The Mechanism of Action of Cyclophosphamide and Its Consequences for the Development of a New Generation of Oxazaphosphorine Cytostatics

Abstract
1. Introduction
2. Summary on Materials and Methods
Animals
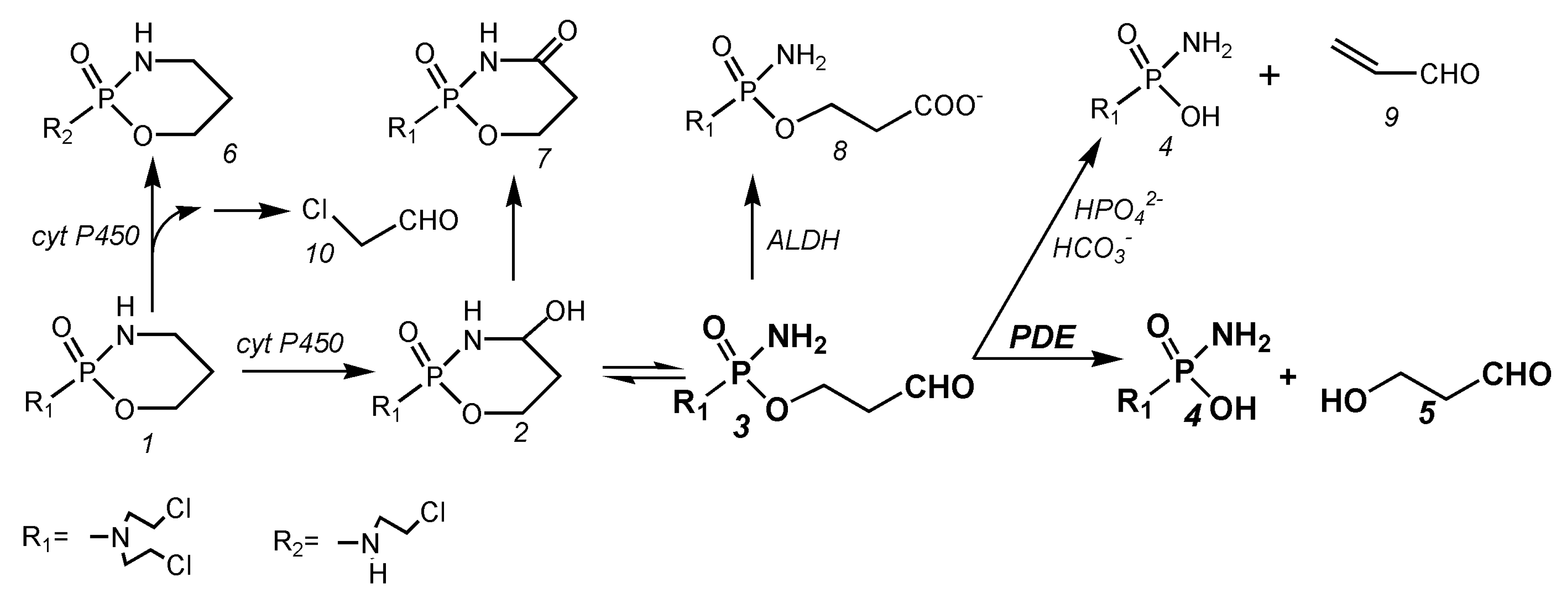
3. Metabolism of Cyclophosphamide
4. Part 1: Causes of Toxicity and Mechanism of Action of Cyclophosphamide
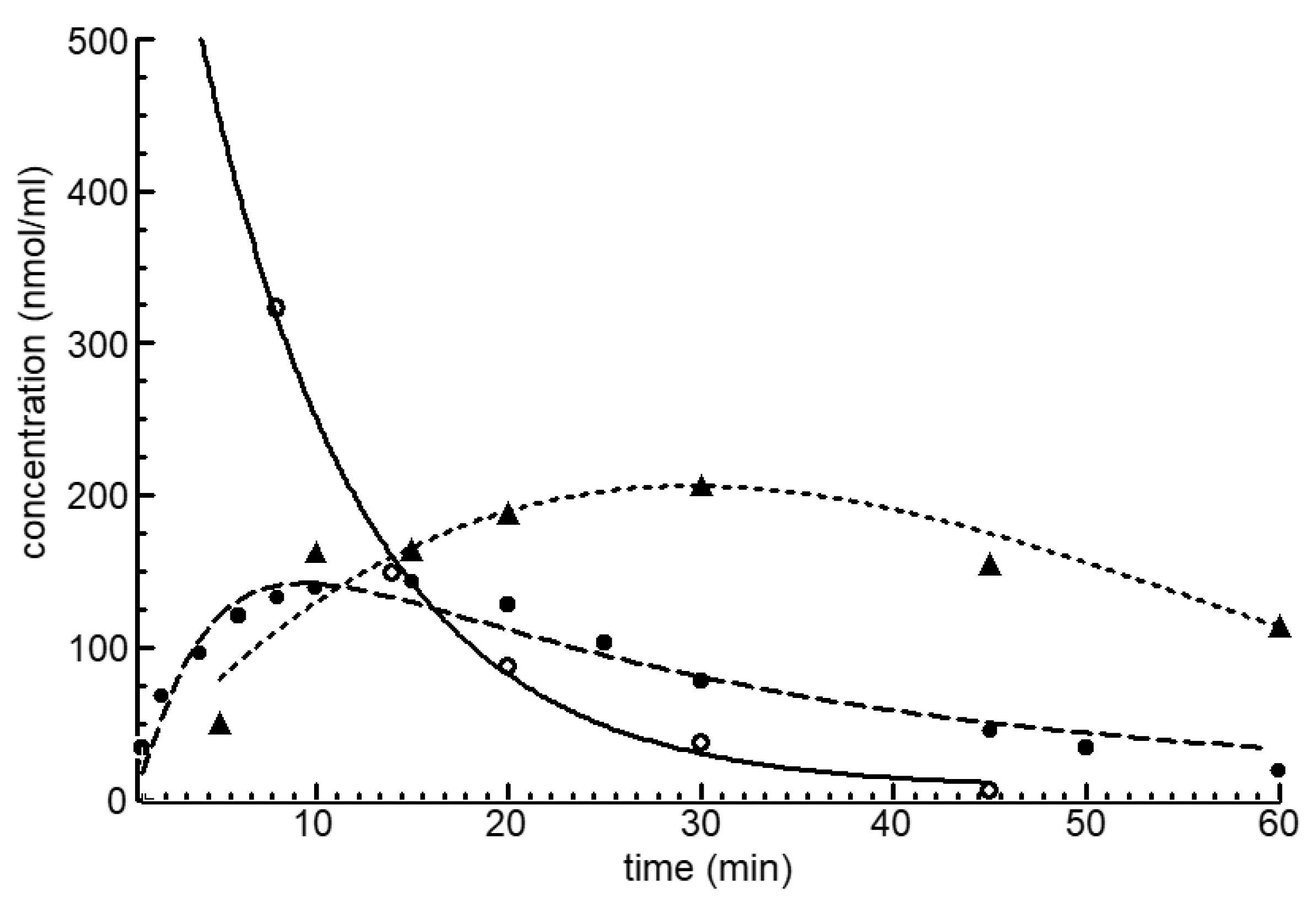
4.1. Balance of the Cyclophosphamide Metabolism
4.2. The Reason of CP Toxicity
4.3. The Mechanism of Action of Cyclophosphamide
4.4. The Special Feature of Cyclophosphamide Compared to Other Alkylating Agents
4.5. Scientific Evidence for the Enhancement of PAM-Induced Apoptosis by HPA
4.6. Reaction Scheme for the Mechanism of Action of Oxazaphosphorine Cytostatics Involving HPA
4.7. Conclusions from the Studies on the Toxicity and the Mechanism of Action of Cyclophosphamide for the Development of a New Generation of Oxazaphosphorine Cytostatics
5. Part 2: Bypassing Toxicity and Increasing Cytotoxic Apoptosis of Oxazaphosphorine Cytostatics
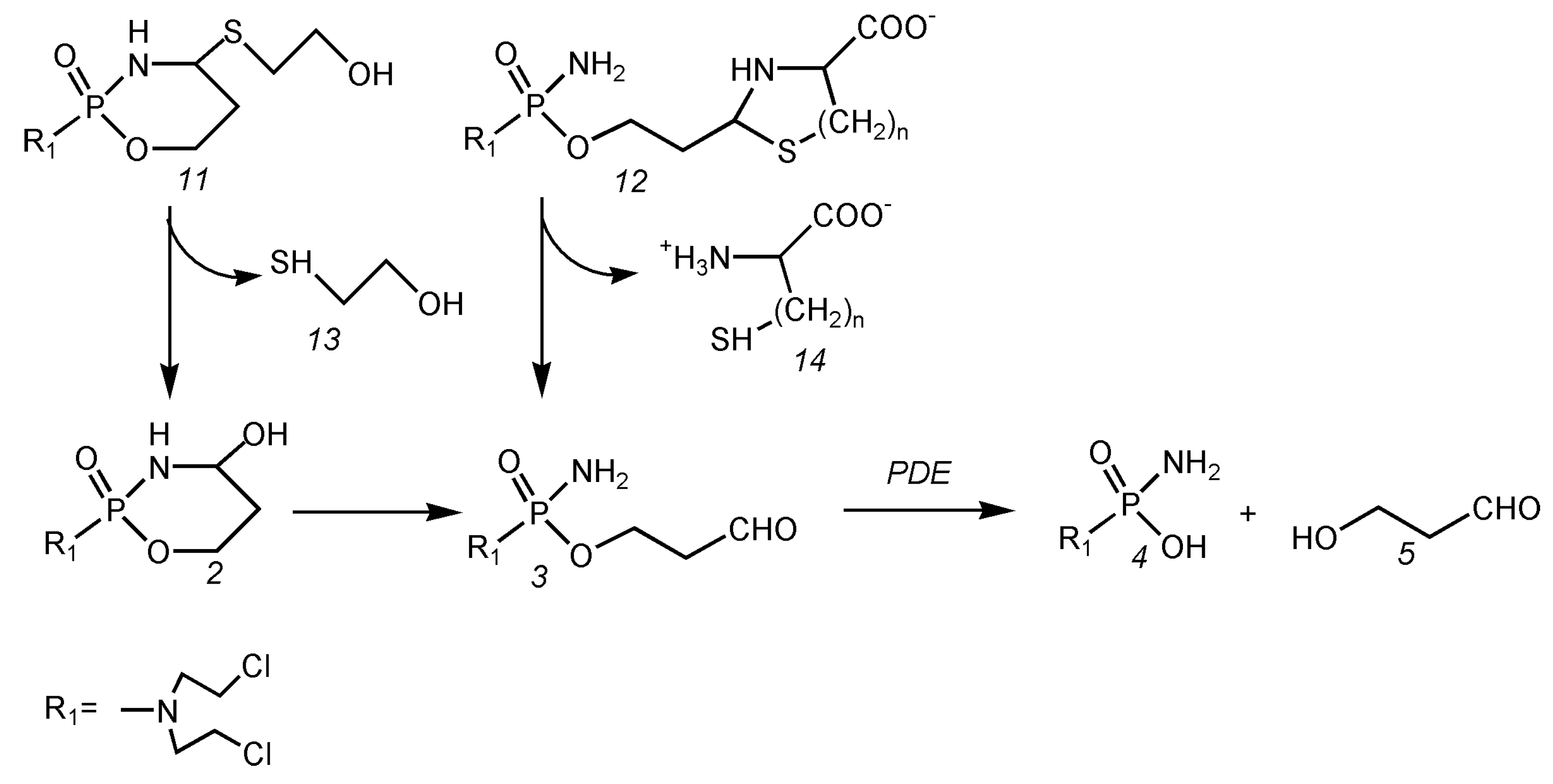
5.1. Decrease in Toxicity by Bypassing the Formation of 4-Hydroxycyclophosphamide in the Formation of Aldophosphamide
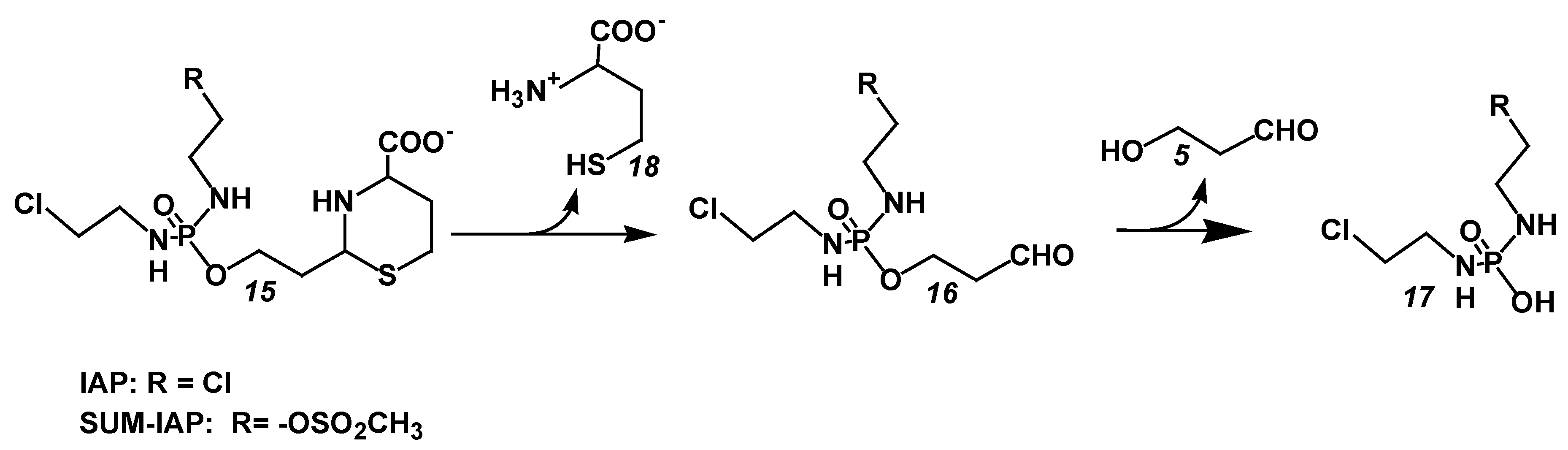
