Multifaceted Protective Role of Glucosamine against Osteoarthritis: Review of Its Molecular Mechanisms




Abstract
1. Introduction
2. Anti-Inflammatory Activities of Glucosamine
3. Antioxidant Properties of Glucosamine
4. Activation of Autophagy by Glucosamine
5. Induction of Tissue Regeneration, Stem Cell Proliferation, and Differentiation by Glucosamine
6. Contradictory Effects of Glucosamine on Joint Health
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Heidari, B. Knee osteoarthritis prevalence, risk factors, pathogenesis and features: Part, I. Casp. J. Intern. Med. 2011, 2, 205–212. [Google Scholar]
- Vina, E.R.; Kwoh, C.K. Epidemiology of osteoarthritis: Literature update. Curr. Opin. Rheumatol. 2018, 30, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Andrianakos, A.A.; Kontelis, L.K.; Karamitsos, D.G.; Aslanidis, S.I.; Georgountzos, A.I.; Kaziolas, G.O.; Pantelidou, K.V.; Vafiadou, E.V.; Dantis, P.C. Prevalence of symptomatic knee, hand, and hip osteoarthritis in Greece. The ESORDIG study. J. Rheumatol. 2006, 33, 2507–2513. [Google Scholar] [PubMed]
- Lee, K.M.; Chung, C.Y.; Sung, K.H.; Lee, S.Y.; Won, S.H.; Kim, T.G.; Choi, Y.; Kwon, S.S.; Kim, Y.H.; Park, M.S. Risk factors for osteoarthritis and contributing factors to current arthritic pain in South Korean older adults. Yonsei Med. J. 2015, 56, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Blagojevic, M.; Jinks, C.; Jeffery, A.; Jordan, K.P. Risk factors for onset of osteoarthritis of the knee in older adults: A systematic review and meta-analysis. Osteoarthr. Cartil. 2010, 18, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Struyf, P.A.; van Heugten, C.M.; Hitters, M.W.; Smeets, R.J. The prevalence of osteoarthritis of the intact hip and knee among traumatic leg amputees. Arch. Phys. Med. Rehabil. 2009, 90, 440–446. [Google Scholar] [CrossRef]
- Troeberg, L.; Nagase, H. Proteases involved in cartilage matrix degradation in osteoarthritis. Biochim. Et Biophys. Acta 2012, 1824, 133–145. [Google Scholar] [CrossRef]
- Mobasheri, A.; Rayman, M.P.; Gualillo, O.; Sellam, J.; van der Kraan, P.; Fearon, U. The role of metabolism in the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2017, 13, 302–311. [Google Scholar] [CrossRef]
- Georgiev, T.; Ivanova, M.; Kopchev, A.; Velikova, T.; Miloshov, A.; Kurteva, E.; Yuzeir, K.; Penkov, M.; Kabakchieva, P.; Rashkov, R.; et al. Cartilage oligomeric protein, matrix metalloproteinase-3, and Coll2-1 as serum biomarkers in knee osteoarthritis: A cross-sectional study. Rheumatol. Int. 2018, 38, 821–830. [Google Scholar] [CrossRef]
- Ma, T.; Zhang, Z.; Song, X.; Bai, H.; Li, Y.; Li, X.; Zhao, J.; Ma, Y.; Gao, L. Combined detection of COMP and CS846 biomarkers in experimental rat osteoarthritis: A potential approach for assessment and diagnosis of osteoarthritis. J. Orthop. Surg. Res. 2018, 13, 230. [Google Scholar] [CrossRef]
- Verma, P.; Dalal, K. Serum cartilage oligomeric matrix protein (COMP) in knee osteoarthritis: A novel diagnostic and prognostic biomarker. J. Orthop. Res. 2013, 31, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, F.A.; Pucinelli, M.L.; da Silva, N.P.; Feldman, D. Serum cartilage oligomeric matrix protein (COMP) levels in knee osteoarthritis in a Brazilian population: Clinical and radiological correlation. Scand. J. Rheumatol. 2007, 36, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Catterall, J.; Dewitt Parr, S.; Fagerlund, K.; Caterson, B. CTX-II is a marker of cartilage degradation but not of bone turnover. Osteoarthr. Cartil. 2013, 21, S77. [Google Scholar] [CrossRef]
- Coyle, C.H.; Henry, S.E.; Haleem, A.M.; O’Malley, M.J.; Chu, C.R. Serum CTXii Correlates With Articular Cartilage Degeneration After Anterior Cruciate Ligament Transection or Arthrotomy Followed by Standardized Exercise. Sports Health 2012, 4, 510–517. [Google Scholar] [CrossRef]
- Blumenfeld, O.; Williams, F.M.; Hart, D.J.; Spector, T.D.; Arden, N.; Livshits, G. Association between cartilage and bone biomarkers and incidence of radiographic knee osteoarthritis (RKOA) in UK females: A prospective study. Osteoarthr. Cartil. 2013, 21, 923–929. [Google Scholar] [CrossRef]
- Hao, H.Q.; Zhang, J.F.; He, Q.Q.; Wang, Z. Cartilage oligomeric matrix protein, C-terminal cross-linking telopeptide of type II collagen, and matrix metalloproteinase-3 as biomarkers for knee and hip osteoarthritis (OA) diagnosis: A systematic review and meta-analysis. Osteoarthr. Cartil. 2019, 27, 726–736. [Google Scholar] [CrossRef]
- Chin, K.Y.; Pang, K.L. Therapeutic Effects of Olive and Its Derivatives on Osteoarthritis: From Bench to Bedside. Nutrients 2017, 9, 1060. [Google Scholar] [CrossRef]
- Sun, J.M.; Sun, L.Z.; Liu, J.; Su, B.H.; Shi, L. Serum interleukin-15 levels are associated with severity of pain in patients with knee osteoarthritis. Dis. Markers 2013, 35, 203–206. [Google Scholar] [CrossRef]
- Snelling, S.J.; Bas, S.; Puskas, G.J.; Dakin, S.G.; Suva, D.; Finckh, A.; Gabay, C.; Hoffmeyer, P.; Carr, A.J.; Lubbeke, A. Presence of IL-17 in synovial fluid identifies a potential inflammatory osteoarthritic phenotype. PLoS ONE 2017, 12, e0175109. [Google Scholar] [CrossRef]
- Lee, Y.M.; Son, E.; Kim, S.H.; Kim, O.S.; Kim, D.S. Anti-inflammatory and anti-osteoarthritis effect of Mollugo pentaphylla extract. Pharm. Biol. 2019, 57, 74–81. [Google Scholar] [CrossRef]
