In Silico Mapping of Essential Residues in the Catalytic Domain of PDE5 Responsible for Stabilization of Its Commercial Inhibitors


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Protein Structure Details
2.2. Molecular Dynamics Simulations Protocol
2.3. Molecular Docking Protocol
3. Results and Discussion
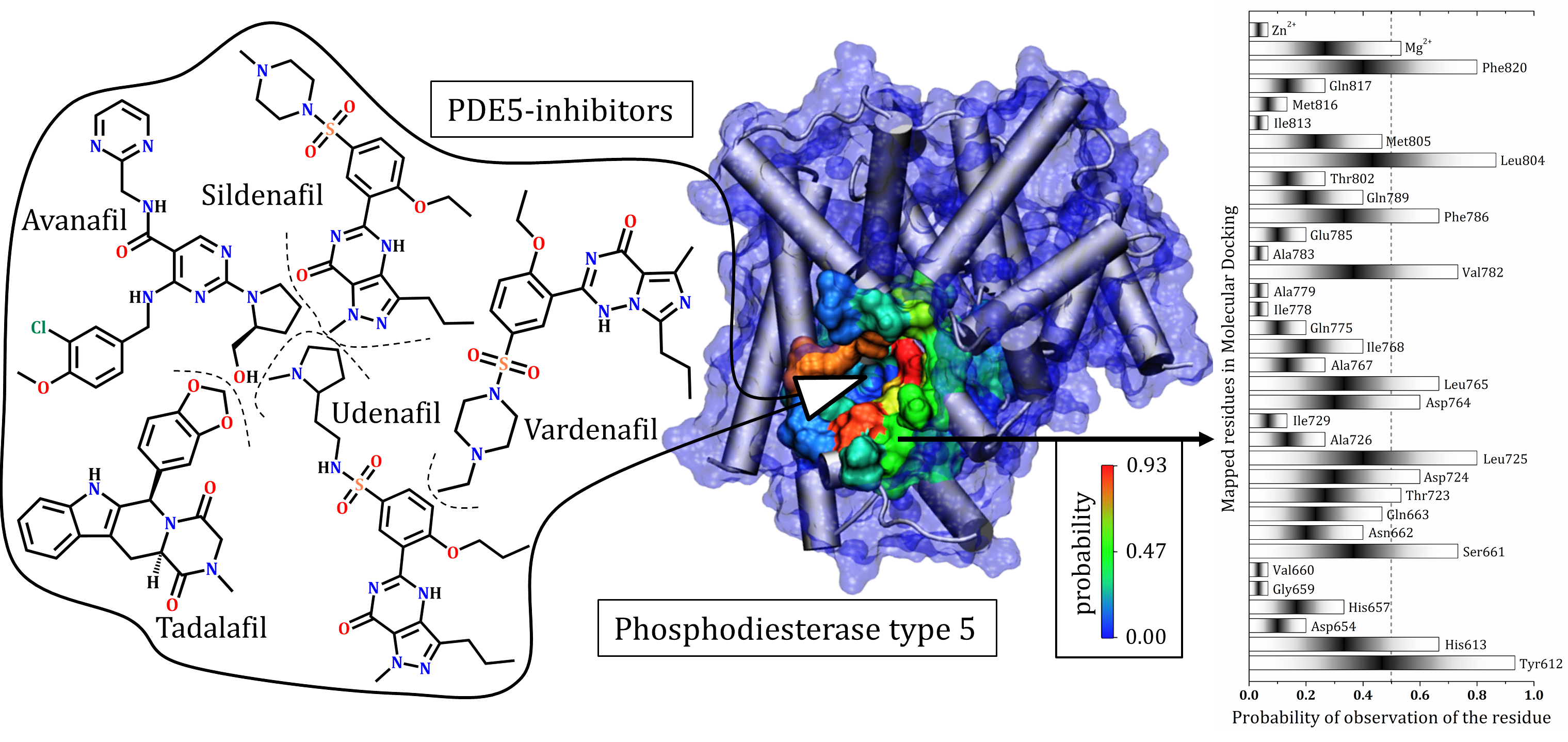
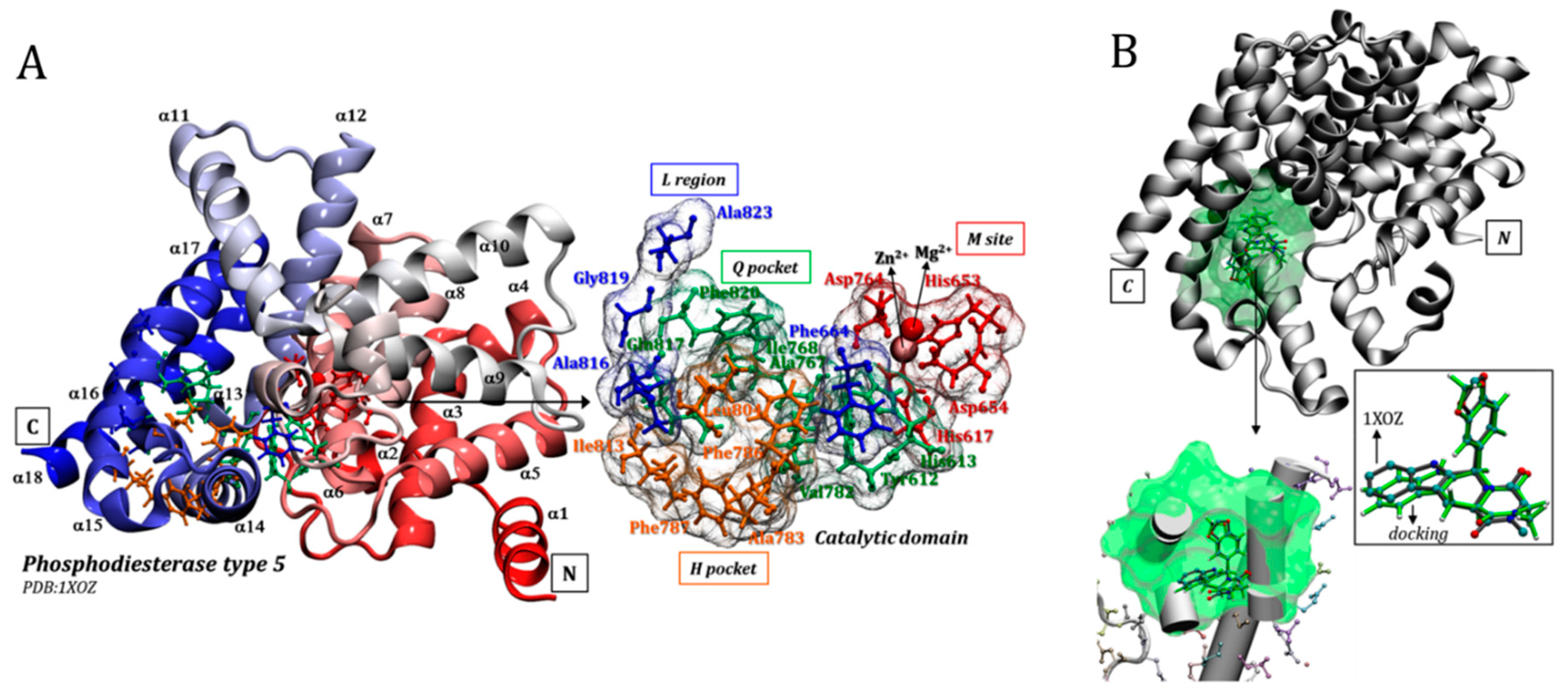
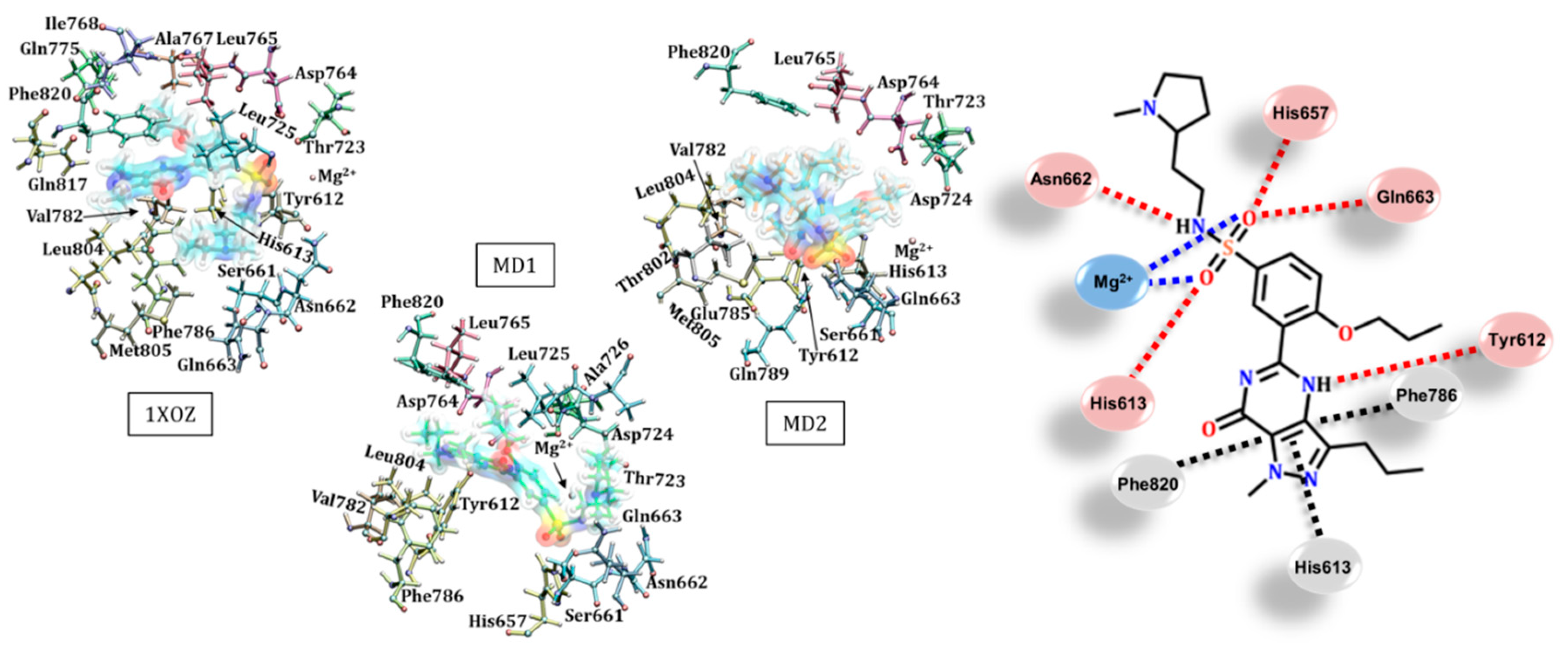
3.1. Catalytic Domain of PDE5 and Docking Validation
3.2. Tadalafil (Cialis®)
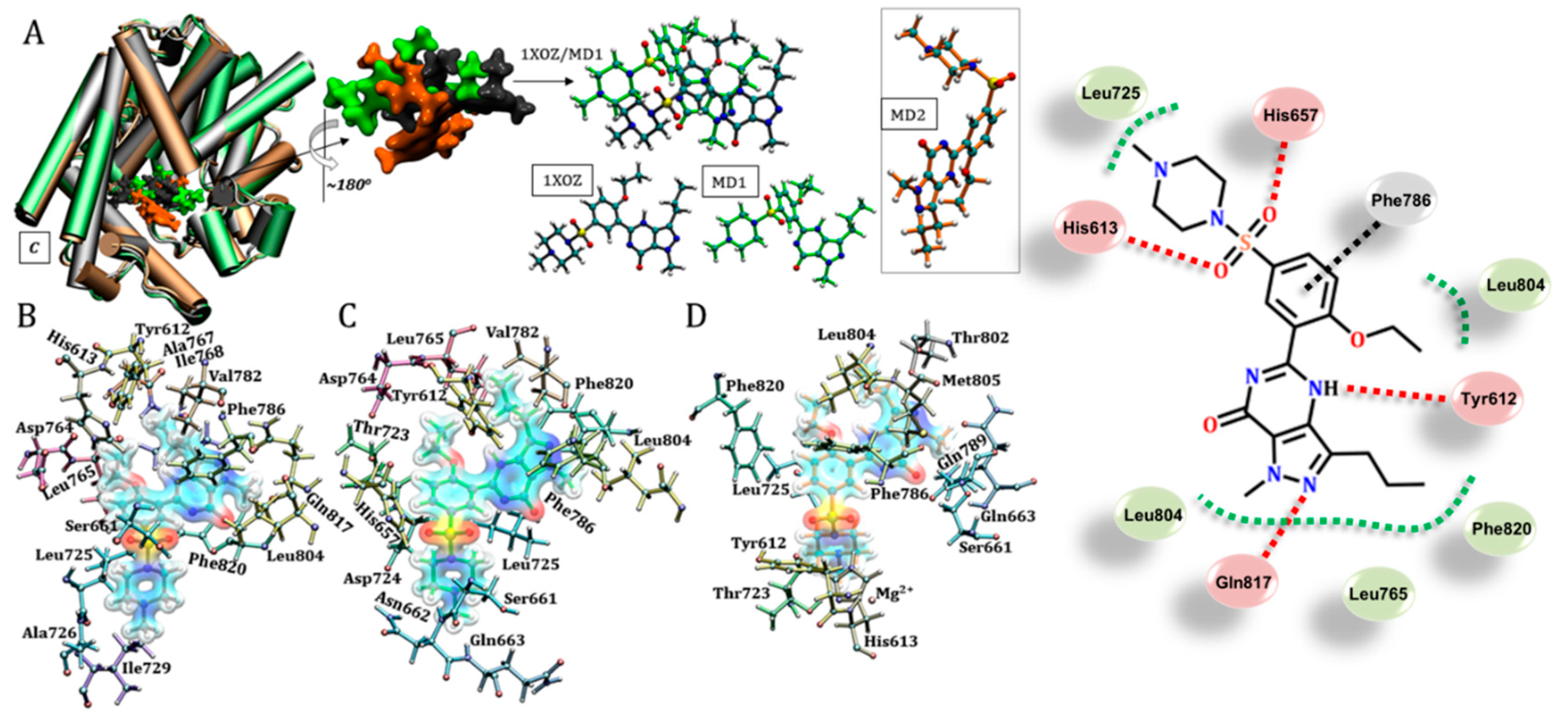
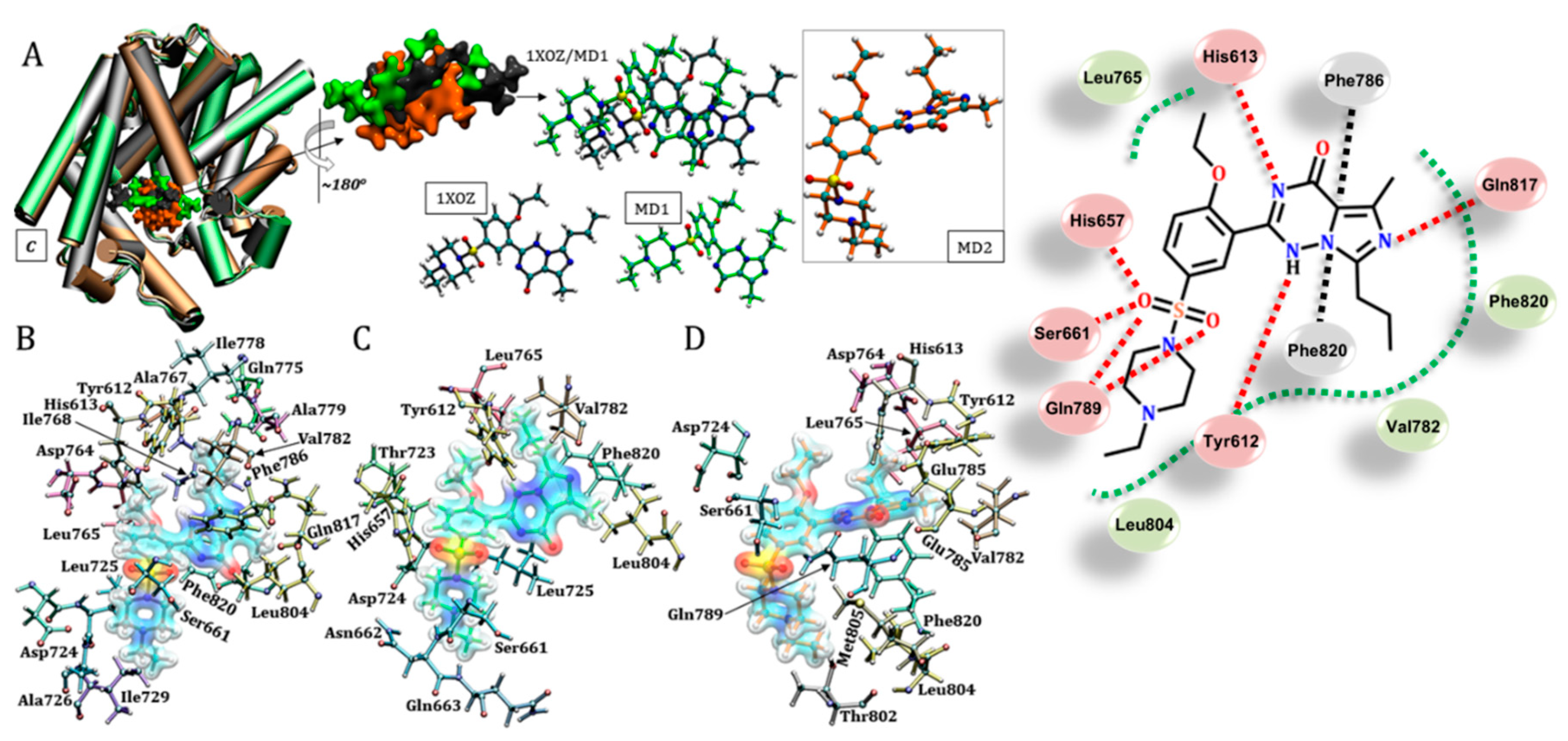
3.3. Sildenafil (Viagra®)
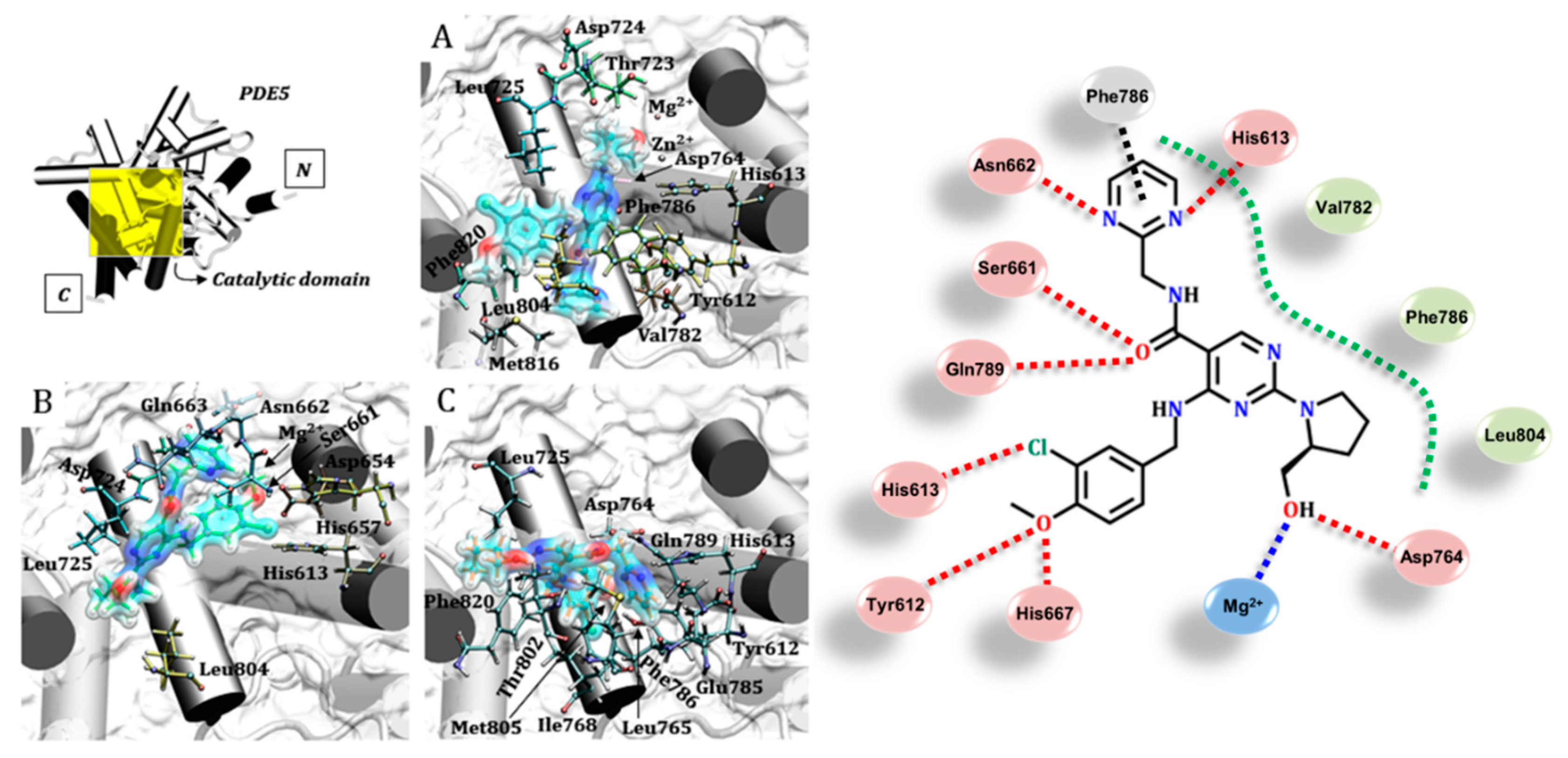
3.4. Avanafil (Stendra®/Spedra®)
3.5. Udenafil (Zydena®)
3.6. Vardenafil (Levitra®)
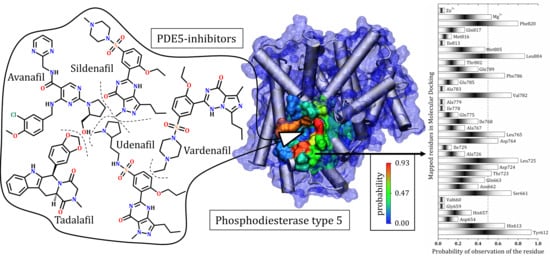
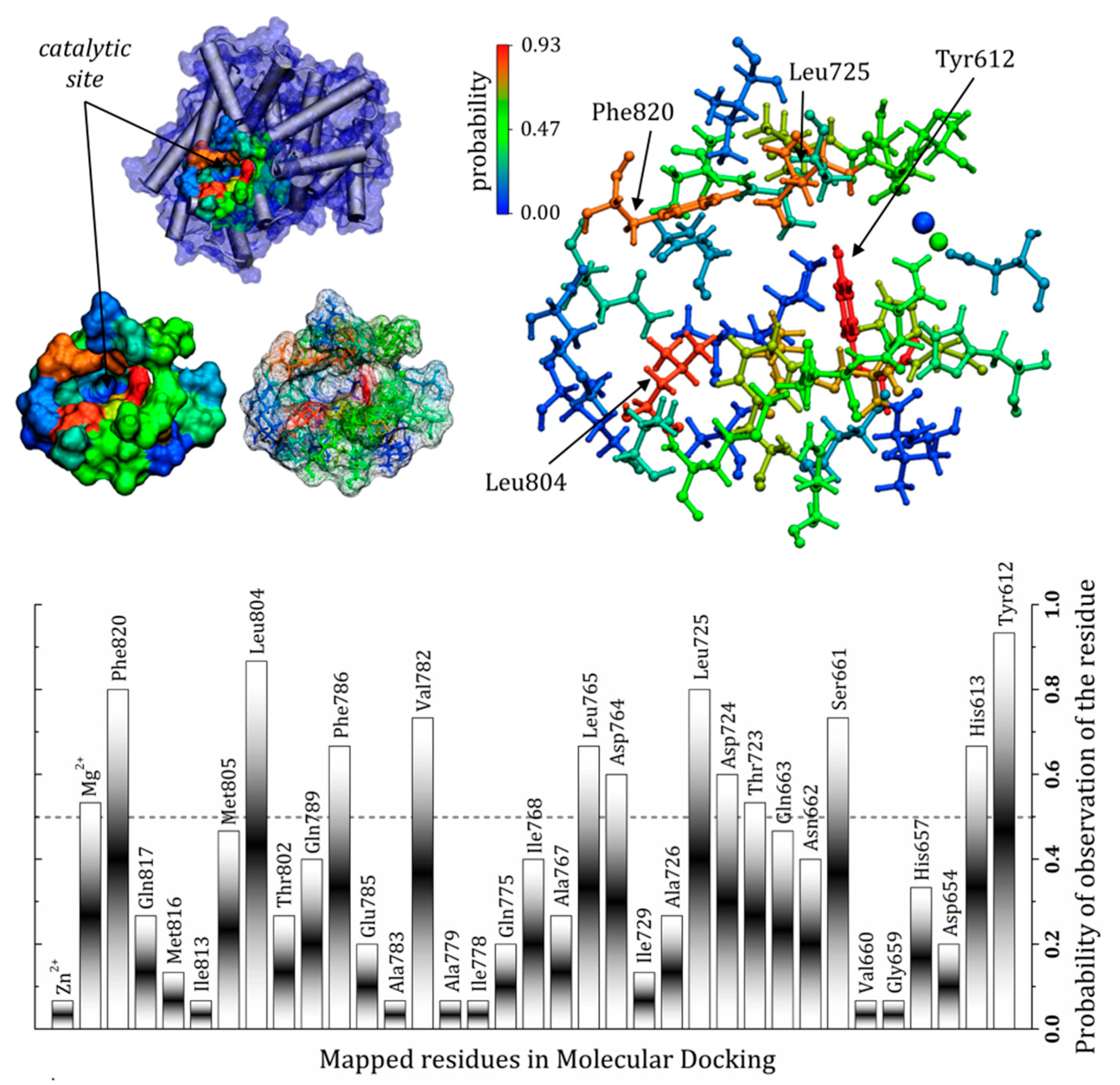
3.7. General Mapping of PDE5–Ligands Interactions in the Catalytic Domain
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Francis, S.H.; Turko, I.V.; Corbin, J.D. Cyclic nucleotide phosphodiesterases: Relating structure and function. Prog. Nucleic Acid Res. Mol. Biol. 2001, 65, 1–52. [Google Scholar] [PubMed]
- Corbin, J.D.; Francis, S.H.; Webb, D.J. Phosphodiesterase type 5 as a pharmacologic target in erectile dysfunction. Urology 2002, 60, 4–11. [Google Scholar] [CrossRef]
- Soderling, S.H.; Beavo, J.A. Regulation of cAMP and cGMP signaling: New phosphodiesterases and new functions. Curr. Opin. Cell Biol. 2000, 12, 174–179. [Google Scholar] [CrossRef]
- Tsai, E.J.; Kass, D.A. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol. Ther. 2009, 122, 216–238. [Google Scholar] [CrossRef]
- Arshad, N.; Visweswariah, S.S. The multiple and enigmatic roles of guanylyl cyclase C in intestinal homeostasis. FEBS Lett. 2012, 586, 2835–2840. [Google Scholar] [CrossRef]