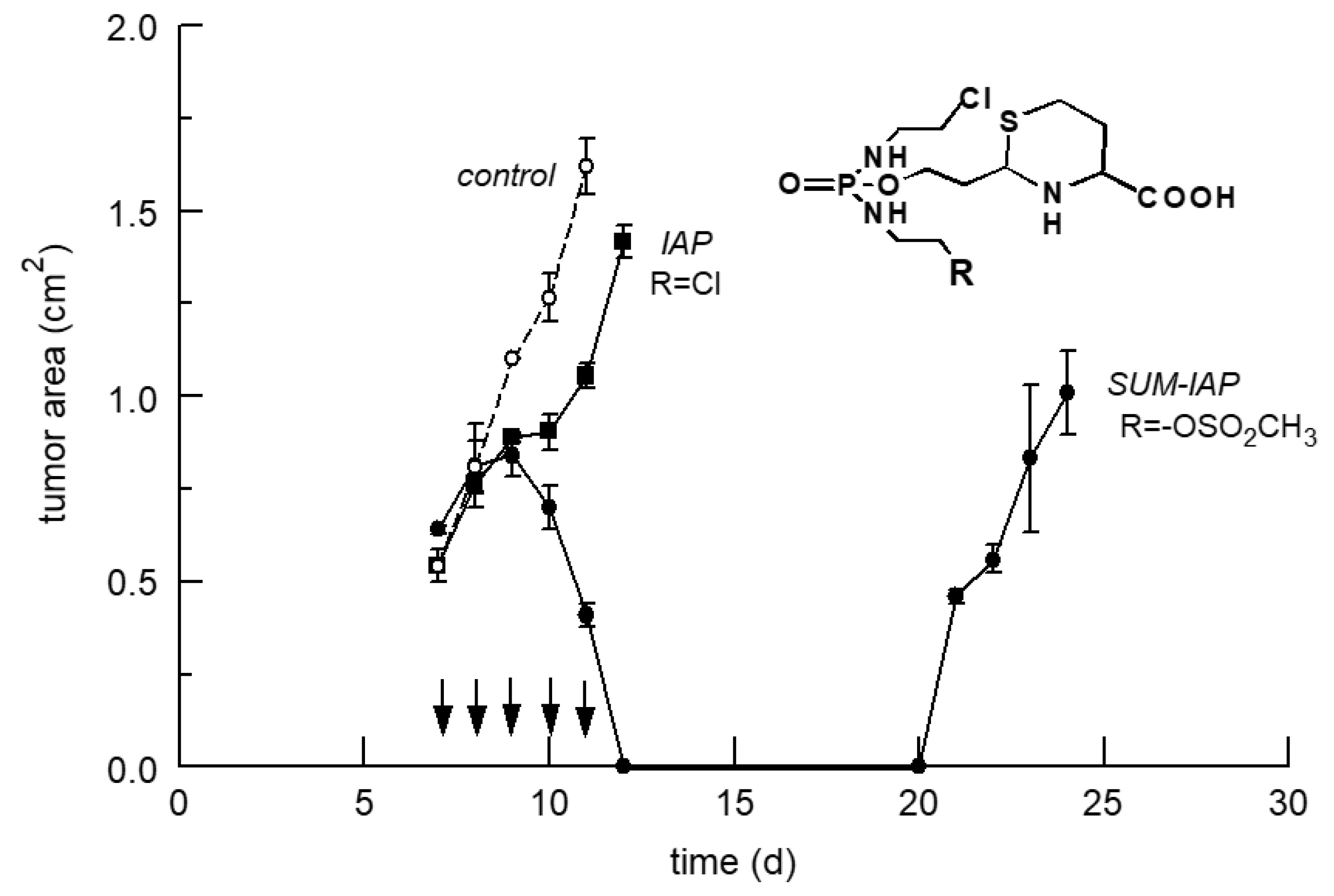
5.2. Increase in the Antitumor Activity of I-Aldoperhydrthiazines by Modulating the Alkylating Function
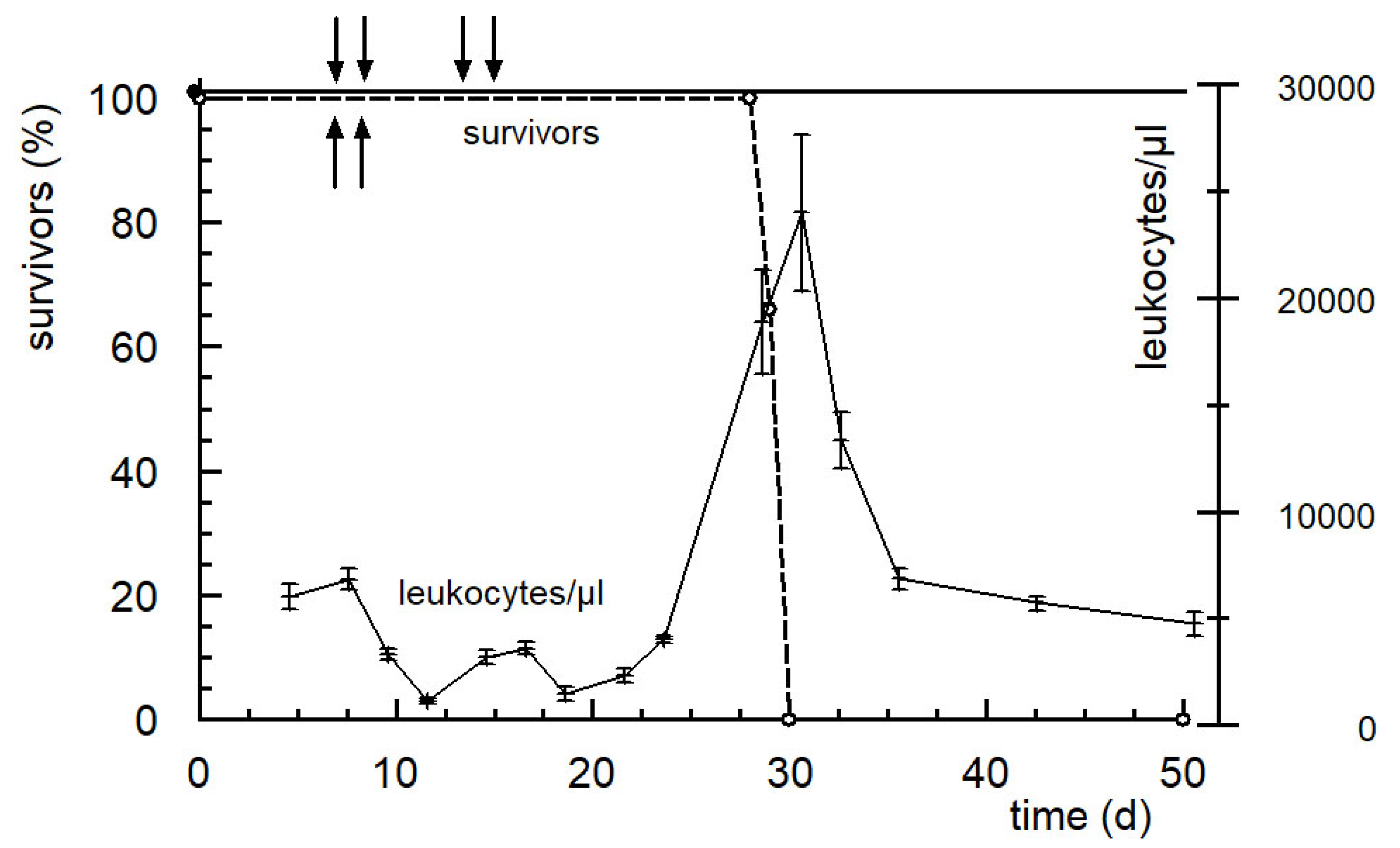
6. The New Generation of Oxazaphosphorine Cytostatics Are Candidates for Immunological Tumor Therapy
Funding
Conflicts of Interest
References
- Meyer, J.; Weinmann, J.P. Phosphamidase Content of Normal and Pathologic Tissues of the Oral Cavity. J. Histochem. Cytochem. 1953, 1, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Tomitka, K.; Takeuchi, T. On the phosphamidase reaction of tumor tissues. Gann 1955, 46, 333–334. [Google Scholar]
- Mazur, L.; Opydo-Chanek, M.; Stojak, M. Glufosfamide as a new oxazaphosphorine anticancer agent. Anti-Cancer Drugs 2011, 22, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Voelcker, G. Causes and possibilities to circumvent cyclophosphamide toxicity. Anti-Cancer Drugs 2020, 31, 617–622. [Google Scholar] [CrossRef]
- Dausman, D. Synthese von N,N-Bis-(2chlorethyl)-Phosphorsäureamid-Derivaten als Substrat für 3′-5′Exonucleasen und Untersuchungen zur Umsetzung von “aktiviertem Cyclophosphamid” mit 1,2 und 1,3 Dinukleophilen. Ph.D. Thesis, Universität Frankfurt am Main, Frankfurt, Germany, 1988. [Google Scholar]