- Rahmati, M.; Mobasheri, A.; Mozafari, M. Inflammatory mediators in osteoarthritis: A critical review of the state-of-the-art, current prospects, and future challenges. Bone 2016, 85, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Ara, R.; Alam, M. Pharmacotherapy for Osteoarthritis: A Review. J. Med. 2011, 12, 142–148. [Google Scholar] [CrossRef]
- Alcaraz, M.J.; Megias, J.; Garcia-Arnandis, I.; Clerigues, V.; Guillen, M.I. New molecular targets for the treatment of osteoarthritis. Biochem. Pharmacol. 2010, 80, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Trijau, S.; Avouac, J.; Escalas, C.; Gossec, L.; Dougados, M. Influence of flare design on symptomatic efficacy of non-steroidal anti-inflammatory drugs in osteoarthritis: A meta-analysis of randomized placebo-controlled trials. Osteoarthr. Cartil. 2010, 18, 1012–1018. [Google Scholar] [CrossRef]
- Monfort, J.; Rotes-Sala, D.; Segales, N.; Montanes, F.J.; Orellana, C.; Llorente-Onaindia, J.; Mojal, S.; Padro, I.; Benito, P. Comparative efficacy of intra-articular hyaluronic acid and corticoid injections in osteoarthritis of the first carpometacarpal joint: Results of a 6-month single-masked randomized study. Jt. Bone Spine Rev. Du Rhum. 2015, 82, 116–121. [Google Scholar] [CrossRef]
- Conrozier, T. Is the Addition of a Polyol to Hyaluronic Acid a Significant Advance in the Treatment of Osteoarthritis? Curr. Rheumatol. Rev. 2018, 14, 226–230. [Google Scholar] [CrossRef]
- Khwaldia, K. Chondroitin and Glucosamine. In Nonvitamin and Nonmineral Nutritional Supplement; Academic Press: London, UK, 2019; pp. 27–35. [Google Scholar]
- Reginster, J.Y.; Bruyere, O.; Neuprez, A. Current role of glucosamine in the treatment of osteoarthritis. Rheumatology 2007, 46, 731–735. [Google Scholar] [CrossRef]
- Durmus, D.; Alayli, G.; Bayrak, I.K.; Canturk, F. Assessment of the effect of glucosamine sulfate and exercise on knee cartilage using magnetic resonance imaging in patients with knee osteoarthritis: A randomized controlled clinical trial. J. Back Musculoskelet. Rehabil. 2012, 25, 275–284. [Google Scholar] [CrossRef]
- Tiraloche, G.; Girard, C.; Chouinard, L.; Sampalis, J.; Moquin, L.; Ionescu, M.; Reiner, A.; Poole, A.R.; Laverty, S. Effect of oral glucosamine on cartilage degradation in a rabbit model of osteoarthritis. Arthritis Rheum. 2005, 52, 1118–1128. [Google Scholar] [CrossRef]
- Jerosch, J. Effects of Glucosamine and Chondroitin Sulfate on Cartilage Metabolism in OA: Outlook on Other Nutrient Partners Especially Omega-3 Fatty Acids. Int. J. Rheumatol. 2011, 2011, 1–17. [Google Scholar] [CrossRef]
- Wen, Z.H.; Tang, C.C.; Chang, Y.C.; Huang, S.Y.; Hsieh, S.P.; Lee, C.H.; Huang, G.S.; Ng, H.F.; Neoh, C.A.; Hsieh, C.S.; et al. Glucosamine sulfate reduces experimental osteoarthritis and nociception in rats: Association with changes of mitogen-activated protein kinase in chondrocytes. Osteoarthr. Cartil. 2010, 18, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Pohlig, F.; Ulrich, J.; Lenze, U.; Muhlhofer, H.M.; Harrasser, N.; Suren, C.; Schauwecker, J.; Mayer-Kuckuk, P.; von Eisenhart-Rothe, R. Glucosamine sulfate suppresses the expression of matrix metalloproteinase-3 in osteosarcoma cells in vitro. BMC Complement. Altern. Med. 2016, 16, 313. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, M.; Sakamoto, K.; Nagaoka, I. Effect of glucosamine on expression of type II collagen, matrix metalloproteinase and sirtuin genes in a human chondrocyte cell line. Int. J. Mol. Med. 2017, 39, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Tiku, M.L.; Narla, H.; Jain, M.; Yalamanchili, P. Glucosamine prevents in vitro collagen degradation in chondrocytes by inhibiting advanced lipoxidation reactions and protein oxidation. Arthritis Res. Ther. 2007, 9, R76. [Google Scholar] [CrossRef] [PubMed]
- Terencio, M.C.; Ferrandiz, M.L.; Carceller, M.C.; Ruhi, R.; Dalmau, P.; Verges, J.; Montell, E.; Torrent, A.; Alcaraz, M.J. Chondroprotective effects of the combination chondroitin sulfate-glucosamine in a model of osteoarthritis induced by anterior cruciate ligament transection in ovariectomised rats. Biomed. Pharmacother. Biomed. Aharmacotherapie 2016, 79, 120–128. [Google Scholar] [CrossRef]
- Selvan, T.; Rajiah, K.; Nainar, M.S.; Mathew, E.M. A clinical study on glucosamine sulfate versus combination of glucosamine sulfate and NSAIDs in mild to moderate knee osteoarthritis. Sci. World J. 2012, 2012. [Google Scholar] [CrossRef]
- Onigbinde, A.T.; Owolabi, A.R.; Lasisi, K.; Isaac, S.O.; Ibikunle, A.F. Symptoms-modifying effects of electromotive administration of glucosamine sulphate among patients with knee osteoarthritis. Hong Kong Physiother. J. 2018, 38, 63–75. [Google Scholar] [CrossRef]
- Frisbie, D.D.; McIlwraith, C.W.; Kawcak, C.E.; Werpy, N.M. Evaluation of intra-articular hyaluronan, sodium chondroitin sulfate and N-acetyl-D-glucosamine combination versus saline (0.9% NaCl) for osteoarthritis using an equine model. Vet. J. 2013, 197, 824–829. [Google Scholar] [CrossRef]
