The Relative Importance of the Small Intestine and the Liver in Phase II Metabolic Transformations and Elimination of p-Nitrophenol Administered in Different Doses in the Rat



Abstract
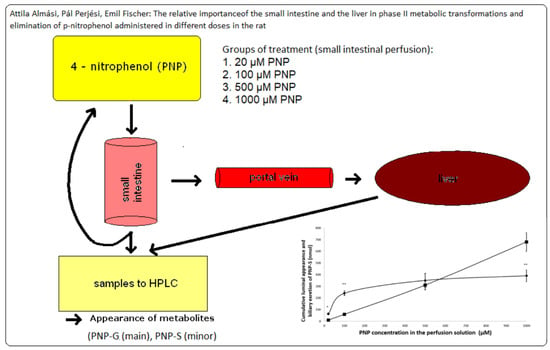
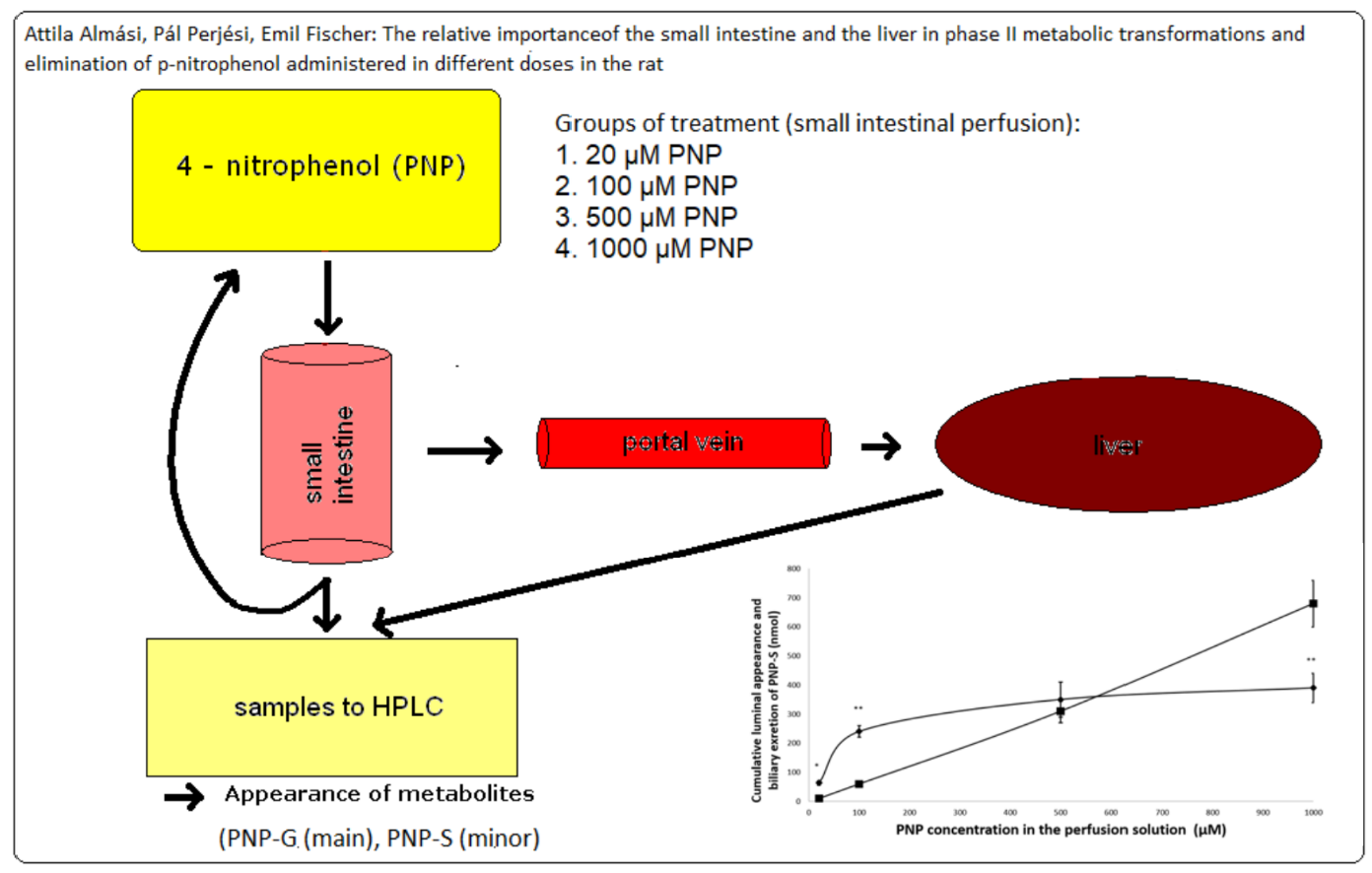{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals and Experimental Procedure
2.3. Sample Analysis
2.3.1. Instrumentation
2.3.2. Analytical Conditions
2.3.3. Calculations, Statistical Analysis
2.3.4. Ethical Approval
3. Results
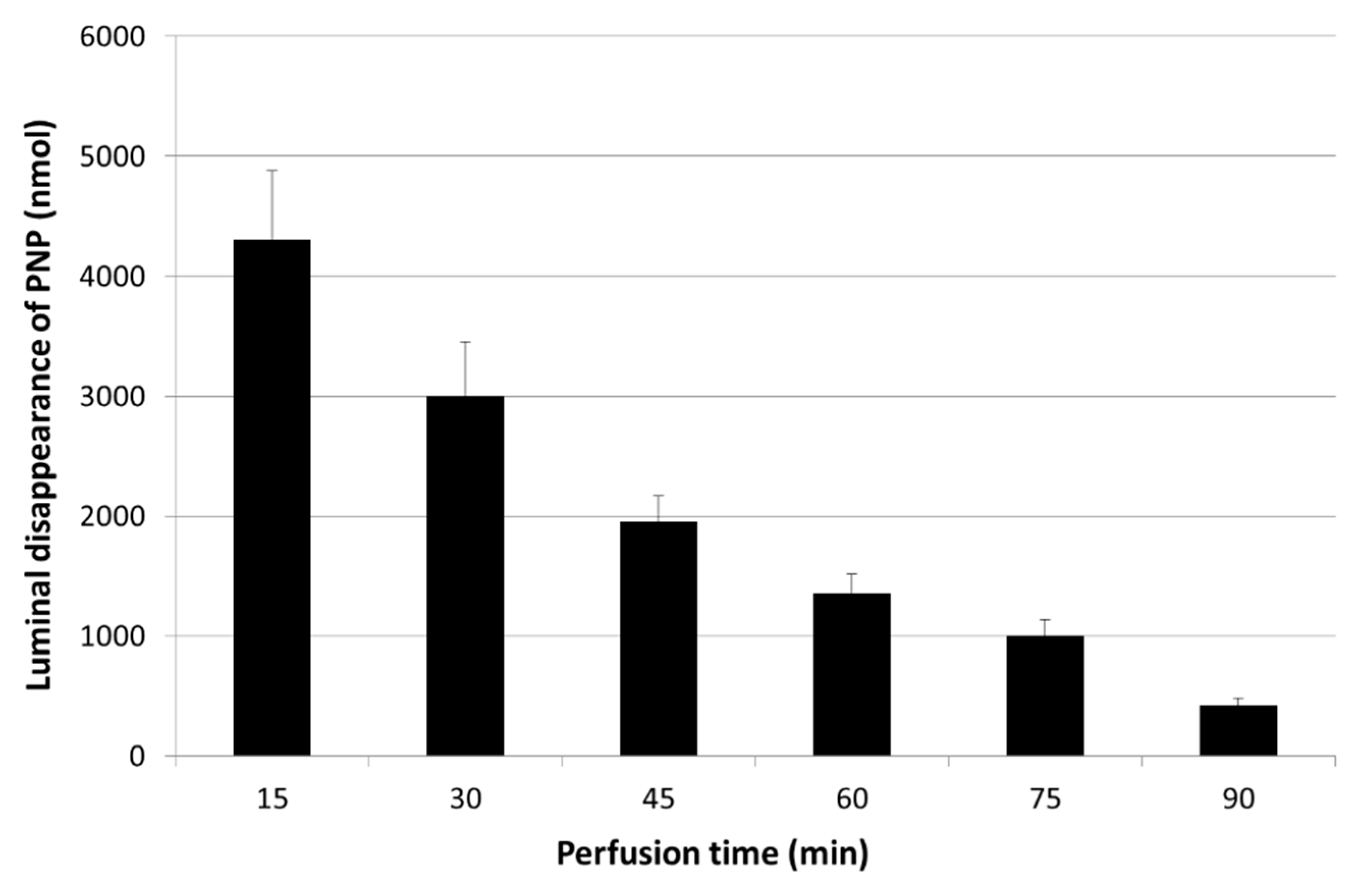
3.1. Luminal Disappearance of p-Nitrophenol (PNP) from the Small Intestine Measured in 90 min Perfusion of 500 µM PNP
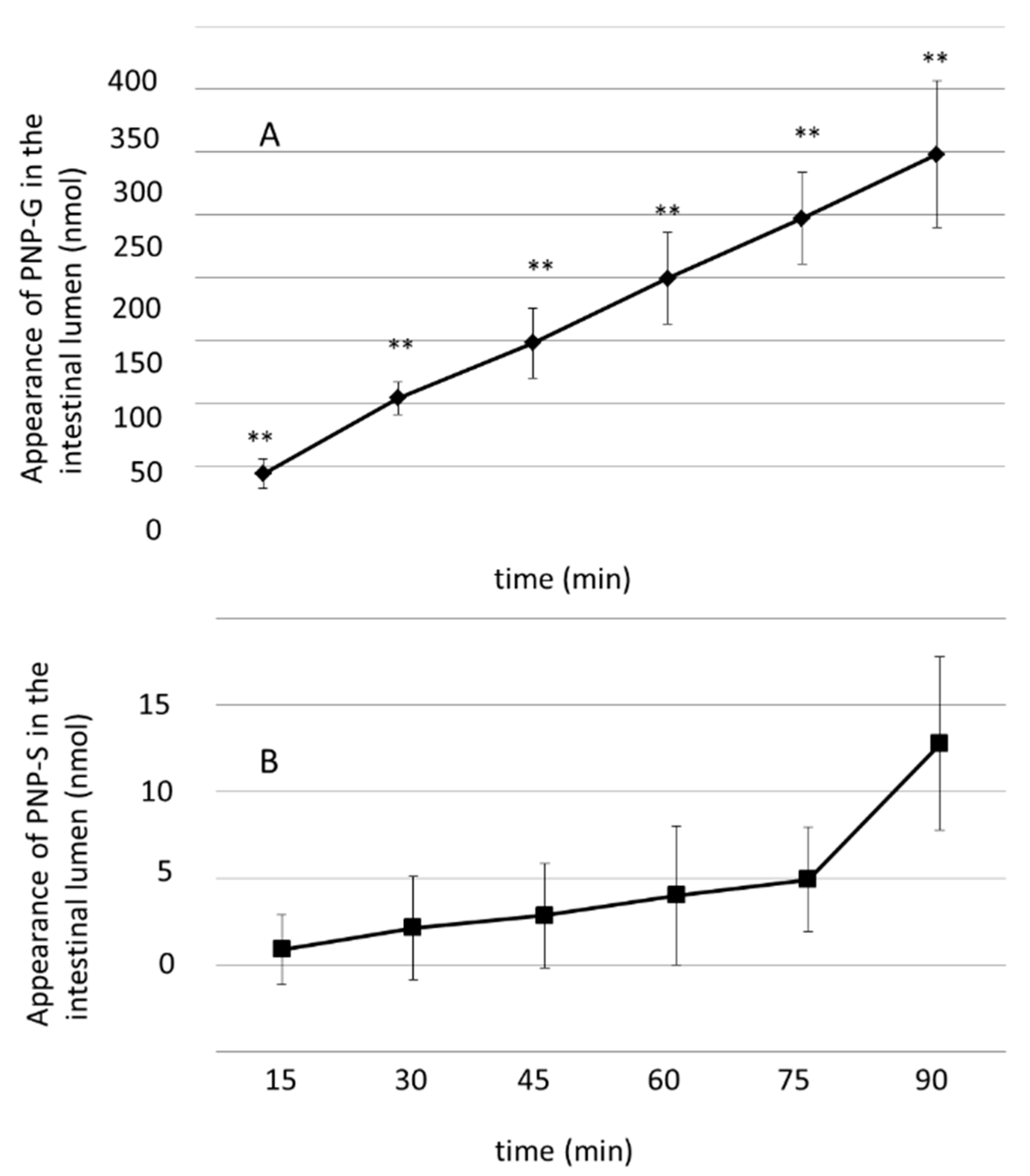
3.2. Luminal Appearance of p-Nitrophenol-Glucuronide (PNP-G) and p-Nitrophenol–Sulfate (PNP-S) in the Small Intestine Measured in 90 min Perfusion of 500 µM PNP
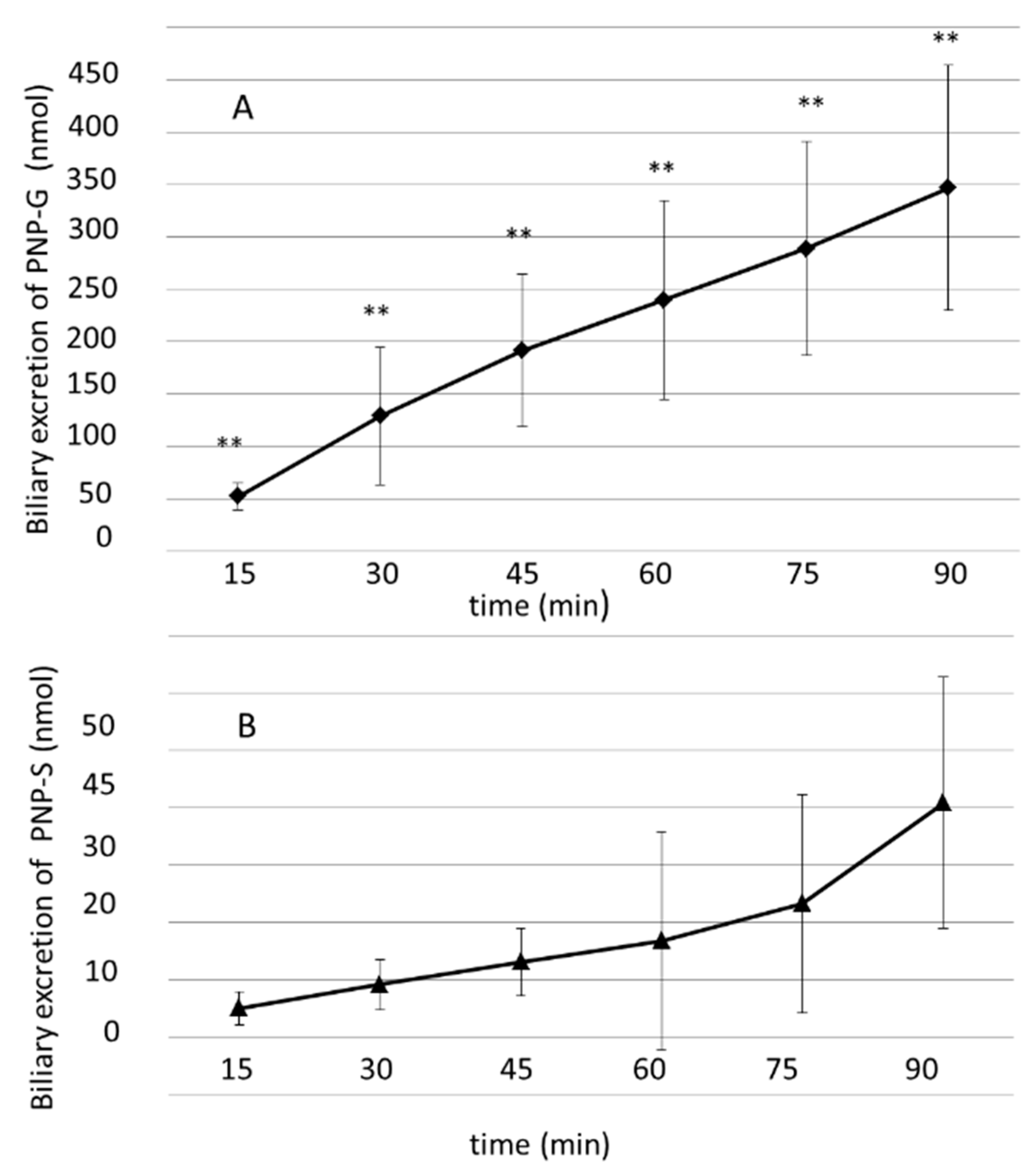
3.3. Biliary Excretion of PNP-G and PNP-S Measured in 90 min Perfusion of 500 µM PNP in the Small Intestine
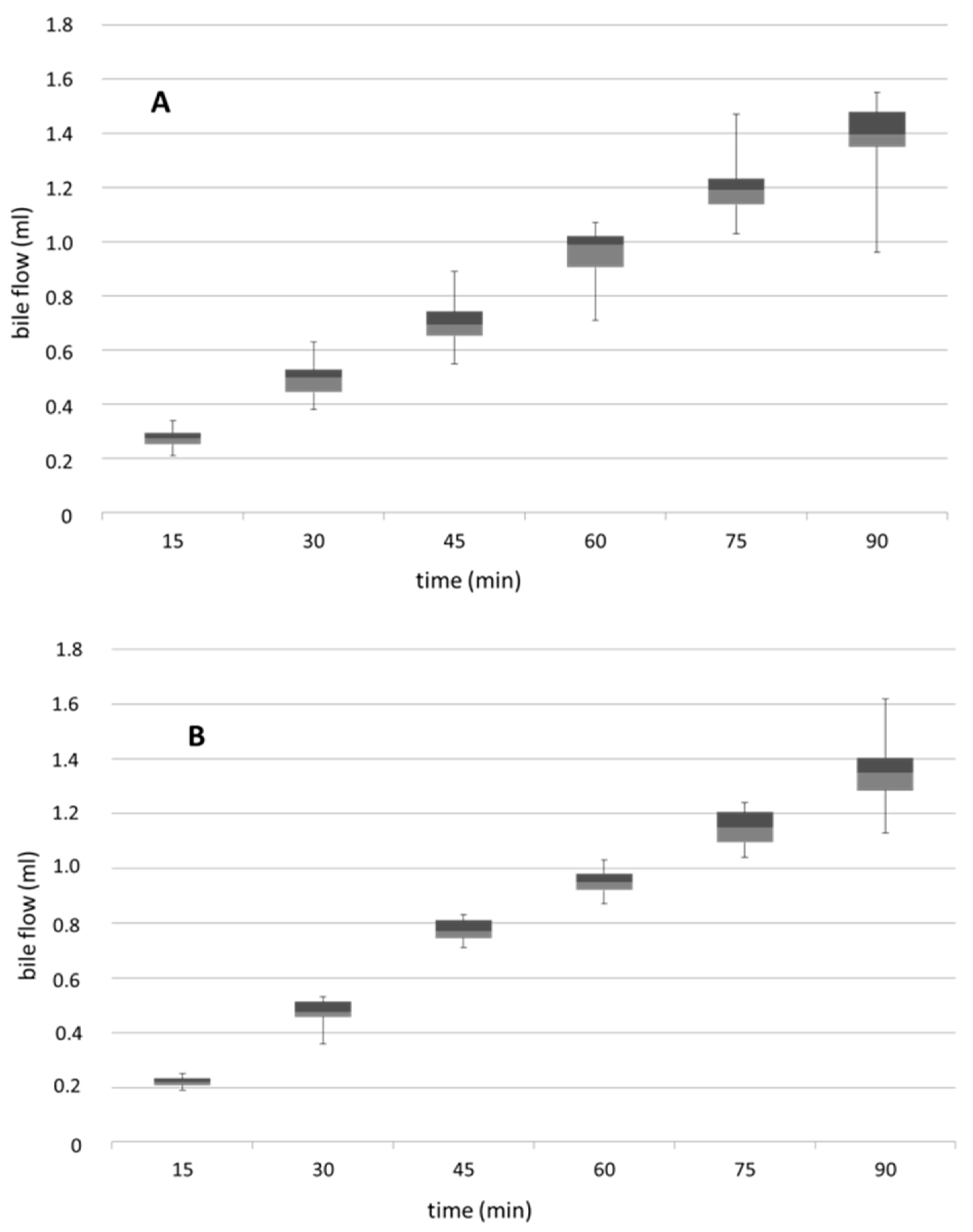
3.4. Biliary Flow Measured in Control and PNP-Perfused Rats Measured in 90 min Perfusion of 500 µM PNP in the Small Intestine
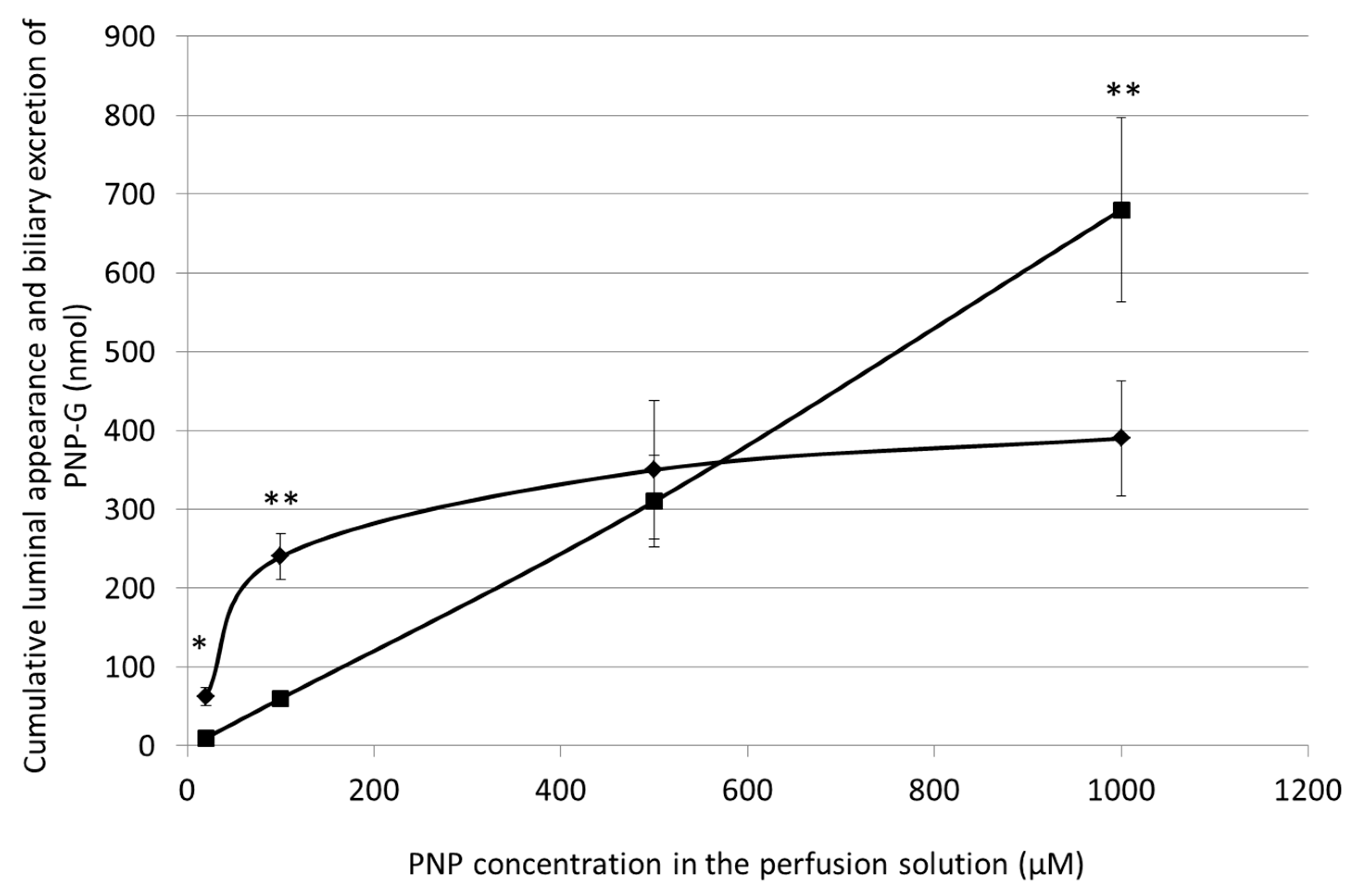
