Voltammetric Behaviour of Drug Molecules as a Predictor of Metabolic Liabilities

 ,
, 
 ,
,  and
and 
Abstract
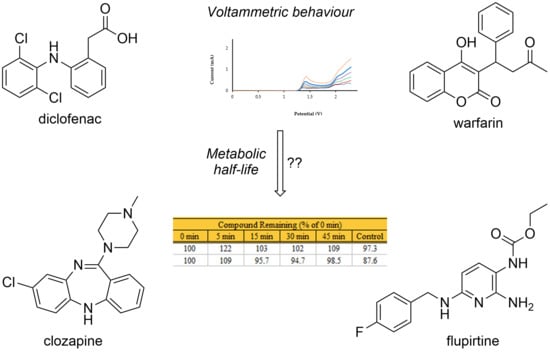
1. Introduction
- Data mining the existing literature for the half-life and oxidation potential values of known drugs;
- Correlating standardised redox measurements with stability measures;
- Analysing examples of short and longer circulating drug molecules to determine their voltammetric parameters.
2. Materials and Methods
2.1. Data Mining
2.2. General Points
2.3. General Procedures for Linear Sweep Voltammetry (LSV)
2.4. Compound Characterisation
2.4.1. 3-Methoxy-4-hydroxyphenylacetic Acid Propyl Ester (2)
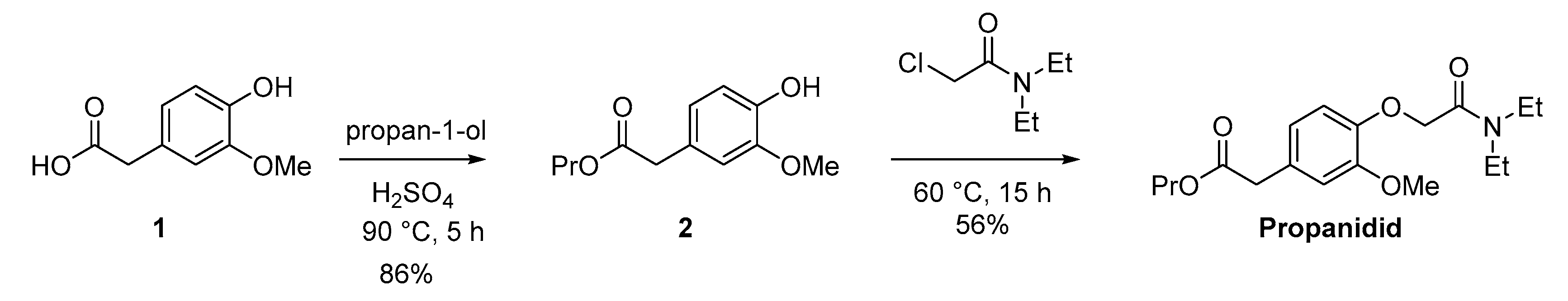
2.4.2. [4-[(Diethylcarbamoyl) methoxy]-3-methoxyphenyl] Acetic Acid Propyl Ester (Propanidid)