- Runhaar, J.; Deroisy, R.; van Middelkoop, M.; Barretta, F.; Barbetta, B.; Oei, E.H.; Vroegindeweij, D.; Giacovelli, G.; Bruyere, O.; Rovati, L.C.; et al. The role of diet and exercise and of glucosamine sulfate in the prevention of knee osteoarthritis: Further results from the PRevention of knee Osteoarthritis in Overweight Females (PROOF) study. Semin. Arthritis Rheum. 2016, 45, S42–S48. [Google Scholar] [CrossRef][Green Version]
- Ucuncu, Y.; Celik, N.; Ozturk, C.; Turkoglu, M.; Cetin, N.; Kockara, N.; Sener, E.; Dundar, C.; Arslan, A.; Dogan, H.; et al. Chondroprotective effects of a new glucosamine combination in rats: Gene expression, biochemical and histopathological evaluation. Life Sci. 2015, 130, 31–37. [Google Scholar] [CrossRef]
- Najima, M.; Shirakawa, T.; Ishii, I.; Okamoto, K. A Study for Evaluating the Effect of the Supplement Containing Glucosamine on Joint Performance and Daily Physical Performance-A Randomized, Double—Blind, Placebo—Controlled, Study Mainly Evaluated by Subjects-oriented Questionnaire. Jpn. Pharmcol. Ther. 2017, 45, 939–955. [Google Scholar]
- Henrotin, Y.; Mobasheri, A.; Marty, M. Is there any scientific evidence for the use of glucosamine in the management of human osteoarthritis? Arthritis Res. Ther. 2012, 14, 201. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.W.; Nicolosi, R.J.; Borzelleca, J.F. Glucosamine effects in humans: A review of effects on glucose metabolism, side effects, safety considerations and efficacy. Food Chem. Toxicol. 2005, 43, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Raynauld, J.P.; Pelletier, J.P.; Abram, F.; Dodin, P.; Delorme, P.; Martel-Pelletier, J. Long-Term Effects of Glucosamine and Chondroitin Sulfate on the Progression of Structural Changes in Knee Osteoarthritis: Six-Year Followup Data From the Osteoarthritis Initiative. Arthritis Care Res. 2016, 68, 1560–1566. [Google Scholar] [CrossRef]
- Nakasone, Y.; Watabe, K.; Watanabe, K.; Tomonaga, A.; Nagaoka, I.; Yamamoto, T.; Yamaguchi, H. Effect of a glucosamine-based combination supplement containing chondroitin sulfate and antioxidant micronutrients in subjects with symptomatic knee osteoarthritis: A pilot study. Exp. Ther. Med. 2011, 2, 893–899. [Google Scholar] [CrossRef]
- Heijink, A.; Gomoll, A.H.; Madry, H.; Drobnic, M.; Filardo, G.; Espregueira-Mendes, J.; Van Dijk, C.N. Biomechanical considerations in the pathogenesis of osteoarthritis of the knee. Knee Surg. Sports Traumatol. Arthrosc. 2012, 20, 423–435. [Google Scholar] [CrossRef]
- Man, G.S.; Mologhianu, G. Osteoarthritis pathogenesis–a complex process that involves the entire joint. J. Med. Life 2014, 7, 37–41. [Google Scholar]
- Wang, M.; Peng, Z.; Vasilev, K.; Ketheesan, N. Investigation of Wear Particles Generated in Human Knee Joints Using Atomic Force Microscopy. Tribol. Lett. 2013, 51, 161–170. [Google Scholar] [CrossRef]
- Cho, H.; Walker, A.; Williams, J.; Hasty, K.A. Study of osteoarthritis treatment with anti-inflammatory drugs: Cyclooxygenase-2 inhibitor and steroids. Biomed Res. Int. 2015, 2015. [Google Scholar] [CrossRef]
- Sokolove, J.; Lepus, C.M. Role of inflammation in the pathogenesis of osteoarthritis: Latest findings and interpretations. Ther. Adv. Musculoskel. Dis. 2013, 5, 77–94. [Google Scholar] [CrossRef]
- Aghazadeh-Habashi, A.; Kohan, M.H.; Asghar, W.; Jamali, F. Glucosamine dose/concentration-effect correlation in the rat with adjuvant arthritis. J. Pharm. Sci. 2014, 103, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, L.; Liu, Y.; Zhang, Y.; Liang, Y.; Mei, Y. Anti-inflammatory effects in a mouse osteoarthritis model of a mixture of glucosamine and chitooligosaccharides produced by bi-enzyme single-step hydrolysis. Sci. Rep. 2018, 8, 5624. [Google Scholar] [CrossRef] [PubMed]
- Waly, N.E.; Refaiy, A.; Aborehab, N.M. IL-10 and TGF-beta: Roles in chondroprotective effects of Glucosamine in experimental Osteoarthritis? Pathophysiology 2017, 24, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Sanches, M.; Assis, L.; Criniti, C.; Fernandes, D.; Tim, C.; Renno, A.C.M. Chondroitin sulfate and glucosamine sulfate associated to photobiomodulation prevents degenerative morphological changes in an experimental model of osteoarthritis in rats. Lasers Med. Sci. 2018, 33, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Cen, X.; Liu, Y.; Wang, S.; Yang, X.; Shi, Z.; Liang, X. Glucosamine oral administration as an adjunct to hyaluronic acid injection in treating temporomandibular joint osteoarthritis. Oral Dis. 2018, 24, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Li, S.; Chen, D. TGF-beta signaling and the development of osteoarthritis. Bone Res. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Zhen, G.; Wen, C.; Jia, X.; Li, Y.; Crane, J.L.; Mears, S.C.; Askin, F.B.; Frassica, F.J.; Chang, W.; Yao, J.; et al. Inhibition of TGF-beta signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat. Med. 2013, 19, 704–712. [Google Scholar] [CrossRef]
- Olivotto, E.; Otero, M.; Marcu, K.B.; Goldring, M.B. Pathophysiology of osteoarthritis: Canonical NF-κB/IKKβ-dependent and kinase-independent effects of IKKα in cartilage degradation and chondrocyte differentiation. RMD Open 2015, 1, e000061. [Google Scholar] [CrossRef]
- Imagawa, K.; de Andrés, M.C.; Hashimoto, K.; Pitt, D.; Itoi, E.; Goldring, M.B.; Roach, H.I.; Oreffo, R.O.C. The epigenetic effect of glucosamine and a nuclear factor-kappa B (NF-kB) inhibitor on primary human chondrocytes–Implications for osteoarthritis. Biochem. Biophys. Res. Commun. 2011, 405, 362–367. [Google Scholar] [CrossRef]