3.5. Cumulative Luminal Appearance and Biliary Excretion of PNP-G Measured in 90 min Perfusion of 20 µM, 100 µM, 500 µM, and 1000 µM PNP in the Small Intestine
3.6. Cumulative Luminal Appearance and Biliary Excretion of PNP-S Measured in 90 min Perfusion of 20 µM, 100 µM, 500 µM, and 1000 µM PNP in the Small Intestine
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Laitinen, M.; Watkins, J.B. Mucosal biotransformations. In Gastrointestinal Toxicology; Rozman, K., Hänninen, O., Eds.; Elsevier Press: Amsterdam, The Netherlands, 1986; pp. 169–192. [Google Scholar]
- Beaumont, K. The importance of gut wall metabolism in determining drug bioavailibility. In Drug Bioavailibility: Estimation of Solubility, Permeability, Absorption and Bioavailability; van de Waterbeemd, H., Lenneräus, H., Artursson, P., Eds.; Willey—VCH Verlag GmbH and Co. KgaA: Weinheim, Germany, 2004; pp. 311–328. [Google Scholar]
- van de Kerkhof, E.G.; de Graaf, I.A.M.; Groothuis, G.M.M. In vitro methods to study intestinal drug metabolism. Curr. Drug Metab. 2007, 8, 658–675. [Google Scholar] [CrossRef] [PubMed]
- Linn, J.H.; Chiba, M.; Baillie, T.A. Is the role of the small intestine in first–pass metabolism overemphasized? Pharm. Rev. 1999, 51, 135–157. [Google Scholar]
- Schwenk, M. Glucuronidation and sulphatation in gastrointestinal tract. Prog. Pharmacol. Clin. Pharmacol. 1989, 7, 155–169. [Google Scholar]
