Development and Characterization of Glimepiride Novel Solid Nanodispersion for Improving Its Oral Bioavailability



Abstract
1. Introduction
2. Materials and Methods
2.1. The Materials
2.2. The Methods
2.2.1. The Experimental Design
2.2.2. The Preparation of GM Solid Dispersion
2.2.3. The Production Yield of GM Solid Dispersion
2.2.4. The Drug Content Uniformity of GM Solid Dispersion
2.2.5. The Determination of Micromeritic Properties of GM Solid Dispersion Powders
The Bulk Density
The Tapped Density
Hausner’s Ratio
Carr’s Index
The Angle of Repose
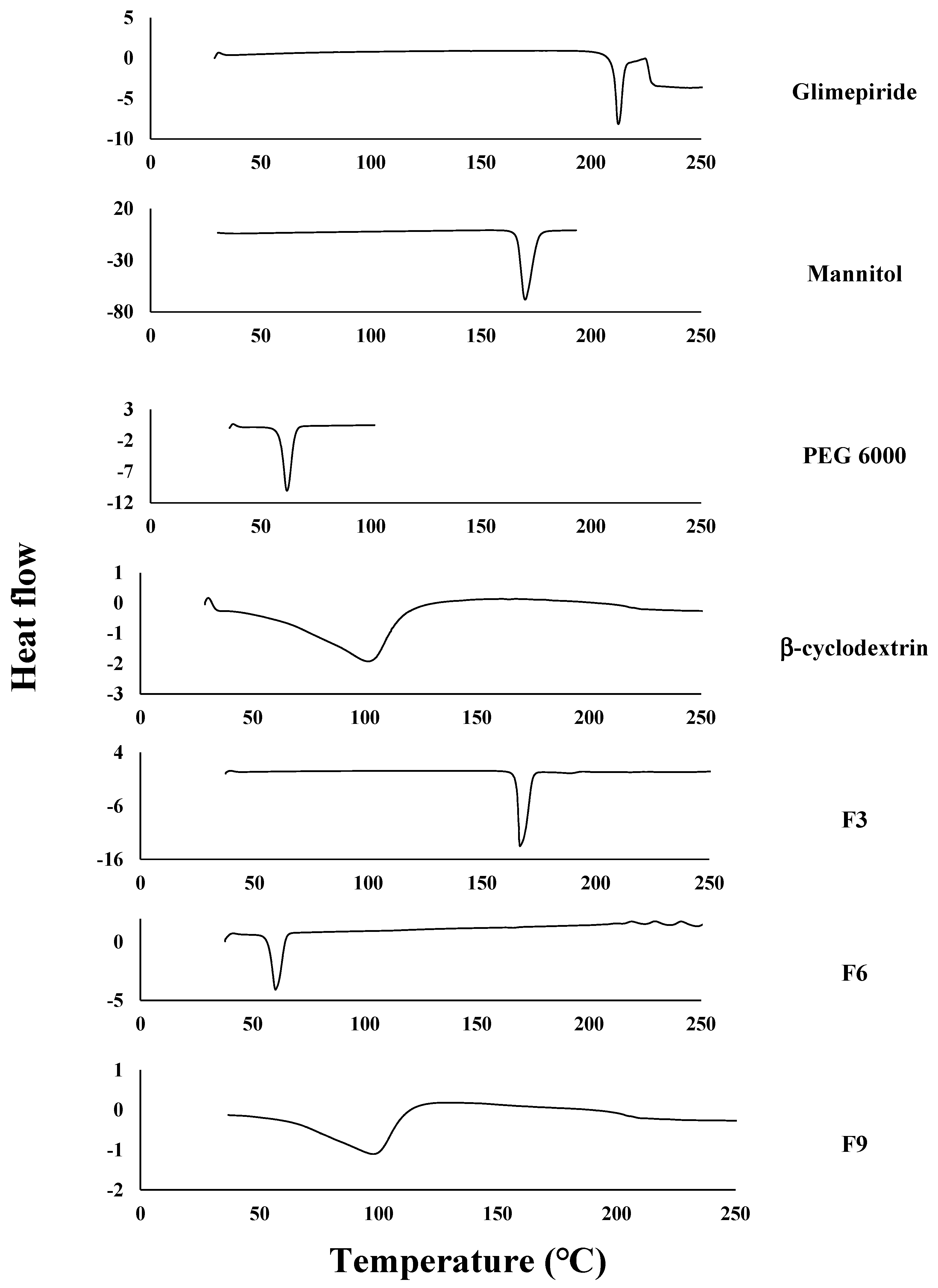
2.2.6. The Differential Scanning Calorimetry (DSC)
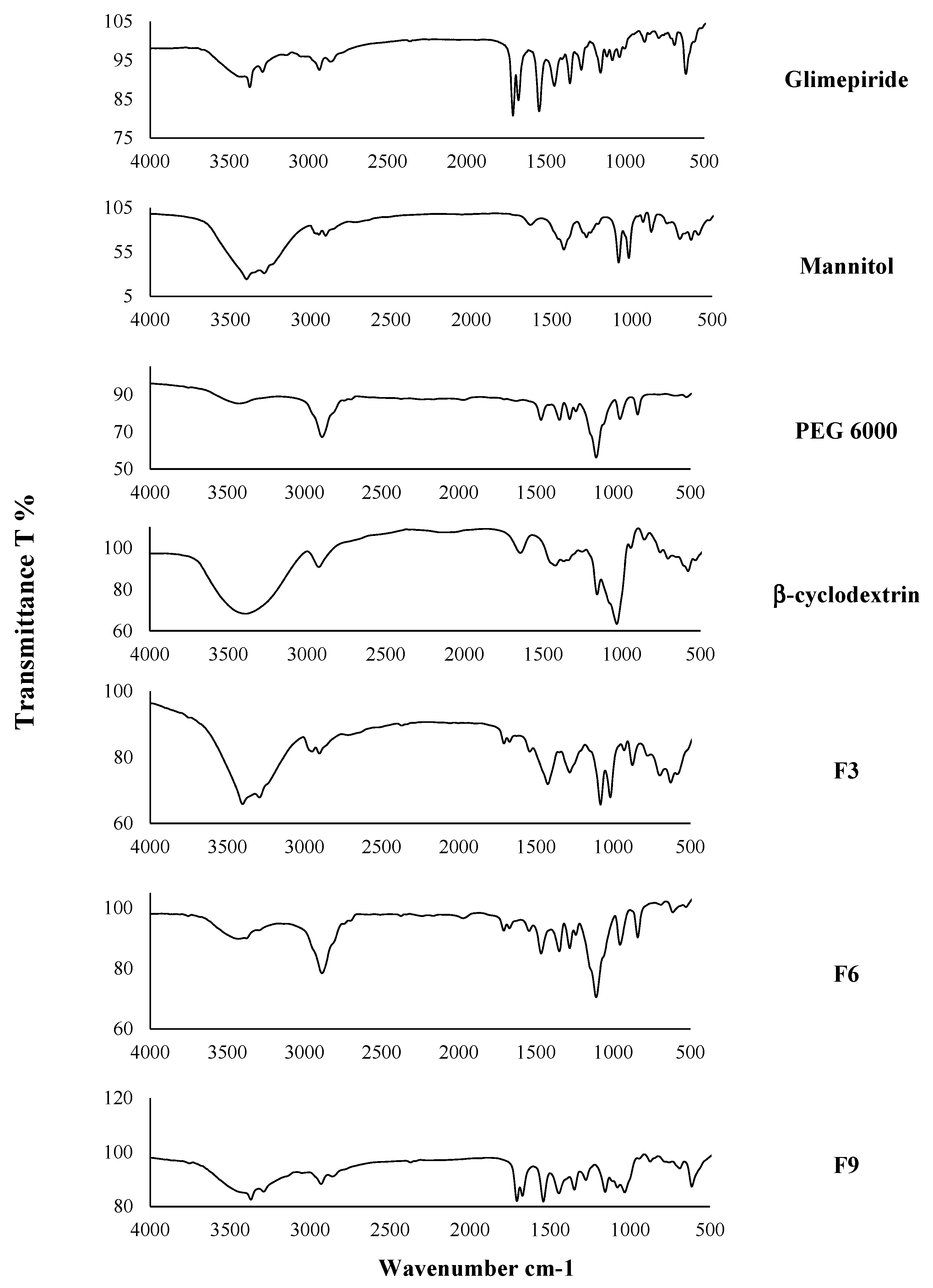
2.2.7. The Infrared Spectroscopy Analysis (IR)
2.2.8. The In-Vitro Release Study of GM from Solid Dispersion
2.2.9. The Selection of Optimized Formulation of GM Solid Dispersion
2.2.10. The scanning Electron Microscopy of the Optimized Formulation (SEM)
2.2.11. The Pharmacological Evaluation of the Antidiabetic Effect of GM Solid Dispersion
2.2.12. The Statistical Analysis
3. Results and Discussion
3.1. The Production Yield % (PY%)
3.2. The Drug Content %(DC%)
3.3. The Micromeritics Properties of GM Solid Dispersion
3.3.1. The Bulk and Tapped Density
3.3.2. Hausner’s Ratio
3.3.3. Carr’s Index (the Compressibility %)
3.3.4. Angle of Repose
3.4. The Differential Scanning Calorimetry (DSC)
3.5. The Infrared Spectroscopy (IR)
3.6. The In-Vitro Release Study of GM From Solid Dispersion
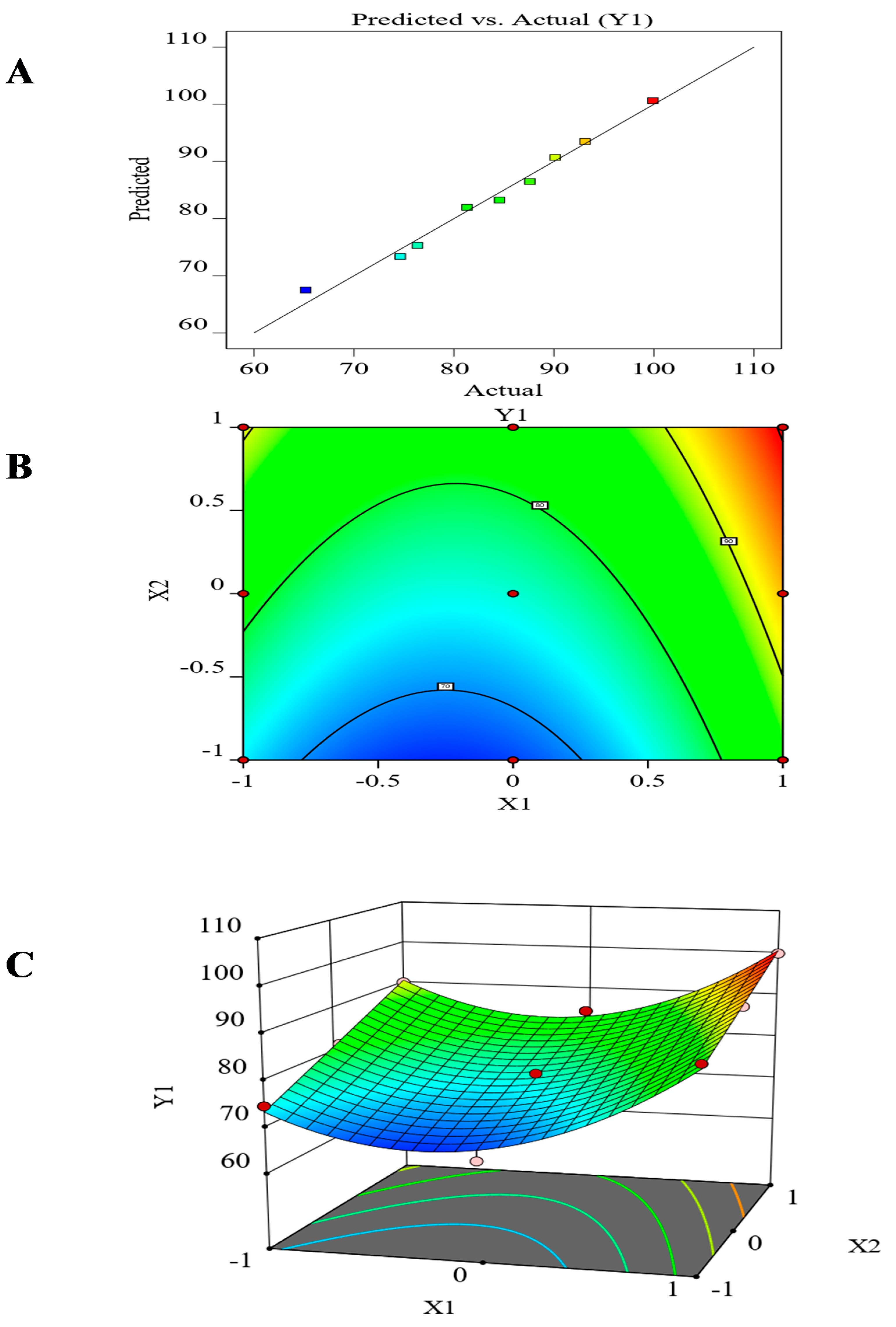
3.7. The Effect of the Formulation Factors in the In-Vitro Release (Y1)
In Vitro Drug Release Kinetics
3.8. The Selection of Optimized Formulation of GM Solid Dispersion
3.9. The Scanning Electron Microscopy of the Optimized Formulation (SEM)
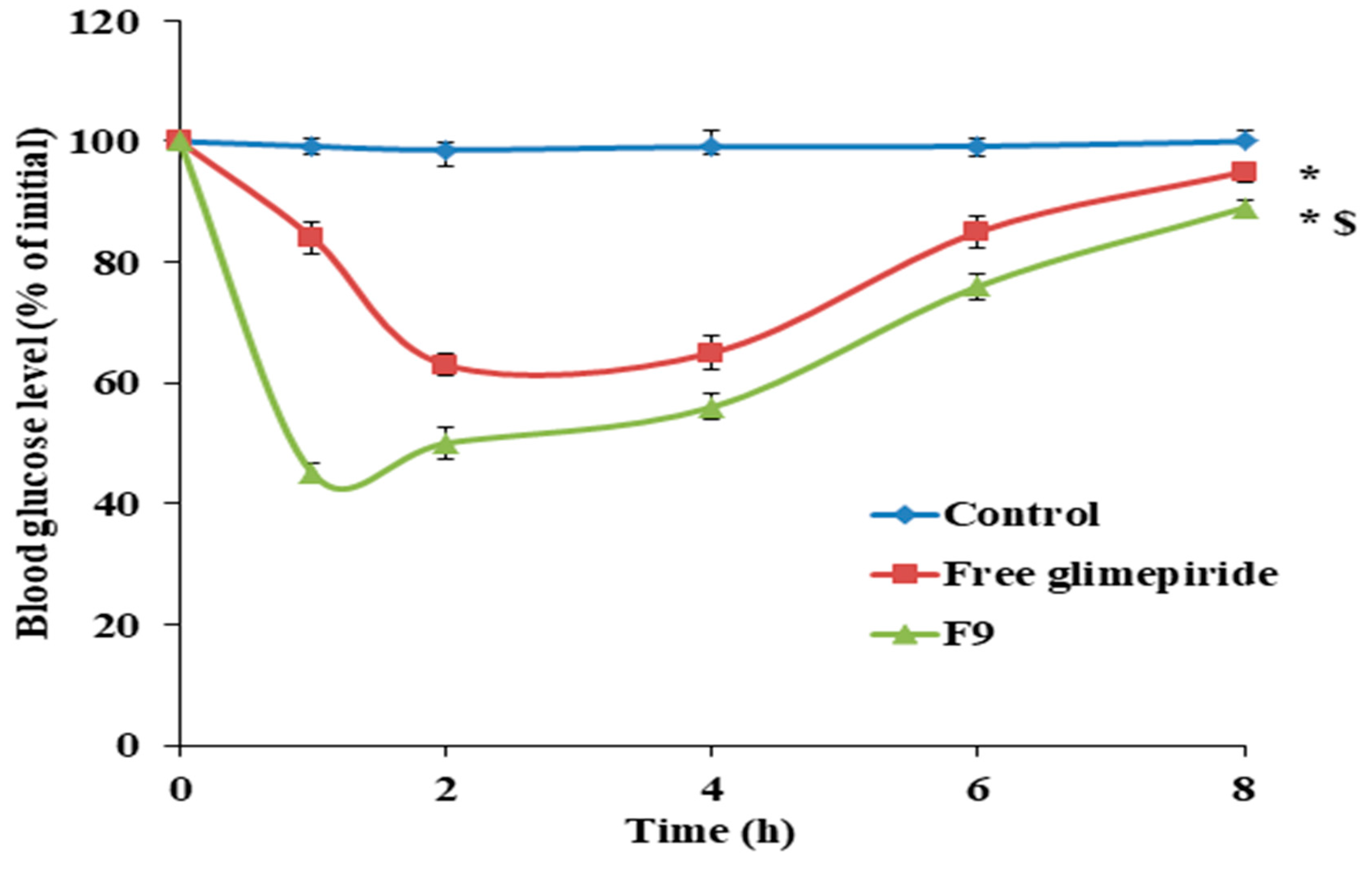
3.10. Antidiabetic Effect of GM Solid Dispersion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Association, A.D. 2. Classification and diagnosis of diabetes: Standards of medical care in diabetes—2018. Diabetes Care 2018, 41, S13–S27. [Google Scholar] [CrossRef]
- Association, A.D. Diagnosis and classification of diabetes mellitus. Diabetes Care 2014, 37, S81–S90. [Google Scholar] [CrossRef] [PubMed]
- Inzucchi, S.E.; Bergenstal, R.M.; Buse, J.B.; Diamant, M.; Ferrannini, E.; Nauck, M.; Peters, A.L.; Tsapas, A.; Wender, R.; Matthews, D.R. Management of hyperglycaemia in type 2 diabetes: A patient-centered approach. Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia 2012, 55, 1577–1596. [Google Scholar] [CrossRef]