- Chiu, H.W.; Li, L.H.; Hsieh, C.Y.; Rao, Y.K.; Chen, F.H.; Chen, A.; Ka, S.M.; Hua, K.F. Glucosamine inhibits IL-1beta expression by preserving mitochondrial integrity and disrupting assembly of the NLRP3 inflammasome. Sci. Rep. 2019, 9, 5603. [Google Scholar] [CrossRef]
- Ziskoven, C.; Jager, M.; Zilkens, C.; Bloch, W.; Brixius, K.; Krauspe, R. Oxidative stress in secondary osteoarthritis: From cartilage destruction to clinical presentation? Orthop. Rev. 2010, 2, e23. [Google Scholar] [CrossRef] [PubMed]
- Panasyuk, A.; Frati, E.; Ribault, D.; Mitrovic, D. Effect of reactive oxygen species on the biosynthesis and structure of newly synthesized proteoglycans. Free Radic. Biol. Med. 1994, 16, 157–167. [Google Scholar] [CrossRef]
- Grover, A.K.; Samson, S.E. Benefits of antioxidant supplements for knee osteoarthritis: Rationale and reality. Nutr. J. 2016, 15, 1. [Google Scholar] [CrossRef]
- Mobasheri, A.; Shakibaei, M.; Biesalski, H.K.; Henrotin, Y. Antioxidants in the Treatment of Osteoarthritis and Bone Mineral Loss; Humana Press: New York, NY, USA, 2013; pp. 275–295. [Google Scholar]
- Chin, K.Y. The spice for joint inflammation: Anti-inflammatory role of curcumin in treating osteoarthritis. Drug Des. Dev. Ther. 2016, 10, 3029–3042. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.-Y.; Ima-Nirwana, S. The Role of Vitamin E in Preventing and Treating Osteoarthritis–A Review of the Current Evidence. Front. Pharm. 2018, 9, 946. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.-Y.; Wong, S.K.; Japar Sidik, F.Z.; Abdul Hamid, J.; Abas, N.H.; Mohd Ramli, E.S.; Afian Mokhtar, S.; Rajalingham, S.; Ima Nirwana, S. The Effects of Annatto Tocotrienol Supplementation on Cartilage and Subchondral Bone in an Animal Model of Osteoarthritis Induced by Monosodium Iodoacetate. Int. J. Environ. Res. Public Health 2019, 16, 2897. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.W.; Jiang, Y.; Shalev, A.; Kowluru, R.; Crook, E.D.; Singh, L.P. An analysis of high glucose and glucosamine-induced gene expression and oxidative stress in renal mesangial cells. Arch. Physiol. Biochem. 2006, 112, 189–218. [Google Scholar] [CrossRef]
- Chen, Y.J.; Huang, Y.S.; Chen, J.T.; Chen, Y.H.; Tai, M.C.; Chen, C.L.; Liang, C.M. Protective effects of glucosamine on oxidative-stress and ischemia/reperfusion-induced retinal injury. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1506–1516. [Google Scholar] [CrossRef]
- Mendis, E.; Kim, M.M.; Rajapakse, N.; Kim, S.K. Sulfated glucosamine inhibits oxidation of biomolecules in cells via a mechanism involving intracellular free radical scavenging. Eur. J. Pharmacol. 2008, 579, 74–85. [Google Scholar] [CrossRef]
- Jamialahmadi, K.; Soltani, F.; Nabavi Fard, M.; Behravan, J.; Mosaffa, F. Assessment of protective effects of glucosamine and N-acetyl glucosamine against DNA damage induced by hydrogen peroxide in human lymphocytes. Drug Chem. Toxicol. 2014, 37, 427–432. [Google Scholar] [CrossRef]
- Chen, J.-T.; Chen, C.-L.; Chen, Y.-H.; Liang, C.-M.; Tai, M.-C. Glucosamine attenuates hydrogen peroxide-induced premature senescence in human retinal pigment epithelial cells In vitro. J. Med. Sci. 2018, 38, 16. [Google Scholar] [CrossRef]
- Fang, C.; Peng, M.; Li, G.; Tian, J.; Yin, D. New Functions of Glucosamine as a Scavenger of the Lipid Peroxidation Product Malondialdehyde. Chem. Res. Toxicol. 2007, 20, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Dai, X.; Shilong, F.; Zhu, M.; Shen, X.; Zhang, K.; Li, S. Antimicrobial and antioxidant capacity of glucosamine-zinc(II) complex via non-enzymatic browning reaction. Food Sci. Biotechnol. 2018, 27, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.L.; Liu, Z.Q. Hybrid of Resveratrol and Glucosamine: An Approach To Enhance Antioxidant Effect against DNA Oxidation. Chem. Res. Toxicol. 2018, 31, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Pavan, R.; Jain, S.; Shraddha; Kumar, A. Properties and therapeutic application of bromelain: A review. Biotechnol. Res. Int. 2012, 2012. [Google Scholar] [CrossRef]
- Grant, L.; McBean, D.E.; Fyfe, L.; Warnock, A.M. A Review of the Biological and Potential Therapeutic Actions of Harpagophytum procumbens. Phytother. Res. 2007, 21, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Gang, D.; Xiaguang, C.; Kanghua, Y.; Aiping, W.; Guangxuan, Z. Combined effect of celecoxib and glucosamine sulfate on inflammatory factors and oxidative stress indicators in patients with knee osteoarthritis. Trop. J. Pharm. Res. 2019, 18, 397–402. [Google Scholar] [CrossRef]
- Xing, R.; Liu, S.; Guo, Z.; Yu, H.; Li, C.; Ji, X.; Feng, J.; Li, P. The antioxidant activity of glucosamine hydrochloride in vitro. Bioorg. Med. Chem. 2006, 14, 1706–1709. [Google Scholar] [CrossRef]
- Wang, X.; Liu, B.; Li, X.; Sun, R. Novel glucosamine hydrochloride-rectorite nanocomposites with antioxidant and anti-ultraviolet activity. Nanotechnology 2012, 23, 495706. [Google Scholar] [CrossRef]
- Xing, R.; Liu, S.; Wang, L.; Cai, S.; Yu, H.; Feng, J.; Li, P. The preparation and antioxidant activity of glucosamine sulfate. Chin. J. Oceanol. Limnol. 2009, 27, 283–287. [Google Scholar] [CrossRef]