- Zimmermann, J.; Bauer, H.H.; Hohorst, H.J.; Voelcker, G. Synthesis of I-aldofosfamide-perhydrothiazines. Arzneimittelforschung 2000, 50, 843–847. [Google Scholar] [CrossRef]
- Peter, G.; Wagner, T.; Hohorst, H.J. Studies on 4-hydroperoxycyclophosphamide (NSC-181815): A simple preparation method and its application for the synthesis of a new class of “activated” sulfur-containing cyclophosphamide (NSC-26271) derivatives. Cancer Treat. Rep. 1976, 60, 429–435. [Google Scholar]
- Voelcker, G.; Haeglsperger, R. Pharmacokinetics of cyclophosphamide and cyclophosphamide metabolites in the mouse and their influence on the therapeutic effect of “activated” cyclophosphamide (4-hydroxycyclophosphamide) (author’s transl). Arzneimittelforschung 1982, 32, 639–647. [Google Scholar]
- Voelcker, G. Enzyme Catalyzed Decomposition of 4-Hydroxycyclophosphamide. Open Conf. Proceeding J. 2017, 8, 44–51. [Google Scholar] [CrossRef]
- Fortmeyer, H.P. Schriftenreihe Versuchstierkunde Nr.8; Paul Patrey Verlag: Berlin, Germany; Hamburg, Germany, 1981. [Google Scholar]
- Voelcker, G.; Pfeiffer, B.; Schnee AHohorst, H.J. Increased Antitumour Activity of mesyl-I-aldophosphamide-perhydrothiazine, in Vivo but Not in Vitro, Compared to I-aldophosphamide-perhydrothiazine. J. Cancer Res. Clin. Oncol. 2000, 126, 74–78. [Google Scholar]