- Tanabe, M.; Motonaga, R.; Terawaki, Y.; Nomiyama, T.; Yanase, T. Prescription of oral hypoglycemic agents for patients with type 2 diabetes mellitus: A retrospective cohort study using a Japanese hospital database. J. Diabetes Investig. 2016, 8, 227–234. [Google Scholar] [CrossRef]
- Simpson, S.H.; Majumdar, S.R.; Tsuyuki, R.T.; Eurich, D.T.; Johnson, J.A. Dose–response relation between sulfonylurea drugs and mortality in type 2 diabetes mellitus: A population-based cohort study. Can. Med Assoc. J. 2006, 174, 169–174. [Google Scholar] [CrossRef]
- González-Ortiz, M.; Guerrero-Romero, J.F.; Violante-Ortiz, R.; Wacher-Rodarte, N.; Martínez-Abundis, E.; Aguilar-Salinas, C.; Islas-Andrade, S.; Arechavaleta-Granell, R.; González-Canudas, J.; Rodríguez-Morán, M.; et al. Efficacy of glimepiride/metformin combination versus glibenclamide/metformin in patients with uncontrolled type 2 diabetes mellitus. J. Diabetes Complicat. 2009, 23, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Gill, B.; Kaur, T.; Kumar, S.; Gupta, G.D. Formulation and evaluation of glimepiride solid dispersion tablets. Asian J. Pharm. 2010, 4, 112–118. [Google Scholar] [CrossRef]
- Wagh, V.T.; Jagtap, V.A.; Shaikh, T.J.; Nandedkar, S.Y. Formulation and evaluation of glimepiride solid dispersion tablets for their solubility enhancement. J. Adv. Sci. Res. 2012, 3, 36–41. [Google Scholar]
- Ning, X.; Sun, J.; Han, X.; Wu, Y.; Yan, Z.; Han, J.; He, Z. Strategies to improve dissolution and oral absorption of glimepiride tablets: Solid dispersion versus micronization techniques. Drug Dev. Ind. Pharm. 2011, 37, 727–736. [Google Scholar] [CrossRef]
- Sharma, M.; Sharma, R.; Jain, D.K.; Saraf, A. Enhancement of oral bioavailability of poorly water soluble carvedilol by chitosan nanoparticles: Optimization and pharmacokinetic study. Int. J. Biol. Macromol. 2019, 135, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J.H. Strategies to address low drug solubility in discovery and development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef]
- Stella, V.J.; Nti-Addae, K.W. Prodrug strategies to overcome poor water solubility. Adv. Drug Deliv. Rev. 2007, 59, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Serajuddin, A.T. Salt formation to improve drug solubility. Adv. Drug Deliv. Rev. 2007, 59, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Blagden, N.; De Matas, M.; Gavan, P.T.; York, P. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Adv. Drug Deliv. Rev. 2007, 59, 617–630. [Google Scholar] [CrossRef]
- Ting, J.M.; Porter, I.W.W.; Mecca, J.M.; Bates, F.S.; Reineke, T.M. Advances in polymer design for enhancing oral drug solubility and delivery. Bioconjug. Chem. 2018, 29, 939–952. [Google Scholar] [CrossRef]