- Calamia, V.; Ruiz-Romero, C.; Rocha, B.; Fernandez-Puente, P.; Mateos, J.; Montell, E.; Verges, J.; Blanco, F.J. Pharmacoproteomic study of the effects of chondroitin and glucosamine sulfate on human articular chondrocytes. Arthritis Res. Ther. 2010, 12, R138. [Google Scholar] [CrossRef] [PubMed]
- Valvason, C.; Musacchio, E.; Pozzuoli, A.; Ramonda, R.; Aldegheri, R.; Punzi, L. Influence of glucosamine sulphate on oxidative stress in human osteoarthritic chondrocytes: Effects on HO-1, p22(Phox) and iNOS expression. Rheumatology 2008, 47, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Mello, D.M.; Nielsen, B.D.; Peters, T.L.; Caron, J.P.; Orth, M.W. Comparison of inhibitory effects of glucosamine and mannosamine on bovine articular cartilage degradation in vitro. Am. J. Vet. Res. 2004, 65, 1440–1445. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Shibakawa, A.; Tanaka, M.; Kato, T.; Nishioka, K. Effects of glucosamine hydrochloride on the production of prostaglandin E2, nitric oxide and metalloproteases by chondrocytes and synoviocytes in osteoarthritis. Clin. Exp. Rheumatol. 2004, 22, 293–299. [Google Scholar]
- Alcaraz, M.J.; Fernandes, P.; Guillen, M.I. Anti-Inflammatory Actions of the Heme Oxygenase-1 Pathway. Curr. Pharm. Des. 2003, 9, 2541–2551. [Google Scholar] [CrossRef]
- Araujo, J.A.; Zhang, M.; Yin, F. Heme oxygenase-1, oxidation, inflammation, and atherosclerosis. Front. Pharmacol. 2012, 3, 119. [Google Scholar] [CrossRef]
- Paine, A.; Eiz-Vesper, B.; Blasczyk, R.; Immenschuh, S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem. Pharmacol. 2010, 80, 1895–1903. [Google Scholar] [CrossRef]
- Ooi, T.C.; Chan, K.M.; Sharif, R. Zinc L-carnosine suppresses inflammatory responses in lipopolysaccharide-induced RAW 264.7 murine macrophages cell line via activation of Nrf2/HO-1 signaling pathway. Immunopharmacol. Immunotoxicol. 2017, 39, 259–267. [Google Scholar] [CrossRef]
- Lepetsos, P.; Papavassiliou, A.G. ROS/oxidative stress signaling in osteoarthritis. Biochim. Et Biophys. Acta 2016, 1862, 576–591. [Google Scholar] [CrossRef]
- Vuolteenaho, K.; Moilanen, T.; Knowles, R.G.; Moilanen, E. The role of nitric oxide in osteoarthritis. Scand. J. Rheumatol. 2007, 36, 247–258. [Google Scholar] [CrossRef]
- Henrotin, Y.E.; Bruckner, P.; Pujol, J.-P.L. The role of reactive oxygen species in homeostasis and degradation of cartilage. Osteoarthr. Cartil. 2003, 11, 747–755. [Google Scholar] [CrossRef]
- Meitzler, J.L.; Antony, S.; Wu, Y.; Juhasz, A.; Liu, H.; Jiang, G.; Lu, J.; Roy, K.; Doroshow, J.H. NADPH oxidases: A perspective on reactive oxygen species production in tumor biology. Antioxid. Redox Signal. 2014, 20, 2873–2889. [Google Scholar] [CrossRef]
- Piperno, M.; Reboul, P.; Hellio Le Graverand, M.P.; Peschard, M.J.; Annefeld, M.; Richard, M.; Vignon, E. Glucosamine sulfate modulates dysregulated activities of human osteoarthritic chondrocytes in vitro. Osteoarthr. Cartil. 2000, 8, 207–212. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef]
- Aigner, T.; Hemmel, M.; Neureiter, D.; Gebhard, P.M.; Zeiler, G.; Kirchner, T.; McKenna, L. Apoptotic Cell Death Is Not a Widespread Phenomenon in Normal Aging and Osteoarthritic Human Articular Knee Cartilage. Arthritis Rheum. 2001, 44, 1304–1312. [Google Scholar] [CrossRef]
- Takayama, K.; Kawakami, Y.; Kobayashi, M.; Greco, N.; Cummins, J.H.; Matsushita, T.; Kuroda, R.; Kurosaka, M.; Fu, F.H.; Huard, J. Local intra-articular injection of rapamycin delays articular cartilage degeneration in a murine model of osteoarthritis. Arthritis Res. Ther. 2014, 16, 482. [Google Scholar] [CrossRef]
- Shintani, T.; Yamazaki, F.; Katoh, T.; Umekawa, M.; Matahira, Y.; Hori, S.; Kakizuka, A.; Totani, K.; Yamamoto, K.; Ashida, H. Glucosamine induces autophagy via an mTOR-independent pathway. Biochem. Biophys. Res. Commun. 2010, 391, 1775–1779. [Google Scholar] [CrossRef]
- Jiang, L.; Jin, Y.; Wang, H.; Jiang, Y.; Dong, J. Glucosamine protects nucleus pulposus cells and induces autophagy via the mTOR-dependent pathway. J. Orthop. Res. 2014, 32, 1532–1542. [Google Scholar] [CrossRef]
- Chen, C.-L.; Chen, Y.-H.; Liang, C.-M.; Tai, M.-C.; Lu, D.-W.; Chen, J.-T. Glucosamine-Induced Autophagy through AMPK–mTOR Pathway Attenuates Lipofuscin-Like Autofluorescence in Human Retinal Pigment Epithelial Cells In Vitro. Int. J. Mol. Sci. 2018, 19, 1416. [Google Scholar] [CrossRef]
- Shintani, T.; Kosuge, Y.; Ashida, H. Glucosamine Extends the Lifespan of Caenorhabditis elegans via Autophagy Induction. J. Appl. Glycosci. 2018, 65, 37–43. [Google Scholar] [CrossRef]
- Kang, Y.H.; Park, S.; Ahn, C.; Song, J.; Kim, D.; Jin, E.J. Beneficial reward-to-risk action of glucosamine during pathogenesis of osteoarthritis. Eur. J. Med. Res. 2015, 20, 89. [Google Scholar] [CrossRef]
- Carames, B.; Kiosses, W.B.; Akasaki, Y.; Brinson, D.C.; Eap, W.; Koziol, J.; Lotz, M.K. Glucosamine activates autophagy in vitro and in vivo. Arthritis Rheum. 2013, 65, 1843–1852. [Google Scholar] [CrossRef]
- Lv, C.; Wang, L.; Zhu, X.; Lin, W.; Chen, X.; Huang, Z.; Huang, L.; Yang, S. Glucosamine promotes osteoblast proliferation by modulating autophagy via the mammalian target of rapamycin pathway. Biomed. Pharmacother. Biomed. Aharmacotherapie 2018, 99, 271–277. [Google Scholar] [CrossRef]