- George, C.F. Drug metabolism by gastrointestinal mucosa. Clin. Pharmacokinet. 1981, 6, 259–274. [Google Scholar] [CrossRef]
- Hartiala, K.J.W. Metabolism of hormones, drugs and other substances by the gut. Physiol. Rev. 1973, 53, 496–534. [Google Scholar] [CrossRef] [PubMed]
- Hänninen, O.; Lindström-Seppä, P.; Pelkonen, K. Role of the gut in xenobiotic metabolism. Arch. Toxicol. 1987, 60, 34–36. [Google Scholar] [CrossRef] [PubMed]
- Lauterbach, F.; Schorn, M.; Sprakties, G.; Sund, R.B. Compartmentalization of intestinal conjugation reactions and conjugate transports: Studies with phenols and p-aminobenzoic acid. Prog. Pharmacol. Clin. Pharmacol. 1982, 7, 231–242. [Google Scholar]
- Sund, R.B.; Lauterbach, F. Drug metabolism and metabolite transport in the small and large intestine: Experiments with 1-Naphthol and phenolphthalein by luminal and contraluminal administration in the isolated guinea pig mucosa. Acta Pharmacol. Toxicol. 1986, 58, 74–83. [Google Scholar] [CrossRef]
- Barnes, S.; Buchina, E.S.; King, R.J.; McBurnett, T.; Taylor, K.B. Bile acid sulfotransferase I from rat liver sulfates bile acids and 3-hydroxy steroids: Purification, N-terminal amino acid sequence and kinetic properties. J. Lipid Res. 1989, 30, 429–440. [Google Scholar]