- Mendes-Silverio, C.B.; Leiria, L.O.S.; Morganti, R.P.; Anhê, G.F.; Marcondes, S.; Mónica, F.Z.; De Nucci, G.; Antunes, E. Activation of Haem-Oxidized Soluble Guanylyl Cyclase with BAY 60-2770 in Human Platelets Lead to Overstimulation of the Cyclic GMP Signaling Pathway. PLoS ONE 2012, 7, e47223. [Google Scholar] [CrossRef]
- Sinðić, A.; Hirsch, J.R.; Velic, A.; Piechota, H.; Schlatter, E.; Sinic, A.; Sin, J.R.H.A. Guanylin and uroguanylin regulate electrolyte transport in isolated human cortical collecting ducts. Kidney Int. 2005, 67, 1420–1427. [Google Scholar] [CrossRef][Green Version]
- Willipinski-Stapelfeldt, B.; Lübberstedt, J.; Stelter, S.; Vogt, K.; Mukhopadhyay, A.K.; Müller, D. Comparative analysis between cyclic GMP and cyclic AMP signalling in human sperm. Mol. Hum. Reprod. 2004, 10, 543–552. [Google Scholar] [CrossRef]
- Bian, K.; Murad, F. sGC-cGMP Signaling: Target for Anticancer Therapy. Adv. Exp. Medicine Biol. 2014, 814, 5–13. [Google Scholar]
- Francis, S.H.; Busch, J.L.; Corbin, J.D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef]
- Murad, F. Nitric Oxide and Cyclic GMP in Cell Signaling and Drug Development. N. Engl. J. Med. 2006, 355, 2003–2011. [Google Scholar] [CrossRef] [PubMed]
- Beavo, J.A. Cyclic nucleotide phosphodiesterases: Functional implications of multiple isoforms. Physiol. Rev. 1995, 75, 725–748. [Google Scholar] [CrossRef] [PubMed]
- Morelli, A.; Filippi, S.; Mancina, R.; Luconi, M.; Vignozzi, L.; Marini, M.; Orlando, C.; Vannelli, G.B.; Aversa, A.; Natali, A.; et al. Androgens Regulate Phosphodiesterase Type 5 Expression and Functional Activity in Corpora Cavernosa. Endocrinology 2004, 145, 2253–2263. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xi, X.; Gu, M.; Feil, R.; Ye, R.D.; Eigenthaler, M.; Hofmann, F.; Du, X. A stimulatory role for cGMP-dependent protein kinase in platelet activation. Cell 2003, 112, 77–86. [Google Scholar] [CrossRef]