- Ricarte, R.G.; Van Zee, N.J.; Li, Z.; Johnson, L.M.; Lodge, T.P.; Hillmyer, M.A. Recent advances in understanding the micro- and nanoscale phenomena of amorphous solid dispersions. Mol. Pharm. 2019, 16, 4089–4103. [Google Scholar] [CrossRef] [PubMed]
- Vidyadhara, S.; Babu, J.R.; Sasidhar, R.L.C.; Ramu, A.; Prasad, S.S.; Tejasree, M. Formulation and evaluation of glimepiride solid dispersions and their tablet formulations for enhanced bioavailability. Pharmanest 2011, 1, 15–20. [Google Scholar]
- Tran, P.; Pyo, Y.C.; Kim, D.H.; Lee, S.E.; Kim, J.K.; Park, J.S. Overview of the manufacturing methods of solid dispersion technology for improving the solubility of poorly water-soluble drugs and application to anticancer drugs. Pharmaceutics 2019, 11, 132. [Google Scholar] [CrossRef]
- Pahovnik, D.; Reven, S.; Grdadolnik, J.; Borstnar, R.; Mavri, J.; Žagar, E. Determination of the interaction between glimepiride and hyperbranched polymers in solid dispersions. J. Pharm. Sci. 2011, 100, 4700–4709. [Google Scholar] [CrossRef]
- Mehta, A.; Vasanti, S.; Tyagi, R.; Shukla, A. Formulation and evaluation of solid dispersions of an anti-diabetic drug. Curr. Trends Biotechnol. Pharm. 2009, 3, 76–84. [Google Scholar]
- Chaudhari, M.D.; Sonawane, R.O.; Zawar, L.; Nayak, S.; Bari, S.B. Solubility and dissolution enhancement of poorly water soluble glimepiride by using solid dispersion technique. Int. J. Pharm. Pharm Sci. 2012, 4, 534–539. [Google Scholar]
- Sahoo, A.C.; Kanungo, S.K.; Dinda, S.C.; Panda, J.; Patra, C.N. Improvement in micromeritic properties and dissolution rate of glimepiride. WJPR 2017, 6, 1545–1560. [Google Scholar] [CrossRef][Green Version]
- Reginald-Opara, J.N.; Attama, A.; Ofokansi, K.; Umeyor, C.; Kenechukwu, F. Molecular interaction between glimepiride and Soluplus®-PEG 4000 hybrid based solid dispersions: Characterisation and anti-diabetic studies. Int. J. Pharm. 2015, 496, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Reven, S.; Homar, M.; Peternel, L.; Kristl, J.; Žagar, E. Preparation and characterization of tablet formulation based on solid dispersion of glimepiride and poly(ester amide) hyperbranched polymer. Pharm. Dev. Technol. 2011, 18, 323–332. [Google Scholar] [CrossRef]
- Singh, A.; Sharma, P.K.; Meher, J.G.; Malviya, R. Evaluation of enhancement of solubility of paracetamol by solid dispersion technique using different polymers concentration. Asian J. Pharm. Clin. Res. 2011, 4, 117–119. [Google Scholar]