- Brand, W.; Boersma, M.G.; Bik, H.; Hoek-van den Hil, E.F.; Vervoort, J.; Barron, D.; Meinl, W.; Glatt, H.; Williamson, G.; van Bladeren, P.J.; et al. Phase II metabolism of hesperetin by individual UDP-glucuronosyltransferases and sulfotransferases and rat and human tissue samples. Drug Metab. Dispos. 2010, 38, 617–625. [Google Scholar] [CrossRef]
- Capiello, M.; Giuliani, L.; Pacifici, G.M. Distribution of UDP-glucuronyltransferase and its endogenous substrate uridine 5′-diphosphoglucuronic acid in human tissues. Eur. J. Pharmacol. 1991, 41, 345–350. [Google Scholar] [CrossRef]
- Danovith, S.H.; Laster, R. The development of arylsulfatase in the small intestine of the rat. Biochem. J. 1969, 114, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Falany, C.N. Molecular enzymology of human cytosolic sulfortransferases. Trends Pharmacol. Sci. 1991, 12, 255–259. [Google Scholar] [CrossRef]
- Inoue, H.; Yokota, H.; Taniyama, H.; Kuwahara, A.; Ogawa, H.; Kato, S.; Yuasa, E. 1-naphthol-β-D-glucuronides formed intraluminally in rat small intestine mucosa and absorbed into the colon. Life Sci. 1999, 65, 1579–1588. [Google Scholar] [CrossRef]