- Wallis, R.M.; Corbin, J.D.; Francis, S.H.; Ellis, P. Tissue distribution of phosphodiesterase families and the effects of sildenafil on tissue cyclic nucleotides, platelet function, and the contractile responses of trabeculae carneae and aortic rings in vitro. Am. J. Cardiol. 1999, 83, 3–12. [Google Scholar] [CrossRef]
- Mullershausen, F.; Russwurm, M.; Thompson, W.J.; Liu, L.; Koesling, D.; Friebe, A. Rapid nitric oxide–induced desensitization of the cGMP response is caused by increased activity of phosphodiesterase type 5 paralleled by phosphorylation of the enzyme. J. Cell Boil. 2001, 155, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Zoraghi, R.; Francis, S.H.; Corbin, J.D. Critical Amino Acids in Phosphodiesterase-5 Catalytic Site That Provide for High-Affinity Interaction with Cyclic Guanosine Monophosphate and Inhibitors. Biochemistry 2007, 46, 13554–13563. [Google Scholar] [CrossRef]
- Boolell, M.; Gepi-Attee, S.; Gingell, J.; Allen, M. Sildenafil, a novel effective oral therapy for male erectile dysfunction. BJU Int. 1996, 78, 257–261. [Google Scholar] [CrossRef]
- Rotella, D.P. Phosphodiesterase 5 inhibitors: Current status and potential applications. Nat. Rev. Drug Discov. 2002, 1, 674–682. [Google Scholar] [CrossRef]
- Haning, H.; Niewöhner, U.; Bischoff, E. Phosphodiesterase type 5 (PDE5) inhibitors. Prog. Med. Chem. 2003, 41, 249–306. [Google Scholar]
- Mergia, E.; Stegbauer, J. Role of Phosphodiesterase 5 and Cyclic GMP in Hypertension. Curr. Hypertens. Rep. 2016, 18, 39. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; Michelakis, E.D. Phosphodiesterase Type 5 Inhibitors for Pulmonary Arterial Hypertension. N. Engl. J. Med. 2009, 361, 1864–1871. [Google Scholar] [CrossRef] [PubMed]