2.4.3. Entacapone
2.4.4. Lidocaine
3. Results
3.1. Comparison of Drug Metabolite OP to Parent Drug OP
3.2. Relationship between Drug OP and Half-Life
3.3. Synthesis of Propanidid
3.4. Voltammetric Study of Propanidid, Entacapone and Lidocaine.
4. Discussion
4.1. Comparing Parent Drugs to Drug Metabolites
4.2. Drug Metabolism Pathways
4.3. New Drug OP Measurements and Stability Inference
4.4. Study limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Raimondi, V.; Ciccarese, F.; Ciminale, V. Oncogenic pathways and the electron transport chain: A dangerous liaison. Br. J. Cancer 2020, 122, 168–181. [Google Scholar] [CrossRef]
- Issa, N.T.; Wathieu, H.; Ojo, A.; Byers, S.W.; Dakshanamurthy, S. Drug Metabolism in Preclinical Drug Development: A Survey of the Discovery Process, Toxicology, and Computational Tools. Curr. Drug Metab. 2017, 18, 556–565. [Google Scholar] [CrossRef]
- Jurva, U.; Weidolf, L. Electrochemical generation of drug metabolites with applications in drug discovery and development. TrAC Trends Anal. Chem. 2015, 70, 92–99. [Google Scholar] [CrossRef]
- Blankert, B.; Kauffmann, J.K. Electroanalytical Methods as Tools for Predictive Drug Metabolism Studies. Rev. Pharm. Biomed. Anal. 2010, 76–83. [Google Scholar] [CrossRef][Green Version]
- Madsen, K.G.; Olsen, J.; Skonberg, C.; Hansen, S.H.; Jurva, U. Development and evaluation of an electrochemical method for studying reactive phase-I metabolites: Correlation to in vitro drug metabolism. Chem. Res. Toxicol. 2007, 20, 821–831. [Google Scholar] [CrossRef]
- Li, Y.; Meng, Q.; Yang, M.; Liu, D.; Hou, X.; Tang, L.; Wang, X.; Lyu, Y.; Chen, X.; Liu, K.; et al. Current trends in drug metabolism and pharmacokinetics. Acta Pharm. Sin. B 2019, 9, 1113–1144. [Google Scholar] [CrossRef] [PubMed]
- Gul, T.; Bischoff, R.; Permentier, H.P. Electrosynthesis methods and approaches for the preparative production of metabolites from parent drugs. TrAC Trends Anal. Chem. 2015, 70, 58–66. [Google Scholar] [CrossRef]
- Rahman, M.H.; Bal, M.K.; Jones, A.M. Metabolism Inspired Electrosynthesis. ChemElectroChem 2019, 6, 4093–4104. [Google Scholar] [CrossRef]
- Bal, M.K.; Banks, C.E.; Jones, A.M. Metabolism mimicry: An electrosynthetic method for the selective deethylation of tertiary benzamides. ChemElectroChem 2019, 6, 4284–4291. [Google Scholar] [CrossRef]
- Wetzel, A.; Jones, A.M. Electrically driven N(sp2)-C(sp2/3) bond cleavage of sulfonamides. ACS Sustain. Chem. Eng. 2020, 8, 3487–3493. [Google Scholar] [CrossRef]
- Tammari, E.; Nezhadali, A.; Lotfi, S.; Mohammadizadeh, M.R. Electrosynthesis of Clozapine Drug Derivative via an EC Electrochemical Mechanism. Anal. Bioanal. Chem. Res. 2017, 4, 319–328. [Google Scholar]
- Alfonso-Súarez, P.; Kolliopoulos, A.V.; Smith, J.P.; Banks, C.E.; Jones, A.M. An Experimentalists Guide to Electrosynthesis: The Shono Oxidation. Tetrahedron Lett. 2015, 56, 6863–6867. [Google Scholar] [CrossRef]
- Barone, M.R.; Jones, A.M. Selective C-H Bond Electro-oxidation of Benzylic Acetates and Alcohols to Benzaldehydes. Org. Biomol. Chem. 2017, 15, 10010–10015. [Google Scholar] [CrossRef]
- Jones, A.M.; Banks, C.E. The Shono-type electroorganic oxidation of unfunctionalised amides. Carbon-carbon bond formation via electrogenerated N-acyliminium ions. Beilstein J. Org. Chem. 2014, 10, 3056–3072. [Google Scholar] [CrossRef]
- Ferro, C.J.; Solkhon, F.; Jalal, Z.; Al-Hamid, A.M.; Jones, A.M. Relevance of physicochemical properties and functional pharmacology data to predict the clinical safety profile of direct oral anticoagulants. Pharmacol. Res. Perspect. 2020, 8, e00603. [Google Scholar] [CrossRef]