- Derfoul, A.; Miyoshi, A.D.; Freeman, D.E.; Tuan, R.S. Glucosamine promotes chondrogenic phenotype in both chondrocytes and mesenchymal stem cells and inhibits MMP-13 expression and matrix degradation. Osteoarthr. Cartil. 2007, 15, 646–655. [Google Scholar] [CrossRef]
- Varghese, S.; Theprungsirikul, P.; Sahani, S.; Hwang, N.; Yarema, K.J.; Elisseeff, J.H. Glucosamine modulates chondrocyte proliferation, matrix synthesis, and gene expression. Osteoarthr. Cartil. 2007, 15, 59–68. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, J.; Dong, J. The potential negative effect of high-dose glucosamine on the chondrocyte: Comment on the article by Carame´s et al. Arthritis Rheumatol. 2014, 66, 228–230. [Google Scholar] [CrossRef]
- de Mattei, M.; Pellati, A.; Pasello, M.; de Terlizzi, F.; Massari, L.; Gemmati, D.; Caruso, A. High doses of glucosamine-HCl have detrimental effects on bovine articular cartilage explants cultured in vitro. Osteoarthr. Cartil. 2002, 10, 816–825. [Google Scholar] [CrossRef]
- Persiani, S.; Rotini, R.; Trisolino, G.; Rovati, L.C.; Locatelli, M.; Paganini, D.; Antonioli, D.; Roda, A. Synovial and plasma glucosamine concentrations in osteoarthritic patients following oral crystalline glucosamine sulphate at therapeutic dose. Osteoarthr. Cartil. 2007, 15, 764–772. [Google Scholar] [CrossRef]
- Hughes, A.; Oxford, A.E.; Tawara, K.; Jorcyk, C.L.; Oxford, J.T. Endoplasmic Reticulum Stress and Unfolded Protein Response in Cartilage Pathophysiology; Contributing Factors to Apoptosis and Osteoarthritis. Int. J. Mol. Sci. 2017, 18, 665. [Google Scholar] [CrossRef]
- Cybulsky, A.V. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat. Rev. Nephrol. 2017, 13, 681–696. [Google Scholar] [CrossRef]
- Qiu, W.; Su, Q.; Rutledge, A.C.; Zhang, J.; Adeli, K. Glucosamine-induced endoplasmic reticulum stress attenuates apolipoprotein B100 synthesis via PERK signaling. J. Lipid Res. 2009, 50, 1814–1823. [Google Scholar] [CrossRef]
- Qiu, W.; Kohen-Avramoglu, R.; Mhapsekar, S.; Tsai, J.; Austin, R.C.; Adeli, K. Glucosamine-induced endoplasmic reticulum stress promotes ApoB100 degradation: Evidence for Grp78-mediated targeting to proteasomal degradation. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 571–577. [Google Scholar] [CrossRef]
- Beriault, D.R.; Werstuck, G.H. The role of glucosamine-induced ER stress in diabetic atherogenesis. Exp. Diabetes Res. 2012, 2012. [Google Scholar] [CrossRef]
- Beriault, D.R.; Dang, V.T.; Zhong, L.H.; Petlura, C.I.; McAlpine, C.S.; Shi, Y.; Werstuck, G.H. Glucosamine induces ER stress by disrupting lipid-linked oligosaccharide biosynthesis and N-linked protein glycosylation. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E48–E57. [Google Scholar] [CrossRef] [PubMed]
- Werstuck, G.H.; Khan, M.I.; Femia, G.; Kim, A.J.; Tedesco, V.; Trigatti, B.; Shi, Y. Glucosamine-Induced Endoplasmic Reticulum Dysfunction Is Associated With Accelerated Atherosclerosis in a Hyperglycemic Mouse Model. Diabetes 2006, 55, 93–101. [Google Scholar] [CrossRef]
- Kung, L.H.W.; Mullan, L.; Soul, J.; Wang, P.; Mori, K.; Bateman, J.F.; Briggs, M.D.; Boot-Handford, R.P. Cartilage endoplasmic reticulum stress may influence the onset but not the progression of experimental osteoarthritis. Arthritis Res. Ther. 2019, 21, 206. [Google Scholar] [CrossRef]
- Li, Y.H.; Tardif, G.; Hum, D.; Kapoor, M.; Fahmi, H.; Pelletier, J.P.; Martel-Pelletier, J. The unfolded protein response genes in human osteoarthritic chondrocytes: PERK emerges as a potential therapeutic target. Arthritis Res. Ther. 2016, 18, 172. [Google Scholar] [CrossRef]
- Igarashi, M.; Sakamoto, K.; Nagaoka, I. Effect of glucosamine, a therapeutic agent for osteoarthritis, on osteoblastic cell differentiation. Int. J. Mol. Med. 2011, 28, 373–379. [Google Scholar] [CrossRef]
- Jung, J.H.; Iwabuchi, K.; Yang, Z.; Loeken, M.R. Embryonic Stem Cell Proliferation Stimulated By Altered Anabolic Metabolism From Glucose Transporter 2-Transported Glucosamine. Sci. Rep. 2016, 6, 28452. [Google Scholar] [CrossRef]
- Agrawal, P.; Pramanik, K.; Biswas, A. Chondrogenic differentiation of mesenchymal stem cells on silk fibroin:chitosan–glucosamine scaffold in dynamic culture. Regen. Med. 2018, 13, 545–558. [Google Scholar] [CrossRef]
- Yao, H.; Xue, J.; Xie, R.; Liu, S.; Wang, Y.; Song, W.; Wang, D.A.; Ren, L. A novel glucosamine derivative with low cytotoxicity enhances chondrogenic differentiation of ATDC5. J. Mater. Sci. Mater. Med. 2017, 28, 170. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Xue, J.; Wang, Q.; Xie, R.; Li, W.; Liu, S.; Cai, J.; Qin, D.; Wang, D.A.; Ren, L. Glucosamine-modified polyethylene glycol hydrogel-mediated chondrogenic differentiation of human mesenchymal stem cells. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 79, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Hwang, N.S.; Varghese, S.; Theprungsirikul, P.; Canver, A.; Elisseeff, J. Enhanced chondrogenic differentiation of murine embryonic stem cells in hydrogels with glucosamine. Biomaterials 2006, 27, 6015–6023. [Google Scholar] [CrossRef] [PubMed]