- Mojarrabi, B.; Mackenzie, P.I. Characterization of two UDP glucuronosyltransferases that are predominantly expressed in human colon. Biochem. Biophys. Res. Commun. 1998, 247, 704–709. [Google Scholar] [CrossRef]
- Tukey, R.H.; Strassburg, C.P. Human UDP-glucuronosyltransferases: Metabolism, expression and disease. Ann. Rev. Pharmacol. Toxicol. 2000, 40, 581–616. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.; Rafiei, A.; Bojcsev, S. Intestinal elimination of p-nitrophenol in the rat. Acta Physiol. Hung. 1995, 83, 355–362. [Google Scholar]
- Bojcsev, S.; Almási, A.; Simon, H.; Perjési, P.; Fischer, E. Investigation of drug metabolism in various segments of small intestine in the rat. Acta Physiol. Hung. 2013, 100, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Almási, A.; Bojcsev, S.; Fischer, T.; Simon, H.; Perjési, P.; Fischer, E. Metabolic enzyme activities and drug excretion in the small intestine and in the liver in the rat. Acta Physiol. Hung. 2013, 100, 478–488. [Google Scholar] [CrossRef]
- Fischer, E.; Almási, A.; Bojcsev, S.; Fischer, T.; Kovács, N.P.; Perjési, P. Effect of experimental diabetes and insulin replacement on intestinal metabolism and excretion of 4-nitrophenol in rats. Can. J. Physiol. Pharmacol. 