- Kumar, S.; Gupta, S.K.; Sharma, P.K. Dissolution rate enhancement of aceclofenac by solid dispersion technique. Asian J. Pharm. Life Sci. 2011, 2231, 4423. [Google Scholar]
- Dabagh, M.A.; Taghipour, B. Investigation of Solid Dispersion Technique in Improvement of Physicochemical Characteristics of Ibuprofen Powder. Iran. J. Pharm. Sci. 2007, 3, 69–76. [Google Scholar]
- Tran, H.T.T.; Park, J.B.; Hong, K.H.; Choi, H.G.; Han, H.K.; Lee, J.; Oh, K.T.; Lee, B.J. Preparation and characterization of pH-independent sustained release tablet containing solid dispersion granules of a poorly water-soluble drug. Int. J. Pharm. 2011, 415, 83–88. [Google Scholar] [CrossRef]
- Shaikh, K.; Patwekar, S.; Payghan, S.; D’Souza, J. Dissolution and stability enhancement of poorly water soluble drug-lovastatin by preparing solid dispersions. Asian J. Biomed. Pharm. Sci. 2011, 1, 24–31. [Google Scholar]
- Kamalakkannan, V.; Puratchikody, A.; Ramanathan, L. Development and characterization of controlled release polar lipid microparticles of candesartan cilexetil by solid dispersion. Res. Pharm. Sci. 2013, 8, 125–136. [Google Scholar]
- Sharma, A.; Jain, C.P.; Tanwar, Y.S. Preparation and characterization of solid dispersions of carvedilol with poloxamer 188. J. Chil. Chem. Soc. 2013, 58, 1553–1557. [Google Scholar] [CrossRef]
- Noolkar, S.B.; Jadhav, N.; Bhende, S.A.; Killedar, S.G. Solid-State Characterization and Dissolution Properties of Meloxicam–Moringa Coagulant–PVP Ternary Solid Dispersions. AAPS PharmSciTech 2013, 14, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Tekade, A.R.; Gattani, S.; Surana, S. Solubility enhancement of lovastatin by modified locust bean gum using solid dispersion techniques. AAPS PharmSciTech 2008, 9, 1262–1269. [Google Scholar] [CrossRef] [PubMed]
- Ghareeb, M.M.; Abdulrasool, A.A.; Hussein, A.A.; Noordin, M.I. Kneading technique for preparation of binary solid dispersion of meloxicam with poloxamer 188. AAPS PharmSciTech 2009, 10, 1206–1215. [Google Scholar] [CrossRef]
- Rajpurohit, V.S.; Rakha, P.; Goyal, S.; DUREJA, H.; Arorac, G.; Nagpal, M. Formulation And Characterization Of Solid Dispersions Of Glimepiride Through Factorial Design. Iran. J. Pharm. Sci. 2011, 7, 7–16. [Google Scholar]
- Qushawy, M.; Prabahar, K.; Abd-Alhaseeb, M.; Swidan, S.; Nasr, A. Preparation and evaluation of carbamazepine solid lipid nanoparticle for alleviating seizure activity in pentylenetetrazole-kindled mice. Molecules 2019, 24, 3971. [Google Scholar] [CrossRef]