- Jalal, Z.; Cabdi, S.; Khan, N.; Dorsch, M.; Gill, N.K.; Toner, F.; Jones, A.M. Sacubitril/Valsartan (Entresto) hospital prescribing in patients with symptomatic chronic HF with reduced ejection fraction: A UK multi-centre study. J. Prescrib. Prac. 2019, 1, 182–192. [Google Scholar] [CrossRef]
- Baranowska, M.; Suliborska, K.; Chrzanowski, W.; Kusznierewicz, B.; Namiesnik, J.; Bartoszek, A. The relationship between standard reduction potentials of catechins and biological activities involved in redox control. Redox Biol. 2018, 17, 355–366. [Google Scholar] [CrossRef]
- De Abreu, D.C.; Ferraz, P.A.D.L.; Goulart, M.O.F. Some Applications of Electrochemistry in Biomedical Chemistry. Emphasis on the Correlation of Electrochemical and Bioactive Properties. J. Braz. Chem. Soc. 2002, 13, 19–35. [Google Scholar] [CrossRef]
- Verduzco-Ramirez, A.; Manzanilla-Davila, S.G.; Morales-Guillen, M.E.; Garcia-Ramos, J.C.; Toledano-Magana, Y.; Marin-Becerra, A.; Flores-Alamo, M.; Ortiz-Frade, L.A.; Olguin-Contreras, L.F.; Ruiz-Azuara, L. Essential Metal-based drugs: Correlation between Redox Potential and Biological Activity of M2+ with a N2O2 Ligand. J. Mex. Chem. Soc. 2017, 61, 109–119. [Google Scholar] [CrossRef]
- Carvalho, M.; Remiao, F.; Milhazes, N.; Borges, F.; Fernandes, E.; Monteiro, M.d.C.; Goncalves, M.J.; Seabra, V.; Amado, F.; Carvalho, F.; et al. Metabolism is required for the expression of ecstasy-induced cardiotoxicity in vitro. Chem. Res. Toxicol. 2007, 17, 623–632. [Google Scholar] [CrossRef]
- Macedo, C.; Branco, P.S.; Ferreira, L.M.; Lobo, A.M.; Capela, J.P.; Fernandes, E.; Bastos, M.d.L.; Carvalho, F. Synthesis and Cyclic Voltammetry Studies of 3,4-Methylenedioxymethamphetamine (MDMA) Human Metabolites. J. Health Sci. 2007, 53, 31–42. [Google Scholar] [CrossRef]
- Coppola, G.M.; Anjaria, H.; Damon, R.E. Correlation of oxidation potential and toxicity in thiobenzamides. Bioorg. Med. Chem. Lett. 1996, 6, 139–142. [Google Scholar] [CrossRef]
- Rodriguez-Cid, L.; Sentellas, S.; Saurina, J. Voltammetric and electrogeneration approaches for the assessment of the oxidative drug metabolism. Anal. Bioanal. Chem. 2018, 410, 2229–2239. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, T.; Hofmann, D.; Klumpp, E. Electrochemistry-mass spectrometry for mechanistic studies and simulation of oxidation processes in the environment. Anal. Bioanal. Chem. 2011, 399, 1859–1868. [Google Scholar] [CrossRef] [PubMed]
- Madsen, K.G.; Skonberg, C.; Jurva, U.; Cornett, C.; Hansen, S.H.; Johansen, T.N.; Olsen, J. Bioactivation of Diclofenac in vitro and in vivo: Correlation to Electrochemical Studies. Chem. Res. Toxicol. 2008, 21, 1107–1119. [Google Scholar] [CrossRef]
- Landsdorp, D.; Vree, T.B.; Janssen, T.J.; Guelen, P.J. Pharmacokinetics of rectal diclofenac and its hydroxy metabolites in man. Int. J. Clin. Pharmacol. Ther. Toxicol. 1990, 28, 298–302. [Google Scholar]
- Winkler, T.E.; Lederer, S.L.; Kim, E.; Ben-Yoav, H.; Kelly, D.L.; Payne, G.F.; Ghodssi, R. Molecular processes in an electrochemical clozapine sensor. Biointerphases 2017, 12, 02B401. [Google Scholar] [CrossRef]
- Yang, M.; Kabulski, J.L.; Wollenberg, L.; Chen, X.; Subramanian, M.; Tracy, T.S.; Lederman, D.; Gannett, P.M.; Wu, N. Electrocatalytic Drug Metabolism by CYP2C9 Bonded to A Self-Assembled Monolayer-Modified Electrode. Drug Metab. Dispos. 2009, 37, 892–899. [Google Scholar] [CrossRef]
- ADVANZ Pharma (1998, 2018). Warfarin 0.5 mg Tablets. Available online: https://www.medicines.org.uk/emc/product/2803/smpc (accessed on 6 October 2020).
- Bristol-Myers Squibb Canada. PrCOUMADIN® Warfarin Sodium Tablets, Bristol-Myers Squibb Std., (Crystalline) Montreal, Canada. 2018. Available online: https://www.bms.com/assets/bms/ca/documents/productmonograph/COUMADIN_EN_PM.pdf (accessed on 28 August 2020).