2015, 93, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Almási, A.; Pinto, É.I.L.N.; Kovács, N.P.; Fischer, T.; Markovics, Z.; Fischer, E.; Perjési, P. Changes in hepatic metabolic enzyme activities and biliary excretion of 4-nitrophenol in streptozotocin induced diabetic rats. Braz. J. Pharm. Sci. 2018, 54, e11347. [Google Scholar] [CrossRef]
- Eadsforth, C.V.; Coveney, D.C. Measurement of phenol in urine using a high-performance liquid chromatographic method. Analyst 1984, 109, 175–176. [Google Scholar] [CrossRef] [PubMed]
- Kothare, A.P.; Zimmerman, C.L. Intestinal metabolism: The role of enzyme localization in phenol metabolite kinetics. Drug Metab. Dispos. 2002, 30, 586–594. [Google Scholar] [CrossRef]
- Kuhn, U.D.; Rost, D.; Müller, M. Para-nitrophenol glucuronidation and sulphatation in rat and human slices. Exp. Toxic. Pathol. 2001, 53, 81–87. [Google Scholar] [CrossRef]
- Gow, P.J.; Ghabrial, H.; Treepongkaruna, S.; Shulkes, A.; Smallwood, R.A.; Morgan, D.J.; Ching, M.S. Conjugation of para-nitrophenol by the isolated perfused neonatal sheep liver. J. Pharm. Sci. 2000, 89, 36–44. [Google Scholar] [CrossRef]
- Toxicological Profile for Nitrophenols: 2-Nitrophenol, 4-Nitrophenol; Agency for Toxic Substances and Disease Registry U.S. Public Health Service: Atlanta, GA, USA, 1992. Available online: https://www.atsdr.cdc.gov/toxprofiles/tp50.pdf (accessed on 30 September 2020).