- de los Santos, C.J.J.; Pérez-Martínez, J.I.; Gómez-Pantoja, M.E.; Moyano, J.R. Enhancement of albendazole dissolution properties using solid dispersions with Gelucire 50/13 and PEG 15000. J. Drug Deliv. Sci. Technol. 2017, 42, 261–272. [Google Scholar] [CrossRef]
- Mahapatra, A.K.; Murthy, P.N.; Patra, R.K.; Mallik, S. Dissolution rate enhancement and solid state characterization of Ritonavir-PEG 4000 solid dispersions. J. Pharm. Adv. Res. 2019, 2, 650–656. [Google Scholar]
- Soni, L.; Ansari, M.; Thakre, N.; Singh, A.; Bhowmick, M.; Rathi, J. Development and in-vitro evaluation of furosemide solid dispersion using different water soluble carriers. Int. J. Res. Dev. Pharm. Life Sci. 2017, 6, 2571–2575. [Google Scholar] [CrossRef]
- Das, S.; Mandal, P. Design, formulation, and evaluation of solid dispersion tablets of poorly water-soluble antidiabetic drug using natural polymer. Asian J. Pharm. Clin. Res. 2019, 12, 195–197. [Google Scholar] [CrossRef]
- Mehenni, L.; Lahiani-Skiba, M.; Ladam, G.; Hallouard, F.; Skiba, M. Preparation and characterization of spherical amorphous solid dispersion with amphotericin B. Pharmaceutics 2018, 10, 235. [Google Scholar] [CrossRef]
- Rao, M.; Mandage, Y.; Thanki, K.; Bhise, S. Dissolution improvement of simvastatin by surface solid dispersion technology. Dissolution Technol. 2010, 17, 27–34. [Google Scholar] [CrossRef]
- Prabahar, K.; Udhumansha, U.; Qushawy, M. Optimization of thiolated chitosan nanoparticles for the enhancement of in vivo hypoglycemic efficacy of sitagliptin in streptozotocin-induced diabetic rats. Pharmaceutics 2020, 12, 300. [Google Scholar] [CrossRef]
- Chavan, R.B.; Lodagekar, A.; Yadav, B.; Shastri, N.R. Amorphous solid dispersion of nisoldipine by solvent evaporation technique: Preparation, characterization, in vitro, in vivo evaluation, and scale up feasibility study. Drug Deliv. Transl. Res. 2020, 10, 903–918. [Google Scholar] [CrossRef]
- Davis, M.T.; Potter, C.B.; Walker, G.M. Downstream processing of a ternary amorphous solid dispersion: The impacts of spray drying and hot melt extrusion on powder flow, compression and dissolution. Int. J. Pharm. 2018, 544, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Browne, E.; Charifou, R.; Worku, Z.A.; Babu, R.P.; Healy, A.M. Amorphous solid dispersions of ketoprofen and poly-vinyl polymers prepared via electrospraying and spray drying: A comparison of particle characteristics and performance. Int. J. Pharm. 2019, 566, 173–184. [Google Scholar] [CrossRef]
- Malviya, R.; Srivastava, P.; Bansal, M.; Sharma, P.K. Improvement of dissolution behavior of paracetamol using solid dispersion technique. Int. J. Pharm. Sci. Res. 2010, 1, 95–99. [Google Scholar]