- Methling, K.; Reszka, P.; Lalk, M.; Vrana, O.; Scheuch, E.; Siegmund, W.; Terhaag, B.; Bednarski, P.J. Investigation of the in vitro metabolism of the analgesic flupirtine. Drug Metab. Dispos. 2009, 37, 479–493. [Google Scholar] [CrossRef]
- Renwick, A.C.; Renwick, A.G.; Flanagan, F.J.; Ferner, R.E. Monitoring of clozapine and norclozapine plasma concentration-time curves in acute overdose. J. Toxicol. Clin. Toxicol. 2000, 38, 325–328. [Google Scholar] [CrossRef]
- Lemmerhirt, C.J.; Rombach, M.; Bodtke, A.; Bednarski, P.J.; Link, A. Oxidation Potentials of N-Modified Derivatives of the Analgesic Flupirtine Linked to Potassium KV7 Channel Opening Activity but Not Hepatocyte Toxicity. ChemMedChem 2014, 10, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Siegmund, W.; Modess, C.; Scheuch, E.; Methling, K.; Keiser, M.; Nassif, A.; Rosskopf, D.; Bednarski, P.J.; Borlak, J.; Terhaag, B. Metabolic activation and analgesic effect of flupirtine in heatlhy subjects, influence of the polymorphic NAT2, UGT1A1 and GSTP1. Br. J. Clin. Pharmacol. 2014, 79, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.A.; Matter, B.A.; Raol, Y.H.; Bourne, D.W.A.; Kelley, R.A.; Kompella, U.B. Brain Distribution and Metabolism of Flupirtine, a Nonopioid Analgesic Drug with Antiseizure Effects, in Neonatal Rats. Pharmaceutics 2018, 10, 281. [Google Scholar] [CrossRef] [PubMed]
- Temocin, Z.; Kim, E.; Li, J.; Panzella, L.; Alfieri, M.L.; Napolitano, A.; Kelly, D.L.; Bentley, W.E.; Payne, G.F. The Analgesic Acetaminophen and the Antipsychotic Clozapine Can Each Redox-Cycle with Melanin. ACS Chem. Neurosci. 2017, 8, 2766–2777. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Blankert, B.; Kauffman, J.-M. Development of amperometric horseradish peroxidase based biosensors for clozapine and for the screening of thiol compounds. Biosens. Bioelectron. 2007, 22, 2707–2711. [Google Scholar] [CrossRef] [PubMed]
- Mylan (1989, 2014). Clozaril 100 mg Tablets. Available online: https://www.medicines.org.uk/emc/product/10290/smpc (accessed on 6 October 2020).
- Novartis. 2004. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2005/019758s054lbl.pdf (accessed on 6 October 2020).
- NHS. Clozapine Guidelines, NHS Foundation Trust, Version 4. 2018. Available online: https://www.southernhealth.nhs.uk/EasysiteWeb/getresource.axd?AssetID=77706&type=full&servicetype=Inline (accessed on 6 October 2020).
- Rosemont Pharmaceuticals Limited (1985, 2013). Promazine Hydrochloride 50 mg/5 mL Oral Syrup. Available online: https://www.medicines.org.uk/emc/product/6698/smpc#:~:text=It%20is%20highly% (accessed on 6 October 2020).
- Berte, M.; Appia, F.T.A.; Sanogo, I.; Ouattara, L. Electrochemical Oxidation of the Paracetamol in its Commercial Formulation on Platinum, and Ruthenium Dioxide Electrodes. Int. J. Electrochem. Sci. 2016, 11, 7736–7749. [Google Scholar] [CrossRef]
- Eisele, A.P.P.; Valezi, C.F.; Sartori, E.R. Exploiting the high oxidation potential of carisoprodol on a boron-doped diamond electrode: An improved method for its simultaneous determination with acetaminophen and caffeine. Analyst 2017, 142, 3514–3521. [Google Scholar] [CrossRef]
- Zentiva. Paracetamol 500mg Soluble Tablets. 2018. Available online: https://www.medicines.org.uk/emc/product/4199/smpc (accessed on 6 October 2020).
- Prescott, L.F. Kinetics and metabolism of paracetamol and phenacetin. Br. J. Clin. Pharmacol. 1980, 10, 291S–298S. [Google Scholar] [CrossRef]
- Medscape. Acetaminophen (OTC). Available online: https://reference.medscape.com/drug/tylenol-acetaminophen-343346#10 (accessed on 29 July 2020).
- Sanofi-Aventis Consumer Healthcare (1998, 2019). New Zealand Data Sheet, Phenergan—Promethazine Hydrochloride. Available online: https://www.medsafe.govt.nz/profs/Datasheet/p/Phenergantabelixir.pdf (accessed on 6 October 2020).
- Ciltas, U.; Yilmaz, B.; Kaban, S.; Akcay, B.K.; Nazik, G. Square Wave Voltammetric Determination of Diclofenac in Pharmaceutical Preparations and Human Serum. Iran. J. Pharm. Res. 2015, 14, 715–722. [Google Scholar]
- Damiri, S.; Oskoei, Y.M.; Fouladgar, M. Highly sensitive voltammetric and impedimetric sensor based on an ionic liquied/cobalt hexacyanoferrate nanoparticle modified multi-walled carbon nanotubes electrode for diclofenac analysis. J. Exper. Nanosci. 2016, 11, 1384–1401. [Google Scholar] [CrossRef]
- Wong, A.; Marestoni, L.D.; Sotomayor, M.D.P.T. Monitoring of diclofenac with biomimetic sensor in batch and FIA systems. J. Braz. Chem. Soc. 2014, 25, 1283–1291. [Google Scholar] [CrossRef]
- NOVARTIS. Voltaren®® (Diclofenac Sodium Enteric-Coated Tablets) Tablets of 75 mg Rx Only Perscribing Information. Caguas, Puerto Rico 00726 Mova Pharmaceuticals Corporation. 2009. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/019201s038lbl.pdf (accessed on 6 October 2020).