- Low, J.E.; Borch, R.F.; Sladek, N.E. Conversion of 4-hydroperoxycyclophosphamide and 4-hydroxycyclophosphamide to phosphoramide mustard and acrolein mediated by bifunctional catalysis. Cancer Res. 1982, 42, 830–837. [Google Scholar]
- Hohorst, H.J.; Bielicki, L.; Voelcker, G. The Enzymatic Basis of Cyclophosphamide Specificity. Adv. Enzym. Regul. 1986, 25, 99–122. [Google Scholar] [CrossRef]
- Brock, N. Comparative pharmacologic study in vitro and in vivo with cyclophosphamide (NSC-26271), cyclophosphamide metabolites, and plain nitrogen mustard compounds. Cancer Treat. Rep. 1976, 60, 301–308. [Google Scholar] [PubMed]
- Brock, N.; Hohorst, H.J. The problem of specificity and selectivity of alkylating cytostatics: Studies on N-2-chlorethylamido-oxazaphosphorines. Z. Krebsforsch 1977, 88, 185–215. [Google Scholar] [CrossRef]
- Schwartz, P.S.; Waxman, D.J. Cyclophosphamide induces caspase 9-dependent apoptosis in 9L tumor cells. Mol. Pharmacol. 2001, 60, 1268–1279. [Google Scholar] [CrossRef] [PubMed]
