
Estrogen Receptor Ligands: A Review (2013–2015)


Abstract
:1. Introduction
1.1. Estrogen
Estrogen Receptor Subtypes
1.2. Potential Clinical Applications of Estrogens
1.2.1. Cancer
1.2.2. Neuropathies
1.2.3. Cardiovascular Disease
1.2.4. Osteoporosis
2. Estrogen Receptor Ligands
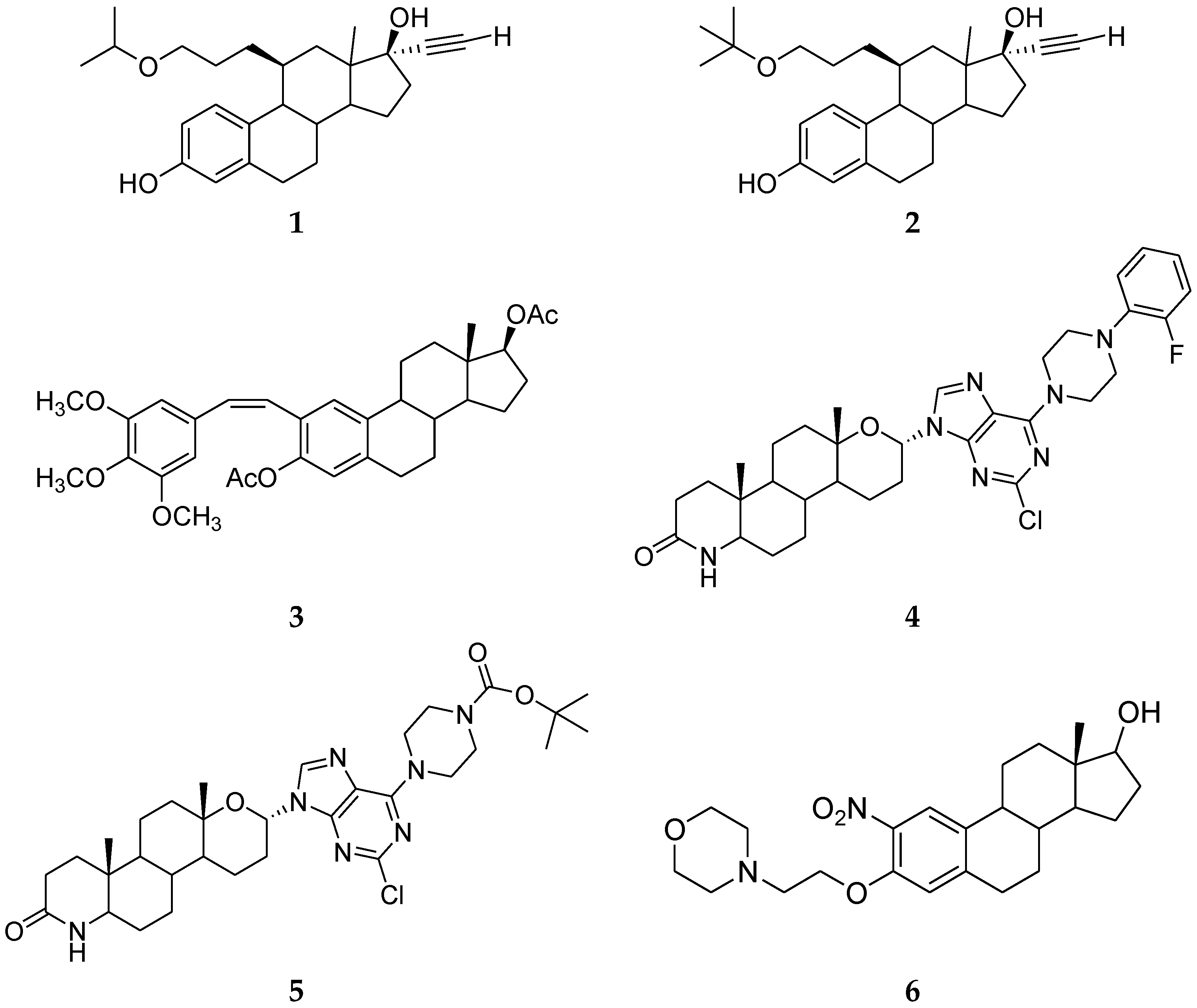
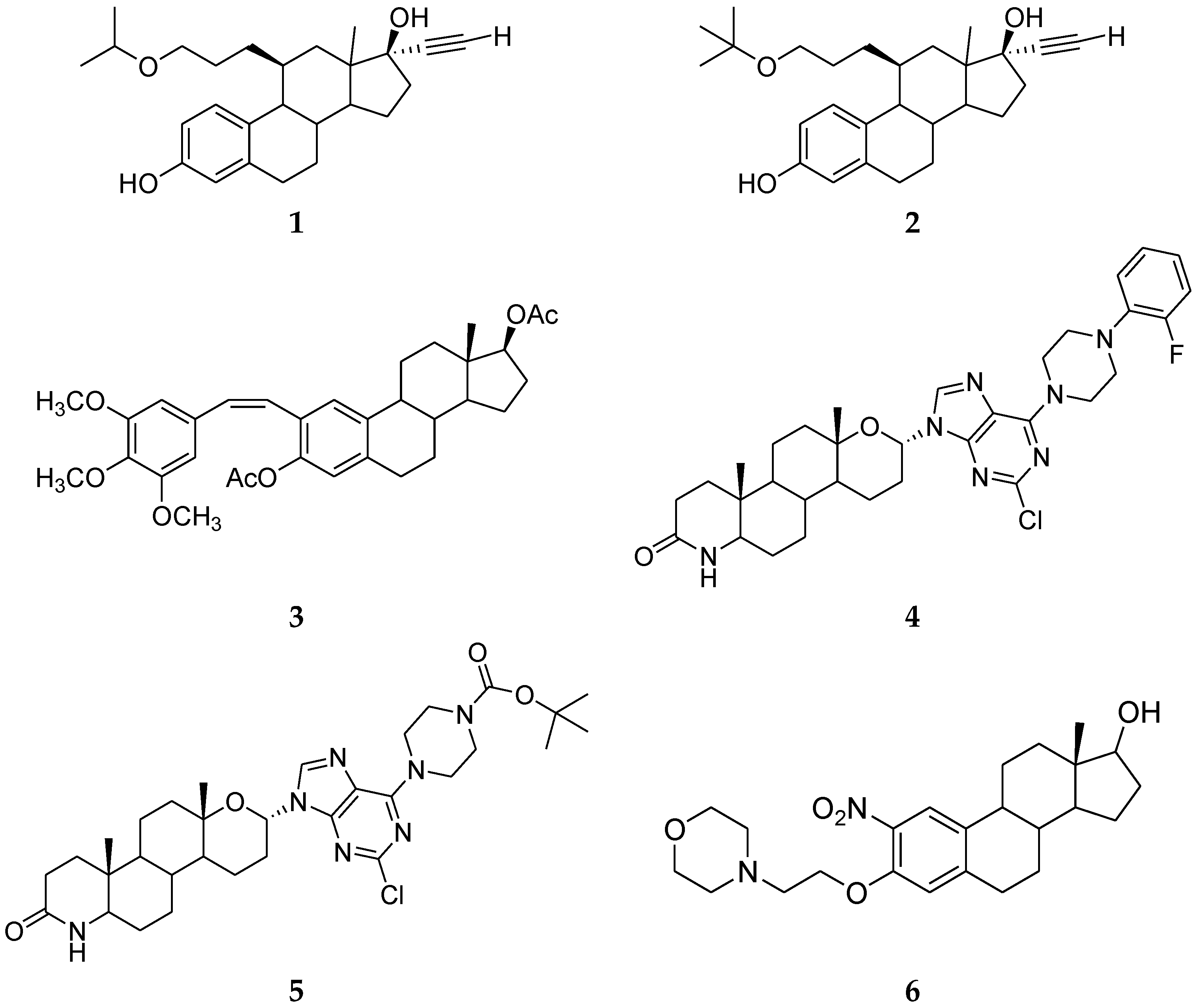
3. Steroidal Ligands
4. Non-Steroidal Ligands