- Pfizer Canada ULC. ARTHROTEC (Diclofenac Sodium and Misoprostol Enteric-Coated Tablets), Action and Clinical Pharmacology. 2020. Available online: https://www.pfizermedicalinformation.ca/en-ca/arthrotec/action-and-clinical-pharmacology# (accessed on 29 July 2020).
- Lecours, M.A.; Eysseric, E.; Yargeau, V.; Lessard, J.; Brisard, G.M.; Segura, P.A. Electrochemistry-High Resolution Mass Spectrometry to Study Oxidation Products of Trimethoprim. Environments 2018, 5, 18. [Google Scholar] [CrossRef]
- Kent Pharmaceuticals Ltd. Trimethoprim 100mg Tablets. 2005. Available online: https://www.medicines.org.uk/emc/product/4059/smpc (accessed on 6 October 2020).
- O’Neill, P.M.; Harrison, A.C.; Storr, R.C.; Hawley, S.R.; Ward, S.A.; Park, B.K. The Effect of Fluorine Substitution on the Metabolism and Antimalarial Activity of Amodiaquine. J. Med. Chem. 1994, 37, 1362–1370. [Google Scholar] [CrossRef] [PubMed]
- Winstanley, P.; Edwards, G.; Ormes, M.; Breckenridge, A. The disposition of amodiaquine in man after oral administration. Br. J. Clin. Pharmacol. 1987, 23, 1–7. [Google Scholar] [CrossRef]
- Msellem, M.; Morris, U.; Soe, A.; Abbas, F.B.; Ali, A.-W.; Barnes, R.; Frumento, P.; Ali, A.S.; Martensson, A.; Bjorkman, A. Increase Sensitivity of Plasmodium falciparum to Artesunate/Amodiaquine Despite 14 Years as First Line Malaria Treatment, Zanzibar. Emerg. Infect. Dis. 2020, 26, 1767–1777. [Google Scholar] [CrossRef] [PubMed]
- Bussy, U.; Ferchaud-Roucher, V.I.; Krempf, T.M.; Silvestre, V.; Boujtita, M. Electrochemical oxidation behavior of Acebutolol and identification of intermediate species by liquid chromatography and mass spectrometry. Electrochim. Acta 2012, 69, 351–357. [Google Scholar] [CrossRef]
- SANOFI (1974, 2019). Sectral 100mg Capsules. Available online: https://www.medicines.org.uk/emc/product/3212/smpc (accessed on 6 October 2020).
- Tadesse, Y.; Tadese, A.; Saini, R.C.; Pal, R. Cyclic Voltammetric Investigation of Caffeine at Anthraquinone Modified Carbon Paste Electrode. Int. J. Electrochem. 2013, 2013, 849327. [Google Scholar] [CrossRef]
- Whitseit, T.L.; Manion, C.V.; Chistensen, H.D. Cardiovascular Effects of Coffee and Caffeine. Am. J. Cardiol. 1984, 53, 919–922. [Google Scholar] [CrossRef]
- Brachtel, D.; Richter, E.E. Absolute bioavailability of caffeine from a tablet formulation. J. Hepatol. 1992, 16, 385. [Google Scholar] [CrossRef]
- Meda Pharmaceuticals Inc. SOMA® COMPOUND (Carisoprodol and Aspirin Tablets, USP) for Oral Use New Jersey, USA. 2009. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/012365s035lbl.pdf (accessed on 6 October 2020).