- Kamarul, T.; Ab-Rahim, S.; Tumin, M.; Selvaratnam, L.; Ahmad, T.S. A preliminary study of the effects of glucosamine sulphate and chondroitin sulphate on surgically treated and untreated focal cartilage damage. Eur. Cells Mater. 2011, 21, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Song, W.; Yao, H.; Hou, S.; Liu, S.; Wang, Y.; Pei, D.; Zhu, X.; Qin, D.; Ren, L. Effects of cholic acid modified glucosamine on chondrogenic differentiation. RSC Adv. 2016, 6, 69586–69594. [Google Scholar] [CrossRef]
- Nakatani, S.; Mano, H.; Im, R.; Shimizu, J.; Wada, M. Glucosamine Regulates Differentiation of a Chondrogenic Cell Line, ATDC5. Biol. Pharm. Bull. 2007, 30, 433–438. [Google Scholar] [CrossRef][Green Version]
- Calamia, V.; Mateos, J.; Fernández-Puente, P.; Lourido, L.; Rocha, B.; Fernández-Costa, C.; Montell, E.; Vergés, J.; Ruiz-Romero, C.; Blanco, F.J. A pharmacoproteomic study confirms the synergistic effect of chondroitin sulfate and glucosamine. Sci. Rep. 2014, 4, 5069. [Google Scholar] [CrossRef]
- Roman-Blas, J.A.; Mediero, A.; Tardio, L.; Portal-Nunez, S.; Gratal, P.; Herrero-Beaumont, G.; Largo, R. The combined therapy with chondroitin sulfate plus glucosamine sulfate or chondroitin sulfate plus glucosamine hydrochloride does not improve joint damage in an experimental model of knee osteoarthritis in rabbits. Eur. J. Pharmacol. 2017, 794, 8–14. [Google Scholar] [CrossRef]
- Kwoh, C.K.; Roemer, F.W.; Hannon, M.J.; Moore, C.E.; Jakicic, J.M.; Guermazi, A.; Green, S.M.; Evans, R.W.; Boudreau, R. Effect of oral glucosamine on joint structure in individuals with chronic knee pain: A randomized, placebo-controlled clinical trial. Arthritis Rheumatol. 2014, 66, 930–939. [Google Scholar] [CrossRef]
- Fransen, M.; Agaliotis, M.; Nairn, L.; Votrubec, M.; Bridgett, L.; Su, S.; Jan, S.; March, L.; Edmonds, J.; Norton, R.; et al. Glucosamine and chondroitin for knee osteoarthritis: A double-blind randomised placebo-controlled clinical trial evaluating single and combination regimens. Ann. Rheum. Dis. 2015, 74, 851–858. [Google Scholar] [CrossRef]
- Cahlin, B.J.; Dahlstrom, L. No effect of glucosamine sulfate on osteoarthritis in the temporomandibular joints--A randomized, controlled, short-term study. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2011, 112, 760–766. [Google Scholar] [CrossRef]
- Runhaar, J.; Rozendaal, R.M.; van Middelkoop, M.; Bijlsma, H.J.W.; Doherty, M.; Dziedzic, K.S.; Lohmander, L.S.; McAlindon, T.; Zhang, W.; Bierma Zeinstra, S. Subgroup analyses of the effectiveness of oral glucosamine for knee and hip osteoarthritis: A systematic review and individual patient data meta-analysis from the OA trial bank. Ann. Rheum. Dis. 2017, 76, 1862–1869. [Google Scholar] [CrossRef]
- Gregori, D.; Giacovelli, G.; Minto, C.; Barbetta, B.; Gualtieri, F.; Azzolina, D.; Vaghi, P.; Rovati, L.C. Association of Pharmacological Treatments With Long-term Pain Control in Patients With Knee Osteoarthritis: A Systematic Review and Meta-analysis. JAMA 2018, 320, 2564–2579. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Machado, G.C.; Eyles, J.P.; Ravi, V.; Hunter, D.J. Dietary supplements for treating osteoarthritis: A systematic review and meta-analysis. Br. J. Sports Med. 2018, 52, 167–175. [Google Scholar] [CrossRef]


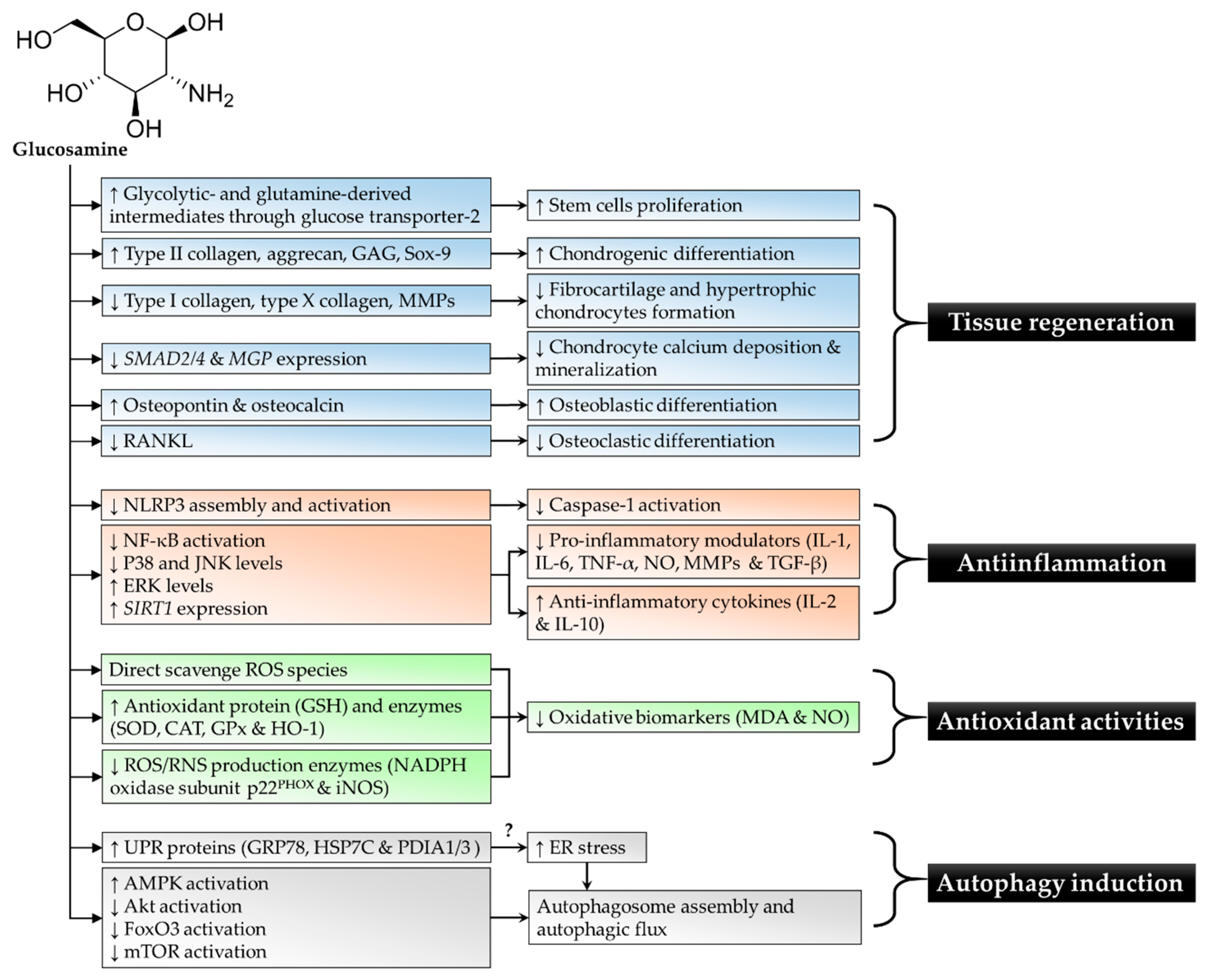{kind=link}
| References | Study Design | Findings |
|---|---|---|
| Anti-inflammation (↓ MMP levels) | ||
| [33] | Osteosarcoma cell lines were treated with 10, 50, and 100 μg/mL glucosamine, and MMP-3 and MMP-9 were assessed | ↓ MMP-3 protein levels in cell lines |
| [129] | 186 different proteins secreted by chondrocytes from patients with osteoarthritis (OA) and drug treatment for 29 days | Glucosamine in combination with chondroitin was more effective in modulating the synthesis of proteoglycans and collagens than glucosamine alone. |
| [34] | Human chondrocytes were treated with glucosamine sulfate (0.1–10 mM). | ↑ the mRNA expression and protein levels of SIRT1 and its downstream gene COL2A1 in chondrocyte ↓ MMP-1 and MMP-9 expression |
| Anti-inflammation (↓ IL and TNF levels) | ||
| [36] | In vitro and in vivo rat models of OA induced by ACLT were treated for 3 months with 175 mg/kg glucosamine sulfate | Protected against cartilage degradation↓ levels of inflammatory proteins in the affected knee ↓ IL-1β and TNF-α |
| [52] | Rats received glucosamine for 16 days orally at doses of 20, 40, 80, or 160 mg/kg/day | ↓ IL-6 and TNF levels ↑ serum nitrite |
| [53] | Mice with OA were treated for 15 days with glucosamine at doses of 40, 80, and 160 mg/kg/day | ↓ joint swelling and OA symptoms ↓ IL-β, IL-6, TNF-α, and IL-2 |
| Rats with OA were treated with 50 or 100 mg/kg glucosamine for 2 months | ↑ cytokine IL-10 and ↓TGF-1 levels | |
| [55] | Wistar rats were treated for 1 month with 500 mg of glucosamine sulfate/kg with chondroitin sulfate | ↓ IL-1β in serum |
| [56] | Patients with temporomandibular joint OA treated for 1 month or 1 year with 40 and 80 mg of glucosamine with hyaluronic acid (vs placebo with hyaluronic acid) | ↓ IL-1β after one month of treatment ↑ TGF-β, ↓ IL-1β, and IL-6 after one year of treatment |
| Antioxidant (direct scavenging of free radicals) | ||
| [80,82] | A cell-free system with the production of superoxide anion and hydroxyl radicals | Scavenge superoxide anion radicals and hydroxyl radicals |
| [80,82] | Cell-free FRAP assay and ferrous ion-chelating assay | A concentration-dependent reducing power but weak ferrous ion-chelating activity |