- Tinel, H.; Hütter, J.; Sandner, P.; Stelte-Ludwig, B.; Stelte-Ludwig, B. Pre-clinical evidence for the use of phosphodiesterase-5 inhibitors for treating benign prostatic hyperplasia and lower urinary tract symptoms. BJU Int. 2006, 98, 1259–1263. [Google Scholar] [CrossRef] [PubMed]
- Corbin, J.D.; Beasley, A.; Blount, M.A.; Francis, S.H. High lung PDE5: A strong basis for treating pulmonary hypertension with PDE5 inhibitors. Biochem. Biophys. Res. Commun. 2005, 334, 930–938. [Google Scholar] [CrossRef]
- Barone, I.; Giordano, C.; Bonofiglio, D.; Catalano, S.; Andò, S. Phosphodiesterase Type 5 as a Candidate Therapeutic Target in Cancers. Curr. Pathobiol. Rep. 2015, 3, 193–201. [Google Scholar] [CrossRef]
- Mónica, F.Z.; De Nucci, G. Tadalafil for the treatment of benign prostatic hyperplasia. Expert Opin. Pharmacother. 2019, 1–9. [Google Scholar] [CrossRef]
- Sung, B.-J.; Hwang, K.Y.; Jeon, Y.H.; Lee, J.I.; Heo, Y.-S.; Kim, J.H.; Moon, J.; Yoon, J.M.; Hyun, Y.-L.; Kim, E.; et al. Structure of the catalytic domain of human phosphodiesterase 5 with bound drug molecules. Nature 2003, 425, 98–102. [Google Scholar] [CrossRef]
- Zenzmaier, C.; Sampson, N.; Pernkopf, D.; Plas, E.; Untergasser, G.; Berger, P. Attenuated Proliferation and Trans -Differentiation of Prostatic Stromal Cells Indicate Suitability of Phosphodiesterase Type 5 Inhibitors for Prevention and Treatment of Benign Prostatic Hyperplasia. Endocrinology 2010, 151, 3975–3984. [Google Scholar] [CrossRef][Green Version]
- Catalano, S.; Campana, A.; Giordano, C.; Győrffy, B.; Tarallo, R.; Rinaldi, A.; Bruno, G.; Ferraro, A.; Romeo, F.; Lanzino, M.; et al. Expression and function of phosphodiesterase type 5 in human breast cancer cell lines and tissues: Implications for targeted therapy. Clin. Cancer Res. 2016, 22, 2271–2282. [Google Scholar] [CrossRef]