- Cleusix, V.; Lacroix, C.; Vollenweider, S.; Duboux, M.; Le Blay, G. Inhibitory activity spectrum of reuterin produced by Lactobacillus reuteri against intestinal bacteria. BMC Microbiol. 2007, 12, 101. [Google Scholar] [CrossRef] [PubMed]
- Iyer, C.; Kosters, A.; Sethi, G.; Kunnumakkara, A.B.; Aggarwal, B.B.; Versalovic, J. Probiotic Lactobacillus reuteri promotes TNF-induced apoptosis in human myeloid leukemia-derived cells by modulation of NF-kappaB and MAPK signalling. Cell. Microbiol. 2008, 10, 1442–1452. [Google Scholar] [CrossRef]
- Wu, M.; Lee, H.; Bellas, R.E.; Schauer, S.L.; Arsura, M.; Katz, D.; FitzGerald, M.J.; Rothstein, T.L.; Sherr, D.H.; Sonenshein, G.E. Inhibition of NF-kappaB/Rel induces apoptosis of murine B cells. EMBO J. 1996, 2, 4682–4690. [Google Scholar] [CrossRef]
- Povirk, L.F.; Shuker, D.E. DNA damage and mutagenesis induced by nitrogen mustards. Mutat. Res. 1994, 318, 205–226. [Google Scholar] [CrossRef]
- Alexander, P.; Mikulski, Z. Differences in the Response of Leukaemia Cells in Tissue Culture to Nitrogen Mustard and to Dimethyl Myleran. Biochem. Pharmacol. 1961, 5, 275–282. [Google Scholar] [CrossRef]
- Sistigu, A.; Viaud, S.; Chaput, N.; Bracci, L.; Proietti, E.; Zitvogel, L. Immunomodulatory effects of cyclophosphamide and implementations for vaccine design. Semin. Immunopathol. 2011, 33, 369–383. [Google Scholar] [CrossRef]