- Mahajan, A.; Surti, N.; Koladiya, P. Solid dispersion adsorbate technique for improved dissolution and flow properties of lurasidone hydrochloride: Characterization using 32 factorial design. Drug Dev. Ind. Pharm. 2018, 44, 463–471. [Google Scholar] [CrossRef]
- Kaur, P.; Singh, S.K.; Garg, V.; Gulati, M.; Vaidya, Y. Optimization of spray drying process for formulation of solid dispersion containing polypeptide-k powder through quality by design approach. Powder Technol. 2015, 284, 1–11. [Google Scholar] [CrossRef]
- Andrade, T.C.; Martins, R.M.; Freitas, L.A.P. Granulation of indomethacin and a hydrophilic carrier by fluidized hot melt method: The drug solubility enhancement. Powder Technol. 2015, 270, 453–460. [Google Scholar] [CrossRef]
- Zaini, E.; Umar, S.; Nurhidayah, N. Improvement of dissolution rate of valsartan by solid dispersion system using D(-) mannitol. Asian J. Pharm. Clin. Res. 2017, 10, 288–290. [Google Scholar] [CrossRef]
- Febriyenti, F.; Rahmi, S.; Halim, A. Study of gliclazide solid dispersion systems using PVP K-30 and PEG 6000 by solvent method. J. Pharm. Bioallied Sci. 2019, 11, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Arora, A.; Aggarwal, G.; Singh, T.G.; Singh, M.; Arora, G.; Nagpal, M. Inclusion complexes of atorvastatin calcium–sulfobutyl ether β cyclodextrin with enhanced hypolipidemic activity. J. Appl. Pharm. Sci. 2019, 9, 60–68. [Google Scholar]
- Kaushik, S.; Pathak, K. Solubility enhancement of glimperide: Development of solid dispersion by solvent melt method, characterization and dosage form development. Pharm. Biomed. Res. 2018, 3, 1–13. [Google Scholar] [CrossRef]
- Rajbanshi, K.; Bajracharya, R.; Shrestha, A.; Thapa, P. Dissolution enhancement of aceclofenac tablet by solid dispersion technique. Int. J. Pharma Sci. Res. 2014, 5, 127–139. [Google Scholar]
- Leonardi, D.; Barrera, M.G.; Lamas, M.C.; Salomon, C.J. Development of prednisone: Polyethylene glycol 6000 fast-release tablets from solid dispersions: Solid-state characterization, dissolution behavior, and formulation parameters. AAPS PharmSciTech 2007, 8, 221–228. [Google Scholar] [CrossRef]
- Abarca, R.L.; Rodríguez, F.J.; Guarda, A.; Galotto, M.J.; Bruna, J.E. Characterization of beta-cyclodextrin inclusion complexes containing an essential oil component. Food Chem. 2016, 196, 968–975. [Google Scholar] [CrossRef]
- Kiran, T.; Shastri, N.; Ramakrishna, S.; Sadanandam, M. Surface solid dispersion of glimepiride for enhancement of dissolution rate. Int. J. Pharm. Tech. Res. 2009, 1, 822–831. [Google Scholar]