- Cahill, K.B.; Quade, J.H.; Carleton, K.L.; Cote, R.H. Identification of Amino Acid Residues Responsible for the Selectivity of Tadalafil Binding to Two Closely Related Phosphodiesterases, PDE5 and PDE6*. J. Boil. Chem. 2012, 287, 41406–41416. [Google Scholar] [CrossRef]
- Kayık, G.; Tüzün, N.Ş.; Durdagi, S. Investigation of PDE5/PDE6 and PDE5/PDE11 selective potent tadalafil-like PDE5 inhibitors using combination of molecular modeling approaches, molecular fingerprint-based virtual screening protocols and structure-based pharmacophore development. J. Enzym. Inhib. Med. Chem. 2017, 32, 311–330. [Google Scholar] [CrossRef] [PubMed]
- Pattis, J.G.; Kamal, S.; Li, B.; May, E.R. Catalytic Domains of Phosphodiesterase 5, 6, and 5/6 Chimera Display Differential Dynamics and Ligand Dissociation Energy Barriers. J. Phys. Chem. B 2019, 123, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Chan-Tack, K.M. Oral sildenafil in erectile dysfunction. Lancet 1998, 352, 1557. [Google Scholar] [CrossRef]
- Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: A new target for the development of specific therapeutic agents. Pharmacol. Ther. 2006, 109, 366–398. [Google Scholar] [CrossRef]
- Zoraghi, R.; Bessay, E.P.; Corbin, J.D.; Francis, S.H. Structural and Functional Features in Human PDE5A1 Regulatory Domain That Provide for Allosteric cGMP Binding, Dimerization, and Regulation. J. Boil. Chem. 2005, 280, 12051–12063. [Google Scholar] [CrossRef]
- Card, G.L.; England, B.P.; Suzuki, Y.; Fong, D.; Powell, B.; Lee, B.; Luu, C.; Tabrizizad, M.; Gillette, S.; Ibrahim, P.N.; et al. Structural Basis for the Activity of Drugs that Inhibit Phosphodiesterases. Structure 2004, 12, 2233–2247. [Google Scholar] [CrossRef]
- Martinez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Chandrasekhar, J.; Impey, R.W.; Jorgensen, W.L.; Madura, J.D.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Xie, X.-Q.S. Exploiting PubChem for virtual screening. Expert Opin. Drug Discov. 2010, 5, 1205–1220. [Google Scholar] [CrossRef] [PubMed]