- Almási, A.; Fischer, E.; Perjési, P. A simple and rapid ion-pair HPLC method for simultaneous quantitation of 4-nitrophenol and its glucuronide and sulfate conjugates. J. Biochem. Biophys. Methods 2006, 69, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Almási, A.; Fischer, E.; Perjési, P. Isocratic ion-pair HPLC method for quantitation of 4-nitrophenol and it’s conjugated metabolites from bile. Sci. Pharm. 2011, 79, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Concise International Chemical Assessment Document 20. In Mononitrophenols; World Health Organization: Geneva, Switzerland, 2000; Available online: http://www.inchem.org/documents/cicads/cicads/cicad_20.htm (accessed on 30 September 2020).
- McConnell, E.L.; Basit, A.W.; Murdan, S. Measurements of rat and mouse gastrointestinal pH, fluid and lymphoid tissue, and implications for in-vivo experiments. J. Pharm. Pharmacol. 2008, 60, 63–70. [Google Scholar] [CrossRef]
- Kuhl, H. Pharmacology of estrogens and progestogens: Influence of different routes of administration. Climacteric 2005, 8 (Suppl. 1), 3–63. [Google Scholar] [CrossRef]
- Kuzma, M.; Fodor, K.; Almási, A.; Mózsik, G.; Past, T.; Perjési, P. Toxicokinetic study of a gastroprotective dose of capsaicin by HPLC-FLD method. Molecules 2019, 24, 2848. [Google Scholar] [CrossRef]
- Peterson, J.J.; Dwyer, J.T.; Jacques, P.F.; McCullough, M.L. Improving the estimation of flavonoid intake for study of health outcomes. Nutr. Rev. 2015, 73, 553–576. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, C.D. Bile flow and bile composition during bile acid depletion and administration. Can. J. Physiol. Pharmacol. 1973, 52, 334–348. [Google Scholar] [CrossRef]
- Kuzma, M.; Fodor, K.; Maász, G.; Avar, P.; Mózsik, G.; Past, T.; Fischer, E.; Perjési, P. A validated HPLC-FLD method for analysis of intestinal absorption and metabolism of capsaicin and dihydrocapsaicin in the rat. J. Pharm. Biomed. Anal. 2015, 103, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Kovács, N.P.; Almási, A.; Garai, K.; Kuzma, M.; Vancea, S.; Fischer, E.; Perjési, P. Investigation of intestinal elimination and biliary excretion of ibuprofen in hyperglycemic rats. Can. J. Physiol. Pharmacol. 2019, 97, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- King, C.; Tang, V.; Ngui, J.; Tephly, T.; Braun, M. Characterization of rat and human UDP-glucuronosyltransferases responsible for the in vitro glucuronidation of diclofenac. Toxicol. Sci. 2001, 61, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.Z.; Huang, B.B.; Gu, T.T.; Tai, T.; Zhou, H.; Jia, Y.M.; Mi, Q.Y.; Zhang, M.R.; Xie, H.G. Human UGT2B7 is the major isoform responsible for the glucuronidation of clopidogrel carboxylate. Biopharm. Drug Dispos. 2018, 39, 88–98. [Google Scholar] [CrossRef]
- Wu, B.; Basu, S.; Meng, S.; Wang, X.; Hu, M. Regioselective sulfation and glucuronidation of phenolics: Insights into the structural basis. Curr. Drug Metab. 2011, 12, 900–916. [Google Scholar] [CrossRef]
- Dong, D.; Ako, R.; Hu, M.; Wu, B. Understanding substrate selectivity of human UDP-glucuronosyltransferases through QSAR modeling and analysis of homologous enzyme. Xenobiotica 2012, 42, 808–820. [Google Scholar] [CrossRef]
- Prueksaritanont, T.; Gorham, L.M.; Hochman, J.H.; Tran, L.O.; Vyas, K.P. Comparative studies of drug metabolising enzymes in dog, monkey and human small intestines and in Caco-2-cells. Drug. Metab. Dispos. 1996, 24, 634–642. [Google Scholar]
- Miles, K.K.; Kessler, F.K.; Smith, P.C.; Ritter, J.K. Characterization of rat intestinal microsomal UDP-glucuronosyltransferase activity toward mycophenolic acid. Drug Metab. Dispos. 2006, 34, 1632–1639. [Google Scholar] [CrossRef]
- Grams, B.; Harms, A.; Braun, S.; Strassburg, C.P.; Manns, M.P.; Obermayer-Straub, P. Distribution and inducibility by 3-methylcholantrene of family 1 UDP glucuronosyltransferases in the rat gastrointestinal tract. Arch. Biochem. Biophys. 2000, 377, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Van der Logt, E.M.; Roelofs, H.M.; Nagengast, F.M.; Peters, W.H. Induction of rat hepatic and intestinal UDP-glucuronosyltransferases by naturally occurring dietary anticarcinogens. Carcinogenesis 2003, 24, 1651–1656. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Chen, Y.; Huang, C.; Chen, G. Caffeine induction of sulfotransferases in rat liver and intestine. J. Appl. Toxicol. 2012, 32, 804–809. [Google Scholar] [CrossRef]
