cyclic histidine peptide |
| GS | Gentamicin sulfate |
| MW | Molecular weight |
| Fmoc | 9-fluorenylmethoxycarbonyl |
| HBTU | O-benzotriazole-N,N,N’,N’-tetramethyl-uronium-hexafluorophosphate |
| HOBt·H2O | 1-hydroxy-benzotriazole hydrate |
| DIPEA | N,N-diisopropylethylamine |
| TFA | 2,2,2-trifluoroacetic acid |
| TFE | 2,2,2-trifluoroethanol |
| TIS | Trisisopropylsilane |
| RP-HPLC | Reversed-phase high-performance liquid chromatography |
| DMSO | Dimethyl sulfoxide |
| TOF | Time of flight |
| OD | Optical density |
| MOE | Molecular Operating Environment |
References
- Vincent, J. Nosocomial infections in adult intensive-care units. Lancet 2003, 361, 2068–2077. [Google Scholar] [CrossRef]
- World Health Organization. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Diamond, G.; Beckloff, N.; Weinberg, A.; Kisich, K. The Roles of Antimicrobial Peptides in Innate Host Defense. Curr. Pharm. Des. 2009, 15, 2377–2392. [Google Scholar] [CrossRef] [PubMed]
- Farrag, H.N.; Metwally, K.; Ikeno, S.; Kato, T. Design and Synthesis of a New Amphipathic Cyclic Decapeptide with Rapid, Stable, and Continuous Antibacterial Effects. Pertanika J. Sci. Technol. 2020, 28, 183–196. [Google Scholar] [CrossRef]
- Lohner, K. New strategies for novel antibiotics: Peptides targeting bacterial cell membranes. Gen. Physiol. Biophys. 2009, 28, 105–116. [Google Scholar] [CrossRef]
- Dutta, S.R.; Gauri, S.S.; Ghosh, T.; Halder, S.K.; DasMohapatra, P.K.; Mondal, K.C.; Ghosh, A.K. Elucidation of structural and functional integration of a novel antimicrobial peptide from Antheraea mylitta. Bioorg. Med. Chem. Lett. 2017, 27, 1686–1692. [Google Scholar] [CrossRef] [PubMed]
- Yeung, A.T.Y.; Gellatly, S.L.; Hancock, R.E.W. Multifunctional cationic host defence peptides and their clinical applications. Cell. Mol. Life Sci. 2011, 68, 2161–2176. [Google Scholar] [CrossRef] [PubMed]
- Madani, F.; Lindberg, S.; Langel, Ü.; Futaki, S.; Gräslund, A. Mechanisms of Cellular Uptake of Cell-Penetrating Peptides. J. Biophys. 2011, 2011, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C. Structural and DNA-binding studies on the bovine antimicrobial peptide, indolicidin: Evidence for multiple conformations involved in binding to membranes and DNA. Nucleic Acids Res. 2005, 33, 4053–4064. [Google Scholar] [CrossRef] [PubMed]
- Gottenbos, B. Antimicrobial effects of positively charged surfaces on adhering Gram-positive and Gram-negative bacteria. J. Antimicrob. Chemother. 2001, 48, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M.; Vogel, H.J. Diversity of antimicrobial peptides and their mechanisms of action. Biochim. Biophys. Acta Biomembr. 1999, 1462, 11–28. [Google Scholar] [CrossRef]
- Bacalum, M.; Radu, M. Cationic Antimicrobial Peptides Cytotoxicity on Mammalian Cells: An Analysis Using Therapeutic Index Integrative Concept. Int. J. Pept. Res. Ther. 2015, 21, 47–55. [Google Scholar] [CrossRef]
- Park, A.J.; Okhovat, J.P.; Kim, J. Antimicrobial peptides. In Clinical and Basic Immunodermatology, 2nd ed.; Springer: Cham, Switzerland, 2017; pp. 81–95. [Google Scholar] [CrossRef]
- Kreutzer, A.G.; Salveson, P.J.; Yang, H. Standard Practices for Fmoc-Based Solid-Phase Peptide Synthesis in the Nowick Laboratory. Retrieved Sept. 2018, 19, 1–14. [Google Scholar]
- Amblard, M.; Fehrentz, J.; Martinez, J.; Subra, G. Methods and Protocols of Modern Solid Phase Peptide Synthesis. Mol. Biotechnol. 2006, 33, 239–254. [Google Scholar] [CrossRef]
- Huber, U.; Majors, R.E. Principles in Preparative HPLC; Agilent Technologies Inc.: Santa Clara, CA, USA, 2007. [Google Scholar]
- Tapeinou, A.; Matsoukas, M.-T.; Simal, C.; Tselios, T. Review cyclic peptides on a merry-go-round; towards drug design. Biopolymers 2015, 104, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Clinical and Laboratory Standards Institute. M100 Performance Standards for Antimicrobial; CLSI: Wayne, PA, USA, 2018; ISBN 1562388045. [Google Scholar]
- Labute, P. LowModeMD—Implicit Low-Mode Velocity Filtering Applied to Conformational Search of Macrocycles and Protein Loops. J. Chem. Inf. Model. 2010, 50, 792–800. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MIC Values (µM) | ||||
|---|---|---|---|---|
| Peptide | P. aeruginosa | E. coli | S. aureus | B. subtilis |
| (1) | 9.3 | 12.5 | 12.5 | 12.5 |
| (2) | 6.25 | 12.5 | 12.5 | 12.5 |
| (3) | 3.1 | 3.1 | 3.1 | 6.25 |
| (4) | 6.25 | 3.1 | 6.25 | 3.1 |
| Gentamicin sulfate [GS] | 6.25 | 3.1 | 6.25 | 1.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farrag, H.N.; Maeda, T.; Kato, T. Design, Synthesis and Antibacterial Studies of Novel Cationic Amphipathic Cyclic Undecapeptides and Their Linear Counterparts against Virulent Bacterial Strains. Sci. Pharm. 2021, 89, 10. https://doi.org/10.3390/scipharm89010010
Farrag HN, Maeda T, Kato T. Design, Synthesis and Antibacterial Studies of Novel Cationic Amphipathic Cyclic Undecapeptides and Their Linear Counterparts against Virulent Bacterial Strains. Scientia Pharmaceutica. 2021; 89(1):10. https://doi.org/10.3390/scipharm89010010
Chicago/Turabian StyleFarrag, Hisham N., Toshinari Maeda, and Tamaki Kato. 2021. "Design, Synthesis and Antibacterial Studies of Novel Cationic Amphipathic Cyclic Undecapeptides and Their Linear Counterparts against Virulent Bacterial Strains" Scientia Pharmaceutica 89, no. 1: 10. https://doi.org/10.3390/scipharm89010010
APA StyleFarrag, H. N., Maeda, T., & Kato, T. (2021). Design, Synthesis and Antibacterial Studies of Novel Cationic Amphipathic Cyclic Undecapeptides and Their Linear Counterparts against Virulent Bacterial Strains. Scientia Pharmaceutica, 89(1), 10. https://doi.org/10.3390/scipharm89010010