- Guerriero, J.L.; Ditsworth, D.; Catanzaro, J.M.; Sabino, G.; Furie, M.B.; Kew, R.R.; Crawford, H.C.; Zong, W.X. DNA alkylating therapy induces tumor regression through an HMGB1-mediated activation of innate immunity. J. Immunol. 2011, 186, 3517–3526. [Google Scholar] [CrossRef] [PubMed]
- van der Most, R.G.; Currie, A.J.; Cleaver, A.L.; Salmons, J.; Nowak, A.K.; Mahendran, S.; Larma, I.; Prosser, A.; Robinson, B.W.; Smyth, M.J.; et al. Cyclophosphamide chemotherapy sensitizes tumor cells to TRAIL-dependent CD8 T cell-mediated immune attack resulting in suppression of tumor growth. PLoS ONE 2009, 4, e6982. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.C.; Blazar, B.R.; Mellor, A.L.; Munn, D.H.; Zhou, G. Chemotherapy rescues tumor-driven aberrant CD4+ T-cell differentiation and restores an activated polyfunctional helper phenotype. Blood 2010, 115, 2397–2406. [Google Scholar] [CrossRef] [PubMed]
- Sharabi, A.; Laronne-Bar-On, A.; Meshorer, A.; Haran-Ghera, N. Chemoimmunotherapy reduces the progression of multiple myeloma in a mouse model. Cancer Prev. Res. 2010, 3, 1265–1276. [Google Scholar] [CrossRef]