- de Magalhães, C.S.; Almeida, D.M.; Barbosa, H.J.C.; Dardenne, L.E. A dynamic niching genetic algorithm strategy for docking highly flexible ligands. Inf. Sci. (N. Y.) 2014, 289, 206–224. [Google Scholar] [CrossRef]
- Seftel, A.D. Phosphodiesterase type 5 inhibitors: Molecular pharmacology and interactions with other phosphodiesterases. Curr. Pharm. Des. 2005, 11, 4047–4058. [Google Scholar] [CrossRef]
- Erickson, J.A.; Jalaie, M.; Robertson, D.H.; Lewis, R.A.; Vieth, M. Lessons in Molecular Recognition: The Effects of Ligand and Protein Flexibility on Molecular Docking Accuracy. J. Med. Chem. 2004, 47, 45–55. [Google Scholar] [CrossRef]
- Spyrakis, F.; BidonChanal, A.; Barril, X.; Luque, F.J. Protein flexibility and ligand recognition: Challenges for molecular modeling. Curr. Top. Med. Chem. 2011, 11, 192–210. [Google Scholar] [CrossRef]
- Huang, S.Y.; Zou, X. Ensemble docking of multiple protein structures: Considering protein structural variations in molecular docking. Proteins Struct. Funct. Genet. 2007, 66, 399–421. [Google Scholar] [CrossRef]
- Osterberg, F.; Morris, G.M.; Sanner, M.F.; Olson, A.J.; Goodsell, D.S. Automated docking to multiple target structures: Incorporation of protein mobility and structural water heterogeneity in AutoDock. Proteins Struct. Funct. Bioinform. 2002, 46, 34–40. [Google Scholar] [CrossRef]
- Zoraghi, R.; Corbin, J.D.; Francis, S.H. Phosphodiesterase-5 Gln817 is critical for cGMP, vardenafil, or sildenafil affinity: Its orientation impacts cGMP but not cAMP affinity. J. Biol. Chem. 2006, 281, 5553–5558. [Google Scholar] [CrossRef]
- de Oliveira, I.P.; Lescano, C.H.; De Nucci, G. Q817G mutation in phosphodiesterase type 5 (PDE-5): Conformational analysis and dissociation profile of the inhibitor Tadalafil. Chem. Biol. Drug Des. 2018, 93, 419–429. [Google Scholar] [CrossRef]