- Wu, B.; Jiang, W.; Yin, T.; Gao, S.; Hu, M. A new strategy to rapidly evaluate kinetics of glucuronide efflux by breast cancer resistance protein (BCRP/ABCG2). Pharm. Res. 2012, 29, 3199–3208. [Google Scholar] [CrossRef]
- Chu, X.Y.; Huskey, S.E.; Braun, M.P.; Sarkadi, B.; Evans, D.C.; Evers, R. Transport of ethinylestradiol glucuronide and ethinylestradiol sulfate by the multidrug resistance proteins MRP1, MRP2, and MRP3. J. Pharmacol. Exp. Ther. 2004, 309, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Jedlitschky, G.; Leier, I.; Buchholz, U.; Hummel-Eisenbeiss, J.; Burchell, B.; Keppler, D. ATP-dependent transport of bilirubin glucuronides by the multidrug resistance protein MRP1 and its hepatocyte canalicular isoform MRP2. Biochem. J. 1997, 327, 305–310. [Google Scholar] [CrossRef]
- Sun, H.; Zhou, X.; Zhang, X.; Wu, B. Decreased Expression of multidrug resistance-associated protein 4 (MRP4/ABCC4) leads to reduced glucuronidation of flavonoids in UGT1A1-overexpressing HeLa cells: The role of futile recycling. J. Agric. Food Chem. 2015, 63, 6001–6008. [Google Scholar] [CrossRef]
- Drozdzik, M.; Busch, D.; Lapczuk, J.; Müller, J.; Ostrowski, M.; Kurzawski, M.; Oswald, S. Protein abundance of clinically relevant drug transporters in the human liver and intestine: A comparative analysis in paired tissue specimens. Clin. Pharmacol. Ther. 2019, 105, 1204–1212. [Google Scholar] [CrossRef]
- Burt, H.J.; Riedmaier, A.E.; Harwood, M.D.; Crewe, H.K.; Gill, K.L.; Neuhoff, S. Abundance of hepatic transporters in Caucasians: A meta-analysis. Drug Metab. Dispos. 2016, 44, 1550–1561. [Google Scholar] [CrossRef]
- Järvinen, E.; Deng, F.; Kidron, H.; Finel, M. Efflux transport of estrogen glucuronides by human MRP2, MRP3, MRP4 and BCRP. J. Steroid Biochem. Mol. Biol. 2018, 178, 99–107. [Google Scholar] [CrossRef]
- Stockdale, M.; Selwyn, M.J. Effects of ring substituents on the activity of phenols as inhibitors and uncouplers of mitochondrial respiration. Eur. J. Biochem. 1971, 21, 565–574. Available online: https://febs.onlinelibrary.wiley.com/doi/pdf/10.1111/j.1432-1033.1971.tb01502.x (accessed on 7 September 2020). [CrossRef] [PubMed]
- Parkinson, A.; Ogilvie, B.W.; Buckley, D.B.; Kazmi, F.; Czerwinski, M.; Parkinson, O. Biotransformation of Xenobiotics. In Casarett and Doull’s Toxicology: The Basic Science of Poisons, 8th ed.; Klaassen, C.D., Ed.; McGraw-Hill Education/Medical: New York, NY, USA, 2013; Chapter 6; ISBN 10 9780071769235. [Google Scholar]
- Collins, S.L.; Patterson, A.D. The gut microbiome: An orchestrator of xenobiotic metabolism. Acta Pharm. Sin. 2020, 10, 19–32. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almási, A.; Perjési, P.; Fischer, E. The Relative Importance of the Small Intestine and the Liver in Phase II Metabolic Transformations and Elimination of p-Nitrophenol Administered in Different Doses in the Rat. Sci. Pharm. 2020, 88, 51. https://doi.org/10.3390/scipharm88040051
Almási A, Perjési P, Fischer E. The Relative Importance of the Small Intestine and the Liver in Phase II Metabolic Transformations and Elimination of p-Nitrophenol Administered in Different Doses in the Rat. Scientia Pharmaceutica. 2020; 88(4):51. https://doi.org/10.3390/scipharm88040051
Chicago/Turabian StyleAlmási, Attila, Pál Perjési, and Emil Fischer. 2020. "The Relative Importance of the Small Intestine and the Liver in Phase II Metabolic Transformations and Elimination of p-Nitrophenol Administered in Different Doses in the Rat" Scientia Pharmaceutica 88, no. 4: 51. https://doi.org/10.3390/scipharm88040051
APA StyleAlmási, A., Perjési, P., & Fischer, E. (2020). The Relative Importance of the Small Intestine and the Liver in Phase II Metabolic Transformations and Elimination of p-Nitrophenol Administered in Different Doses in the Rat. Scientia Pharmaceutica, 88(4), 51. https://doi.org/10.3390/scipharm88040051