- Wang, G.; Huynh, K.; Barhate, R.; Rodrigues, W.; Moore, C.; Coulter, C.; Vincent, M.; Soares, J. Validation of a New Homogeneous Immunoassay for the Detection of Carisoprodol in Urine. J. Anal. Toxicol. 2011, 35, 108–112. [Google Scholar] [CrossRef]
- Nousiainen, U. The effects of phenobarbital, bis-p-nitrophenyl phosphate and disulfiram on the hydrolysis of propanidid in wistar rats. Gen. Pharm. 1984, 15, 397–402. [Google Scholar] [CrossRef]
- Forsberg, M.; Lehtonen, M.; Heikkinen, M.; Savolainen, J.; Järvinen, T.; Männistö, P.T. Pharmacokinetics and Pharmacodynamics of Entacapone and Tolcapone after Acute and Repeated Administration: A Comparative Study in the Rat. J. Pharmacol. Exper. Therap. 2003, 304, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Collinsworth, K.A.; Kalman, S.M.; Harrison, D.C. The Clinical Pharmacology of Lidocaine as an Antiarrhythmic Drug. Circulation 1974, 50, 1217–1230. [Google Scholar] [CrossRef] [PubMed]
- Hiltmaan, R.; Wollweber, H.; Hoffmeister, F.; Wirth, W. 3-Methoxy-4-Carbadmidomethyoxy-Phenylacetic Acid Esters. U.S. Patent 3086978, 23 April 1963. [Google Scholar]
- Gieshoff, T.; Kehl, A.; Schollmeyer, D.; Moeller, K.D.; Waldvogel, S.R. Insights into the Mechanism of Anodic N-N Bond Formation by Dehydrogenative Coupling. J. Am. Chem. Soc. 2017, 139, 12317–12324. [Google Scholar] [CrossRef]
- Randles, J.E.B. Kinetics of rapid electrode reactions. Discuss. Faraday Soc. 1947, 1, 11. [Google Scholar] [CrossRef]
- Randles, J.E.B. A cathode ray polarograph. Part II.—The current-voltage curves. Trans. Faraday Soc. 1948, 44, 327. [Google Scholar] [CrossRef]
- Sevcik, A. Oscillographic polarography with periodical triangular voltage. Collect. Czechoslov. Chem. Commun. 1948, 13, 349. [Google Scholar] [CrossRef]
- Laviron, E.; Roullier, L.; Degrand, C. A multilayer model for the study of space distributed redox modified electrodes: Part II. Theory and application of linear potential sweep voltammetry for a simple reaction. J. Electroanal. Chem. Interfacial Electrochem. 1980, 112, 11–23. [Google Scholar] [CrossRef]
- Pardiñas, A.F.; Nalmpanti, M.; Pocklington, A.J.; Legge, S.E.; Medway, C.; King, A.; Jansesn, J.; Helthuis, M.; Zammit, S.; MacCabe, J.M.J.; et al. Pharmacogenomic Variants and Drug Interactions Identified Through the Genetic Analysis of Clozapine Metabolism. Am. J. Psychiatry 2019, 176, 477–486. [Google Scholar] [CrossRef]
- Siegmund, W. Pharmacokinetics, Metabolism and Analgesic Effects of Flupirtine. In Clinical Trial NCT01676246; 2012. Available online: https://clinicaltrials.gov/ct2/show/NCT01676246 (accessed on 6 October 2020).
- Harish, S.; Bhuvana, K.; Bengalorkar, G.M.; Kumar, T.N. Flupirtine: Clinical pharmacology. J. Anaesthesiol. Clin. Pharmacol. 2012, 28, 172–177. [Google Scholar] [CrossRef]
- Agarwal, A.; Bui, A.D. Oxidation-reduction potential as a new marker for oxidative stress: Correlation to male infertility. Investig. Clin. Urol. 2017, 58, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Baranczewski, P.; Stanczak, A.; Kautiainen, A.; Sandin, P.; Edlund, P.O. Introduction to early in vitro identification of metabolites of new chemical entities in drug discovery and development. Pharmacol. Rep. 2006, 58, 341–352. [Google Scholar]
- Irving, R.M.; Elfarra, A.A. Role of Reactive Metabolites in the Circulation in Extrahepatic Toxicity. Expert Opin. Drug Metab. Toxicol. 2012, 8, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Curry, S.H. Chapter 8—Pharmacokinetics of antipsychotic drugs. Antipsychotic Drugs and Their Side-Effects; Barnes, T.R.E., Ed.; Academic Press: San Diego, CA, USA, 1993; pp. 127–144. [Google Scholar]
- Zabirowicz, E.S.; Gan, T.J. 34—Pharmacology of Postoperative Nausea and Vomiting. In Pharmacology and Physiology for Anesthesia, 2nd ed.; Hemmings, H.C., Egan, T.D., Eds.; Elsevier: Philadelphia, PA, USA, 2019; pp. 671–692. [Google Scholar]