- Dill, K.A.; Maccallum, J.L. The Protein-Folding Problem, 50 Years On. Science 2012, 338, 1042–1046. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Y.; Huai, Q.; Cai, J.; Zoraghi, R.; Francis, S.H.; Corbin, J.D.; Robinson, H.; Xin, Z.; Lin, G.; et al. Multiple conformations of phosphodiesterase-5: Implications for enzyme function and drug development. J. Biol. Chem. 2006, 281, 21469–21479. [Google Scholar] [CrossRef] [PubMed]
- Masche, U.P. Avanafil. Pharma Krit. 2016, 38, 13–14. [Google Scholar] [CrossRef]
- Sanford, M. Avanafil: A Review of Its Use in Patients with Erectile Dysfunction. Drugs Aging 2013, 30, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Burke, R.M.; Evans, J.D. Avanafil for treatment of erectile dysfunction: Review of its potential. Vasc. Health Risk Manag. 2012, 8, 517–523. [Google Scholar]
- Kotera, J.; Mochida, H.; Inoue, H.; Noto, T.; Fujishige, K.; Sasaki, T.; Kobayashi, T.; Kojima, K.; Yee, S.; Yamada, Y.; et al. Avanafil, a Potent and Highly Selective Phosphodiesterase-5 Inhibitor for Erectile Dysfunction. J. Urol. 2012, 188, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.G.; Kim, J.J. Udenafil: Efficacy and tolerability in the management of erectile dysfunction. Ther. Adv. Urol. 2013, 5, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-Y.; Li, Z.; Cai, Y.-H.; Feng, L.-J.; Wu, Y.; Li, X.; Luo, H.-B. The Molecular Basis for the Selectivity of Tadalafil toward Phosphodiesterase 5 and 6: A Modeling Study. J. Chem. Inf. Model. 2013, 53, 3044–3053. [Google Scholar] [CrossRef]
- Corbin, J.; Francis, S.; Zoraghi, R. Tyrosine-612 in PDE5 contributes to higher affinity for vardenafil over sildenafil. Int. J. Impot. Res. 2006, 18, 251–257. [Google Scholar] [CrossRef]