- Alves, L.D.S.; Soares, M.F.D.L.R.; De Albuquerque, C.T.; Da Silva, É.R.; Vieira, A.C.C.; Fontes, D.A.F.; Figueirêdo, C.B.M.; Soares-Sobrinho, J.L.; Neto, P.J.R. Solid dispersion of efavirenz in PVP K-30 by conventional solvent and kneading methods. Carbohydr. Polym. 2014, 104, 166–174. [Google Scholar] [CrossRef]
- Ganesan, P.; Soundararajan, R.; Shanmugam, U.; Ramu, V. Development, characterization and solubility enhancement of comparative dissolution study of second generation of solid dispersions and microspheres for poorly water soluble drug. Asian J. Pharm. Sci. 2015, 10, 433–441. [Google Scholar] [CrossRef]
- Mamatha, T.; Anitha, N.; Qureshi, H.K. Development of nevirapine tablets by direct compression method using solid dispersion technique. J. Pharm. Res. 2017, 16, 72–79. [Google Scholar] [CrossRef]
- Pagadala, A. Formulation and evaluation of solid dispersion of glimepiride in to sustained release. Glob. J. Pharm. Pharm. Sci. 2016, 1, 1–5. [Google Scholar] [CrossRef]
- Li, H.; Pan, T.; Cui, Y.; Li, X.; Gao, J.; Yang, W.; Shen, S. Improved oral bioavailability of poorly water-soluble glimepiride by utilizing microemulsion technique. Int. J. Nanomed. 2016, 11, 3777–3788. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Independent Factors | Low (−1) | Medium (0) | High (1) |
|---|---|---|---|
| X1 = Type of carrier | Mannitol | PEG 6000 | β-Cyclodextrin |
| X2 = Drug: Carrier ratio | 1:1 | 1:3 | 1:6 |
| Dependent Variables | Goal | ||
| Y1 = The drug release (%) | Maximize | ||
| Formulation No. | Variable X1 | Variable X2 |
|---|---|---|
| F1 | −1 | −1 |
| F2 | −1 | 0 |
| F3 | −1 | 1 |
| F4 | 0 | −1 |
| F5 | 0 | 0 |
| F6 | 0 | 1 |
| F7 | 1 | −1 |
| F8 | 1 | 0 |
| F9 | 1 | 1 |
| Formulation No. | PY % | DC % |
|---|---|---|
| F1 | 99.5 ± 2.5 | 98.1 ± 1.5 |
| F2 | 98.5 ± 3.1 | 97.4 ± 2.6 |
| F3 | 99.1 ± 1.5 | 99.6 ± 2.1 |
| F4 | 98.4 ± 2.8 | 96.8 ± 2.9 |
| F5 | 99.8 ± 2.2 | 99.3 ±3.7 |
| F6 | 99.2 ± 3.9 | 97.2 ± 3.2 |
| F7 | 99.7 ± 2.1 | 98.7 ± 2.7 |
| F8 | 98.7 ± 2.3 | 98.3 ± 2.4 |
| F9 | 98.8 ± 3.7 | 99.5 ± 1.8 |
| Formulation No. | Bulk Density (g/cm3) | Tapped Density (g/cm3) | Hausenr’s Ratio | Carr’s Index (%) | Angle of Repose |
|---|---|---|---|---|---|
| F1 | 0.54 ± 0.03 | 0.61 ± 0.02 | 1.13 ± 0.03 | 11.48 ± 0.34 | 17.45 ± 0.82 |
| F2 | 0.46 ± 0.02 | 0.51 ± 0.01 | 1.11 ± 0.01 | 9.80 ± 0.54 | 14.26 ± 0.91 |
| F3 | 0.47 ± 0.02 | 0.54 ± 0.01 | 1.15 ± 0.02 | 12.96 ± 0.65 | 18.45 ± 0.75 |
| F4 | 0.51 ± 0.01 | 0.57 ± 0.01 | 1.12 ± 0.01 | 10.53 ± 0.15 | 15.85 ± 0.64 |
| F5 | 0.49 ± 0.03 | 0.58 ± 0.02 | 1.18 ± 0.01 | 15.52 ± 0.95 | 19.45 ± 0.54 |