- Heylmann, D.; Bauer, M.; Becker, H.; van Gool, S.; Bacher, N.; Steinbrink, K.; Kaina, B. Human CD4+CD25+ regulatory T cells are sensitive to low dose cyclophosphamide: Implications for the immune response. PLoS ONE 2013, 8, e83384. [Google Scholar] [CrossRef]
- Voelcker, G. Immunostimulating and cancer-reductive experimental therapy with the oxazaphosphorine cytostatic SUM-IAP. Anti-Cancer Drugs 2018, 29, 411–415. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bioavailability | |||
|---|---|---|---|
| Compound | nmol/g | % | |
| cyclophosphamide | 358 | 100 | |
| OHCP + ALDO | 328 | 92 | 100 |
| KCP + CARB | 266 | 74 | 80 |
| protein binding therap., toxic. reactions | 62 | 17 | 20 |
| LD50 (µmol/kg) | |||||
|---|---|---|---|---|---|
| OHCP (µmol/kg) | 437 | 480 | 531 | 708 | ~530 |
| mortality | 0/7 | 1/8 | 5/10 | 10/10 | |
| PAM (µmol/kg) | 548 | 685 | 805 | 973 | ~810 |
| mortality | 0/6 | 0/6 | 11/21 | 6/6 |
| Dosage (mmol/kg) | LD50 (mmol/kg) | ||||
|---|---|---|---|---|---|
| 4-(S-ethanol)-CP (11 Figure 4) | 0.53 | 0.64 | 0.78 | 0.94 | ~0.7 |
| mortality | 0/5 | 2/5 | 3/5 | 5/5 | |
| aldophosphamid thiazolidine (12, n = 1 Figure 4) | 4.9 | 5.9 | 8.9 | ~6 | |
| mortality | 0/5 | 3/7 | 5/5 | ||
| 4aldophosphamid perhydrothiazine (12, n = 2 Figure 4) | 4.5 | 5.5 | ~5 | ||
| mortality | 0/5 | 5/5 | |||