- Hellstrom, W.J.G. Vardenafil: A new approach to the treatment of erectile dysfunction. Curr. Urol. Rep. 2003, 4, 479–487. [Google Scholar] [CrossRef]
- Palit, V.; Eardley, I. An update on new oral PDE5 inhibitors for the treatment of erectile dysfunction. Nat. Rev. Urol. 2010, 7, 603–609. [Google Scholar] [CrossRef]
- Corbin, J.D.; Rannels, S.R.; Francis, S.H. Phosphodiesterase-5 Inhibition BT–Heart Disease and Erectile Dysfunction; Kloner, R.A., Ed.; Humana Press: Totowa, NJ, USA, 2004; pp. 117–130. [Google Scholar] [CrossRef]
- Oliveira, I.P.; Martínez, L. Molecular basis for competitive solvation of the Burkholderia cepacia lipase by sorbitol and urea. Phys. Chem. Chem. Phys. 2016, 18, 21797–21808. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Oliveira, I.P.; Lescano, C.H.; De Nucci, G. In Silico Mapping of Essential Residues in the Catalytic Domain of PDE5 Responsible for Stabilization of Its Commercial Inhibitors. Sci. Pharm. 2019, 87, 29. https://doi.org/10.3390/scipharm87040029
de Oliveira IP, Lescano CH, De Nucci G. In Silico Mapping of Essential Residues in the Catalytic Domain of PDE5 Responsible for Stabilization of Its Commercial Inhibitors. Scientia Pharmaceutica. 2019; 87(4):29. https://doi.org/10.3390/scipharm87040029
Chicago/Turabian Stylede Oliveira, Ivan Pires, Caroline Honaiser Lescano, and Gilberto De Nucci. 2019. "In Silico Mapping of Essential Residues in the Catalytic Domain of PDE5 Responsible for Stabilization of Its Commercial Inhibitors" Scientia Pharmaceutica 87, no. 4: 29. https://doi.org/10.3390/scipharm87040029
APA Stylede Oliveira, I. P., Lescano, C. H., & De Nucci, G. (2019). In Silico Mapping of Essential Residues in the Catalytic Domain of PDE5 Responsible for Stabilization of Its Commercial Inhibitors. Scientia Pharmaceutica, 87(4), 29. https://doi.org/10.3390/scipharm87040029