- Muta, K.; Fukami, T.; Nakajima, M. A proposed mechanism for the adverse effects of acebutolol: CES2 and CYP2C19-mediated metabolism and antinuclear antibody production. Biochem. Pharmacol. 2015, 98, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Miners, J.O.; Birkett, D.J. Cytochrome P4502C9: An enzyme of major importance in human drug metabolism. Br. J. Clin. Pharmacol. 1998, 45, 525–538. [Google Scholar] [CrossRef]
- Mahadevan, S.B.K.; McKiernan, P.J.; Davies, P.; Kelly, D.A. Paracetamol induced hepatotoxicity. Arch. Dis. 2006, 91, 598–603. [Google Scholar] [CrossRef]
- Athersuch, T.J.; Antoine, D.J.; Boobis, A.R.; Coen, M.; Daly, A.K.; Passamai, L.; Nicholson, J.K.; Wilson, I.D. Paracetamol metabolism, hepatotoxicity, biomarkers and therapeutic interventions: A perspective. Toxicol. Res. 2018, 7, 347–357. [Google Scholar] [CrossRef]
- Spreux-Varoquax, O.; Chapalain, J.P.; Cordonnier, P.; Advenier, C. Determination of trimethoprim, sulfphamethoxazole and its N4-acetyl metabolite in biological fluids by high-performance liquid chromatography. J. Chromat. B Biomed. Appl. 1983, 274, 187–199. [Google Scholar] [CrossRef]
- Hoppu, K.; Arjomaa, P. Difference in Trimethoprim Pharmacokinetics between Children and Adults. Chemotherapy 1984, 30, 283–287. [Google Scholar] [CrossRef]
- Winnie, N.G.; Lobach, A.R.M.; Zhu, X.; Chen, X.; Liu, F.; Metushi, I.G.; Sharma, A.; Li, J.; Cai, P.; Ip, J.M.; et al. Uetrecht. Animal Models of Idiosyncratic Drug Reactions. Adv. Pharmacol. 2012, 63, 81–135. [Google Scholar]
- Ginsburg, H.; Famin, O.; Zhang, J.; Krugliak, M. Inhibition of glutathione-dependent degradation of heme by chloroquine and amodiaquine as a possible basis for their antimalarial mode of action. Biochem. Pharmacol. 1998, 56, 1305–1313. [Google Scholar] [CrossRef]
- Tian, D.-D.; Natesan, S.; White, J.R.M., Jr.; Paine, F. Effects of Common CYP 1A2 Genotypes and Other Key Factors on Intraindividual Variation in the Caffeine Metabolic Ratio: An Exploratory Analysis. Clin. Transl. Sci. 2019, 12, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Institute of Medicine (US) Committee on Military Nutrition Research. Caffeine for the Sustainment of Mental Task Performance: Formulations for Military Operations; National Academies Press (US): Washington, DC, USA, 2001. Available online: https://www.ncbi.nlm.nih.gov/books/NBK223802/ (accessed on 13 October 2020).
- Gonzalez, L.A.; Gatch, M.B.; Forster, M.J.; Dillon, G.H. Abuse Potential of Soma: The GABA(A) Receptor as a Target. Mol. Cell. Pharmacol. 2009, 1, 180–186. [Google Scholar]
- Cenani, A.; Brosnan, R.J.; Knych, H.K. In vitro and in vivo GABAA Receptor Interaction of the Propanidid Metabolite 4-(2-[Diethylamino]-2-Oxoethoxy)-3-Methoxy-Benzeneacetic Acid. Pharmacology 2019, 103, 10–16. [Google Scholar] [CrossRef]
- Wikberg, T.; Vuorela, A.; Ottoila, P.; Taskinen, J. Identification of major metabolites of the catechol-O-methyltransferase inhibitor entacapone in rats and humans. Drug Metab. Dispos. 1993, 21, 81–92. [Google Scholar] [PubMed]
- Shoghi-Kalkhoran, M.; Faridbod, F.; Norouzi, P.; Ganjali, M.R. Praseodymium molybdate nanoplates/reduced graphene oxide nanocomposite based electrode for simultaneous electrochemical determination of entacapone, levodopa and carbidopa. J. Mater. Sci: Mater. Electron. 2018, 29, 20–31. [Google Scholar] [CrossRef]
- Jain, R.; Yadav, R.K.; Dwivedi, A. Square-wave adsorptive stripping voltammetric behaviour of entacapone at HMDE and its determination in the presence of surfactants. Colloids Surf. A 2010, 359, 25–30. [Google Scholar] [CrossRef]
- Baghayeri, M.; Tehrani, M.B.; Amiri, A.; Maleki, B.; Farhadi, S. A novel way for detection of antiparkinsonism drug entacapone via electrodeposition of silver nanoparticles/functionalized multi-walled carbon nanotubes as an amperometric sensor. Mater. Sci. Eng. C 2016, 66, 77–83. [Google Scholar] [CrossRef]