| [71] | A cell-free system as measured by electron spin resonance spectroscopy | Directly scavenge the superoxide anion, hydroxyl radical, and carbon-centered radicals. Glucosamine sulfate is more potent than hydrochloride form |
| Antioxidant (upregulation of antioxidant protein/enzymes levels) | ||
| [83] | Normal human articular chondrocytes were treated with glucosamine sulfate (10 mM) | ↓ IL-1β-stimulated SOD2 upregulation |
| [71] | SW1353 chondrocytes were treated with glucosamine sulfate and hydrochloride (50–1000 μg/mL) | ↑ GSH level upon glucosamine sulfate treatment but not glucosamine hydrochloride |
| [84] | Primary human osteoarthritic chondrocytes were treated with glucosamine sulfate (1 and 10 mM) | ↑ HO-1 mRNA and protein level |
| Antioxidant (suppression of free radical production) | ||
| [84] | Primary human osteoarthritic chondrocytes were treated with glucosamine sulfate (1 and 10 mM) | ↓ NADPH oxidase subunit p22Phox level |
| [85] | Articular cartilage explant discs cultures were co-incubated with LPS and glucosamine hydrochloride (0.5–10 mg/mL) | ↓ NO production |
| [86] | Primary non-OA chondrocytes were treated with glucosamine hydrochloride (100 μg/mL) | ↓ NO production (partially) |
| [84] | Primary human osteoarthritic chondrocytes were treated with glucosamine sulfate (1 and 10 mM) | ↓ IL-1β-stimulated NO production and iNOS upregulation |
| Activation of the autophagy | ||
| [103,104] | Human chondrocytes were treated with glucosamine (10–100 mM) for 2 and 24 h | High doses ↓cartilage degradation↓ peroxisomal oxidation |
| [104] | Human articular cartilage chondrocytes were treated with glucosamine (0.1–10 mM) for 8 and 24 h | ↑ LC3-II/LC3-I ratio ↑ autophagy flux Inhibited Akt/FoxO3/mTOR phosphorylation |
| GFP-LC3-transgenic mice were treated with glucosamine (250 mg/kg body weight/day) via oral and intraperitoneal administration for 7 days | ↑ autophagy in the mice liver and knee joint cartilage | |
| [105] | Human hFOB1.19 osteoblasts were treated with glucosamine (0.2–1 mM) up to 48 h | ↑ LC3 II and Beclin 1 ↓ SQSTM1/p62 Inhibited mTOR phosphorylation |
| [83] | Normal human articular chondrocytes were treated with glucosamine sulfate (10 mM) and then stimulated with IL-1β (10 ng/mL) | ↑ GRP78, HSP7C and PDIA1/3 protein levels |
| Tissue regeneration (stem cell proliferation and differentiation) | ||
| [121] | Low glucose-isolated ESCs that highly expressed glucose transporter 2 were treated with glucosamine (0.8 and 2 mM) for 4 days | ↑ ESCs proliferation |
| [106] | hMSCs were treated with glucosamine hydrochloride (100 μM and 1 mM) for 11 days | ↑ chondrogenesis (↑ type II collagen, aggrecan, and sulfated GAG levels) |
| [106] | Primary normal and OA chondrocytes were treated with glucosamine hydrochloride (100 μM and 1 mM) for 11 days | ↑ chondrogenic phenotypes with the restoration of type II collagen, aggrecan, and sulfated GAG levels Partially blocked IL-1β-mediated downregulation of type II collagen and aggrecan genes expression and inhibited the MMP-13 gene expression |
| [125] | 3D-culture of murine ESCs were treated with 2 mM glucosamine, together with glycoproteins, hyaluronic acid, chondroitin sulfate, and heparin sulfate for 21 days | ↑ ESC organoids size ↑ chondrogenesis (↑ GAG, aggrecan, type II collagen and Sox-9 genes expression and ↓ type X collagen) |
| [128] | Chondrogenic ATDC5 cells were treated with 4.6 mM glucosamine for 5 and 35 days | ↓ calcium deposition and mineralization with ↑ sulfated GAG level and ↓ SMAD2/4 and MGP genes expression |
| [120] | MC3T3-E1 osteoblasts were treated with glucosamine (0.1 mM and 1 mM) and N-acetyl glucosamine for 3 and 21 days | ↑ osteoblastic differentiation (↑ osteopontin and osteocalcin levels) ↓ osteoclastic differentiation (↓ RANKL level) |
| [126] | Male New Zealand white rabbits with surgical-induced knee joint cartilage damage were received ACI surgical repair treatment with or without the oral gavage of glucosamine sulphate (120 mg/day) for 3 and 6 months | Glucosamine improved the hyaline cartilage regeneration on ACI repair sites with ↑ proteoglycans, type II collagen and GAG expression |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Saadi, H.M.; Pang, K.-L.; Ima-Nirwana, S.; Chin, K.-Y. Multifaceted Protective Role of Glucosamine against Osteoarthritis: Review of Its Molecular Mechanisms. Sci. Pharm. 2019, 87, 34. https://doi.org/10.3390/scipharm87040034
Al-Saadi HM, Pang K-L, Ima-Nirwana S, Chin K-Y. Multifaceted Protective Role of Glucosamine against Osteoarthritis: Review of Its Molecular Mechanisms. Scientia Pharmaceutica. 2019; 87(4):34. https://doi.org/10.3390/scipharm87040034
Chicago/Turabian StyleAl-Saadi, Hiba Murtadha, Kok-Lun Pang, Soelaiman Ima-Nirwana, and Kok-Yong Chin. 2019. "Multifaceted Protective Role of Glucosamine against Osteoarthritis: Review of Its Molecular Mechanisms" Scientia Pharmaceutica 87, no. 4: 34. https://doi.org/10.3390/scipharm87040034
APA StyleAl-Saadi, H. M., Pang, K.-L., Ima-Nirwana, S., & Chin, K.-Y. (2019). Multifaceted Protective Role of Glucosamine against Osteoarthritis: Review of Its Molecular Mechanisms. Scientia Pharmaceutica, 87(4), 34. https://doi.org/10.3390/scipharm87040034