| F6 | 0.45 ± 0.04 | 0.55 ± 0.02 | 1.22 ± 0.02 | 18.18 ± 0.47 | 23.44 ± 0.63 |
| F7 | 0.42 ± 0.03 | 0.50 ± 0.01 | 1.19 ± 0.02 | 16.00 ± 0.45 | 20.91 ± 0.68 |
| F8 | 0.37 ± 0.05 | 0.43 ± 0.02 | 1.16 ± 0.03 | 13.95 ± 0.12 | 18.67 ± 0.42 |
| F9 | 0.40 ± 0.04 | 0.48 ± 0.01 | 1.20 ± 0.03 | 16.67 ± 0.38 | 22.86 ± 0.46 |
| Source | Sum of Squares | DF | Mean Square | F-Ratio | p-Value |
|---|---|---|---|---|---|
| Model | 881.07 | 5 | 176.21 | 42.21 | 0.0056 |
| A-(X1) | 198.95 | 1 | 198.95 | 47.66 | 0.0062 |
| B-(X2) | 371.15 | 1 | 371.15 | 88.90 | 0.0025 |
| AB | 2.53 | 1 | 2.53 | 0.6056 | 0.4932 |
| A 2 | 308.43 | 1 | 308.43 | 73.88 | 0.0033 |
| B 2 | 0.0093 | 1 | 0.0093 | 0.0022 | 0.9652 |
| Residual | 12.52 | 3 | 4.17 | ||
| Correlation Total | 893.59 | 8 | |||
| Formula No. | Correlation Coefficient (r) | |||||
|---|---|---|---|---|---|---|
| Zero | First | Second | Diffusion | H-C | B-L | |
| F1 | 0.9916 | −0.9981 | 0.9946 | 0.9959 | 0.9971 | 0.9973 |
| F2 | 0.9910 | −0.9971 | 0.9860 | 0.9927 | 0.9965 | 0.9964 |
| F3 | 0.9893 | −0.9987 | 0.9884 | 0.9952 | 0.9977 | 0.9976 |
| F4 | 0.9956 | −0.9975 | 0.9951 | 0.9906 | 0.9968 | 0.9964 |
| F5 | 0.9932 | −0.9964 | 0.9899 | 0.9876 | 0.9956 | 0.9957 |
| F6 | 0.9902 | −0.9970 | 0.9842 | 0.9933 | 0.9966 | 0.9965 |
| F7 | 0.9856 | −0.9977 | 0.9968 | 0.9963 | 0.9951 | 0.9961 |
| F8 | 0.9712 | −0.9927 | 0.9918 | 0.9904 | 0.9879 | 0.9877 |
| F9 | 0.8797 | −0.9909 | 0.8293 | 0.9454 | 0.9881 | 0.9654 |
| Free glimepiride | 0.9922 | −0.9930 | 0.9917 | 0.9868 | 0.9926 | 0.9722 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qushawy, M.; Nasr, A.; Swidan, S.; Mortagi, Y. Development and Characterization of Glimepiride Novel Solid Nanodispersion for Improving Its Oral Bioavailability. Sci. Pharm. 2020, 88, 52. https://doi.org/10.3390/scipharm88040052
Qushawy M, Nasr A, Swidan S, Mortagi Y. Development and Characterization of Glimepiride Novel Solid Nanodispersion for Improving Its Oral Bioavailability. Scientia Pharmaceutica. 2020; 88(4):52. https://doi.org/10.3390/scipharm88040052
Chicago/Turabian StyleQushawy, Mona, Ali Nasr, Shady Swidan, and Yasmin Mortagi. 2020. "Development and Characterization of Glimepiride Novel Solid Nanodispersion for Improving Its Oral Bioavailability" Scientia Pharmaceutica 88, no. 4: 52. https://doi.org/10.3390/scipharm88040052
APA StyleQushawy, M., Nasr, A., Swidan, S., & Mortagi, Y. (2020). Development and Characterization of Glimepiride Novel Solid Nanodispersion for Improving Its Oral Bioavailability. Scientia Pharmaceutica, 88(4), 52. https://doi.org/10.3390/scipharm88040052