- Nouri-Nigjeh, E.; Permentier, H.P.; Bischoff, R.; Bruins, A.P. Lidocaine Oxidation by Electrogenerated Reactive Oxygen Species in the Light of Oxidative Drug Metabolism. Anal. Chem. 2010, 82, 7625–7633. [Google Scholar] [CrossRef] [PubMed]
- Halbert, M.K.; Baldwin, R.P. Determination of lidocaine and active metabolites in blood serum by liquid chromatography with electrochemical detection. J. Chromatogr. B. Biomed. Appl. 1984, 306, 269–277. [Google Scholar] [CrossRef]
- Jurva, U.; Wikström, H.V.; Bruins, A.P. In vitro mimicry of metabolic oxidation reactions by electrochemistry/mass spectrometry. Rapid Commun. Mass Spectrom. 2000, 14, 529–533. [Google Scholar] [CrossRef]
- Gul, T.; Bischoff, R.; Permentier, H.P. Optimization of reaction parameters for the electrochemical oxidation of lidocaine with a Design of Experiments approach. Electrochim. Acta 2015, 171, 23–28. [Google Scholar] [CrossRef]
- Oliveira, R.T.S.; Salazar-Banda, G.R.; Ferreira, V.S.; Oliveira, S.C.; Avaca, L.A. Electroanalytical Determination of Lidocaine in Pharmaceutical Preparations Using Boron-Doped Diamond Electrodes. Electroanalysis 2007, 19, 1189–1194. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug/Compound | OP (mV) | Mean OP (mV) | Std. OP (mV) | Mean t1/2 (h) | Reference (s) |
|---|---|---|---|---|---|
4′-OH-DCL | 167 | - | - | 4.3 | OP [25] t1/2 [26] |
5-OH-DCL | 236 | - | - | 2.5 | OP [25] t1/2 [26] |
1,1′-ferrocenedimethanol | 261 | - | - | 48.0 | OP [27] t1/2 [27] |
Warfarin | 280 | - | 485 | 40.0 | OP [28] t1/2 [29,30] |
D13223 | 347 | - | 635 | 10.7 | OP [31] t1/2 [27] |
Norcolzapine | 390 | - | - | 22.5 | OP [27] t1/2 [32] |
Flupirtine 1 | 535 | 442.5 | 823 | 8.0 | OP [31,33] t1/2 [34,35] |
| Flupirtine 2 | 350 | 638 | |||
Clozapine 1 | 600 | 475.3 | - | 11.9 | OP [11,25,27,36,37] t1/2 [32,38,39,40] |
| Clozapine 2 | 440 | 645 | |||
| Clozapine 3 | 381 | - | |||
| Clozapine 4 | 480 | - | |||
Promazine | 530 | - | 735 | 30.0 | OP [4] t1/2 [41] |
Paracetamol 1 | 392 | 595.5 | - | 2.2 | OP [5,25,42,43] t1/2 [44,45,46] |
| Paracetamol 2 | 400 | - | |||
| Paracetamol 3 | 610 | 898 | |||
| Paracetamol 4 | 980 | 1185 | |||
Promethazine  | 670 | - | 875 | 9.5 | OP [4] t1/2 [47] |
Diclofenac 1 | 870 | 700.0 | 1158 | 1.7 | OP [48,49,50] t1/2 [26,51,52] |
| Diclofenac 2 | 630 | 827 | |||
| Diclofenac 3 | 600 | 799 | |||
Trimethoprim 1 | 1000 | 950.0 | - | 9.0 | OP [5,53] t1/2 [54] |
| Trimethoprim 2 | 900 | - | |||
Amodiaquine | 970 | - | - | 5.1 | OP [55] t1/2 [56,57] |
Acebutolol | 1002 | - | - | 9.0 | OP [58] t1/2 [59] |
Caffeine | 1500 | - | 1705 | 4.6 | OP [60] t1/2 [61,62] |
Carisoprodol | 2003 | - | 2208 | 2.0 | OP [43] t1/2 [63,64] |
| Additional drugs studied | |||||
Propanidid | 1450 | - | 1655 | 0.1 (5.9 min) | t1/2 [65] |
Entacapone | 1250 | - | 1455 | 0.4 (24 min) | t1/2 [66] |
Lidocaine | 1150 | - | 1355 | 1.6 (97.5 min) | t1/2 [67] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuchigami, H.; Bal, M.K.; Brownson, D.A.C.; Banks, C.E.; Jones, A.M. Voltammetric Behaviour of Drug Molecules as a Predictor of Metabolic Liabilities. Sci. Pharm. 2020, 88, 46. https://doi.org/10.3390/scipharm88040046
Fuchigami H, Bal MK, Brownson DAC, Banks CE, Jones AM. Voltammetric Behaviour of Drug Molecules as a Predictor of Metabolic Liabilities. Scientia Pharmaceutica. 2020; 88(4):46. https://doi.org/10.3390/scipharm88040046
Chicago/Turabian StyleFuchigami, Hikari, Mandeep K. Bal, Dale A. C. Brownson, Craig E. Banks, and Alan M. Jones. 2020. "Voltammetric Behaviour of Drug Molecules as a Predictor of Metabolic Liabilities" Scientia Pharmaceutica 88, no. 4: 46. https://doi.org/10.3390/scipharm88040046
APA StyleFuchigami, H., Bal, M. K., Brownson, D. A. C., Banks, C. E., & Jones, A. M. (2020). Voltammetric Behaviour of Drug Molecules as a Predictor of Metabolic Liabilities. Scientia Pharmaceutica, 88(4), 46. https://doi.org/10.3390/scipharm88040046