| Dosage (mg/kg) | ILS (%) 1 | Long Time Survivors 2 | |
|---|---|---|---|
| Ifosfamide | 175 d1-4 | 233 | 0/5 |
| 350 d1-2 | 289 | 0/5 | |
| IAP (R = Cl, 15 Figure 5) | 750 d1-4 | 244 | 2/6 |
| 1500 d1-2 | 400 | 4/5 |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voelcker, G. The Mechanism of Action of Cyclophosphamide and Its Consequences for the Development of a New Generation of Oxazaphosphorine Cytostatics. Sci. Pharm. 2020, 88, 42. https://doi.org/10.3390/scipharm88040042
Voelcker G. The Mechanism of Action of Cyclophosphamide and Its Consequences for the Development of a New Generation of Oxazaphosphorine Cytostatics. Scientia Pharmaceutica. 2020; 88(4):42. https://doi.org/10.3390/scipharm88040042
Chicago/Turabian StyleVoelcker, Georg. 2020. "The Mechanism of Action of Cyclophosphamide and Its Consequences for the Development of a New Generation of Oxazaphosphorine Cytostatics" Scientia Pharmaceutica 88, no. 4: 42. https://doi.org/10.3390/scipharm88040042
APA StyleVoelcker, G. (2020). The Mechanism of Action of Cyclophosphamide and Its Consequences for the Development of a New Generation of Oxazaphosphorine Cytostatics. Scientia Pharmaceutica, 88(4), 42. https://doi.org/10.3390/scipharm88040042