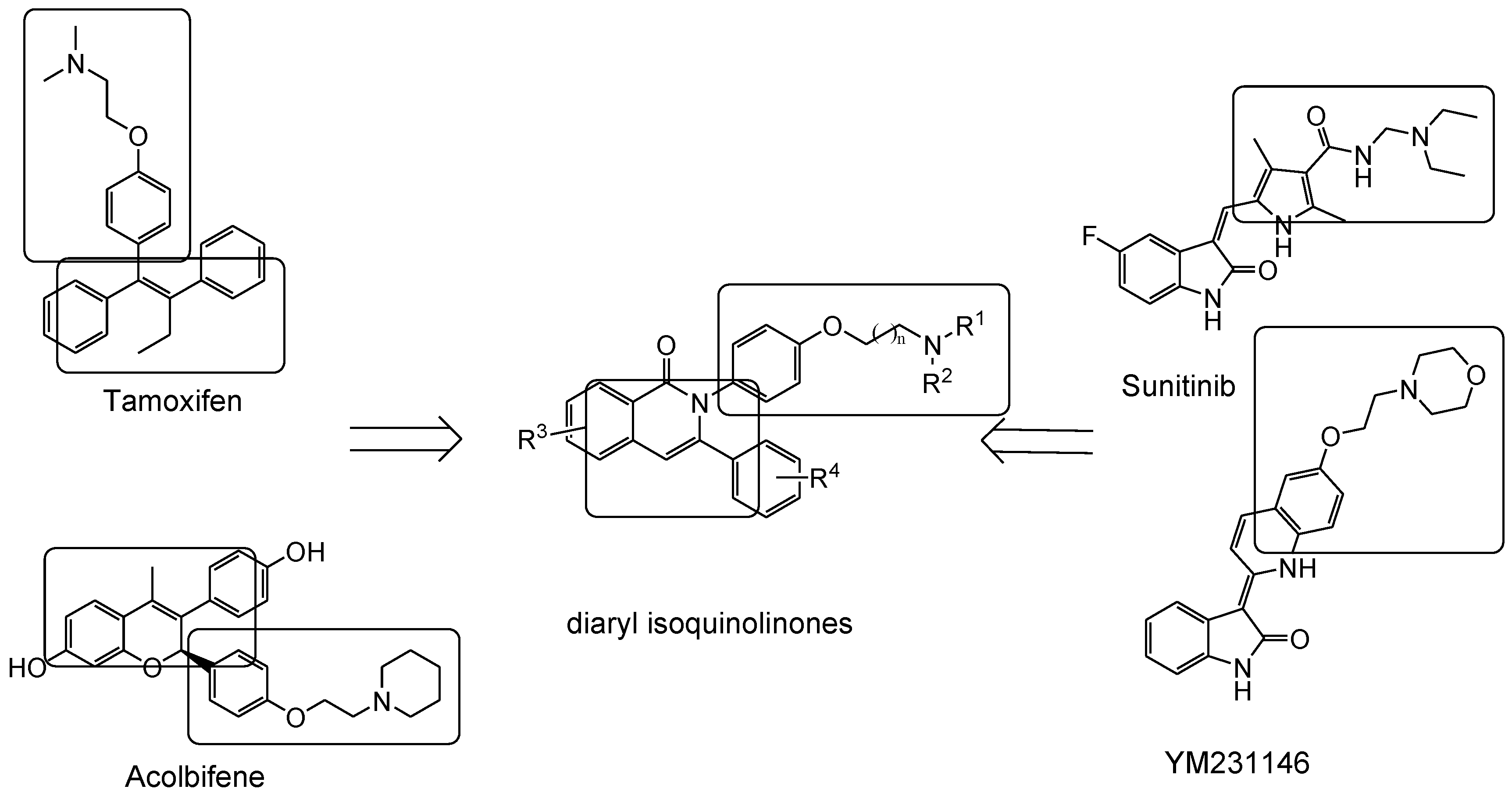
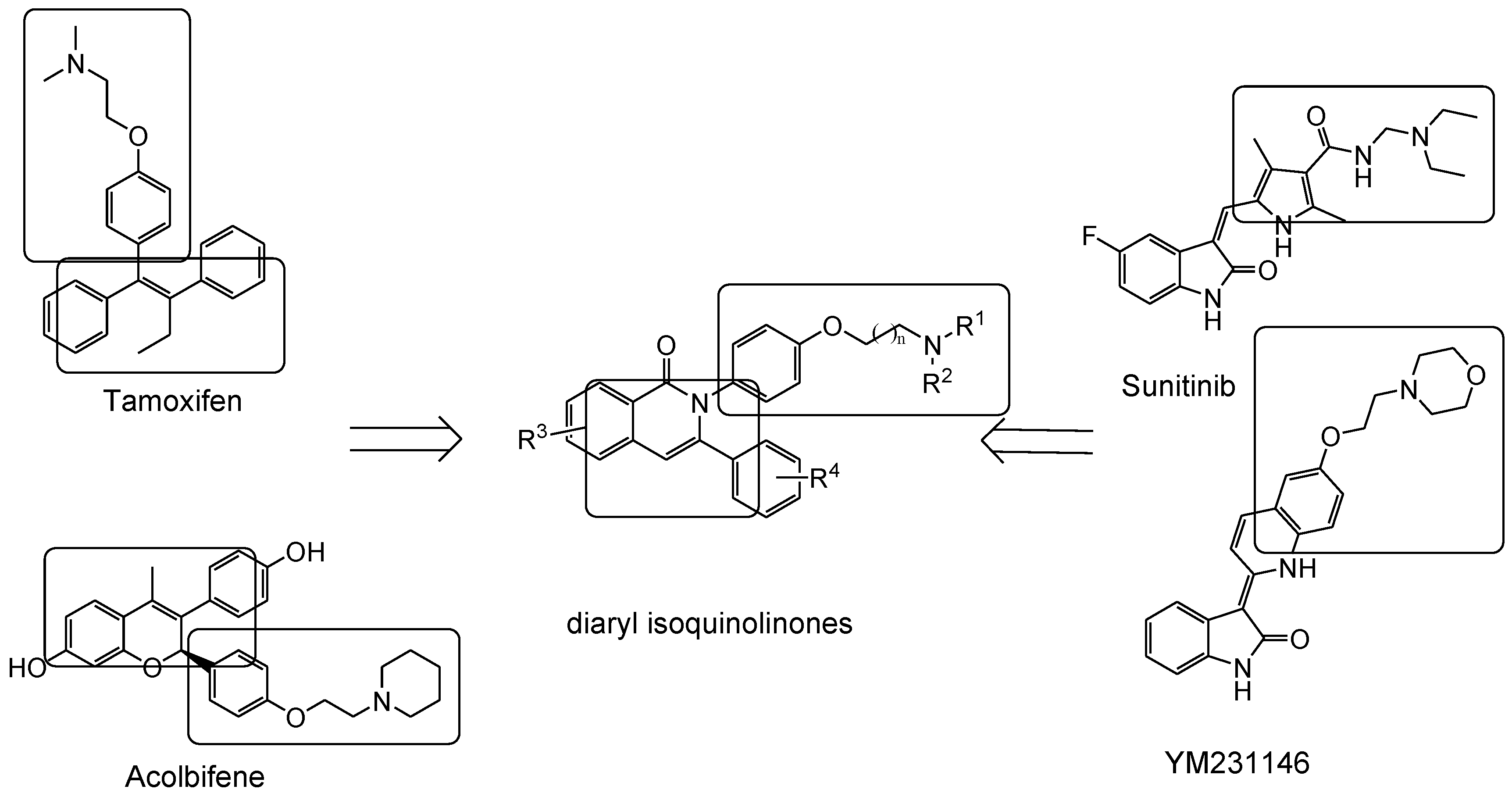
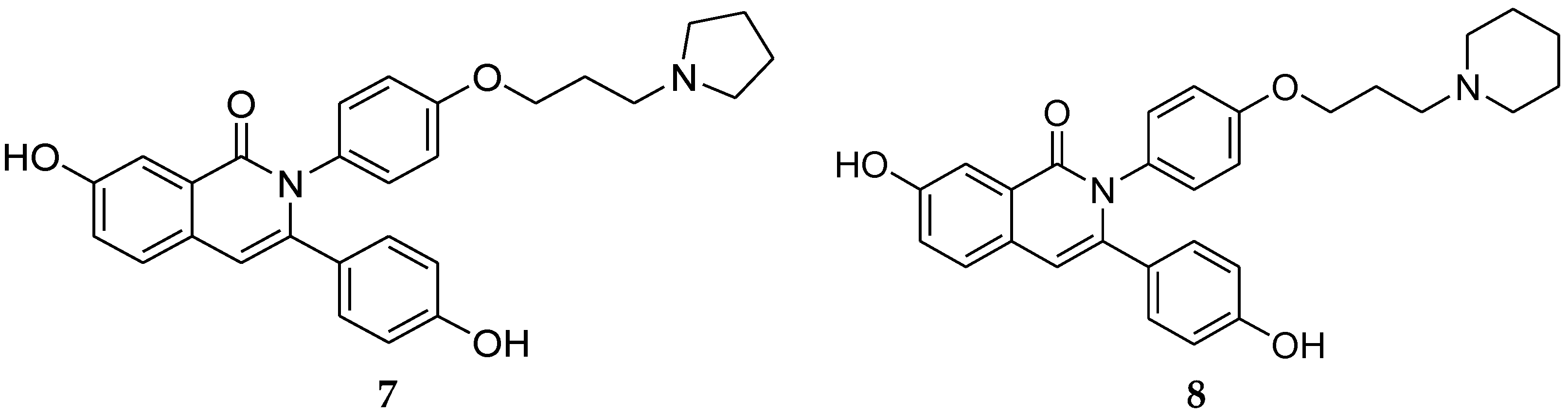
4.1. 2,3-Diaryl Isoquinolinone
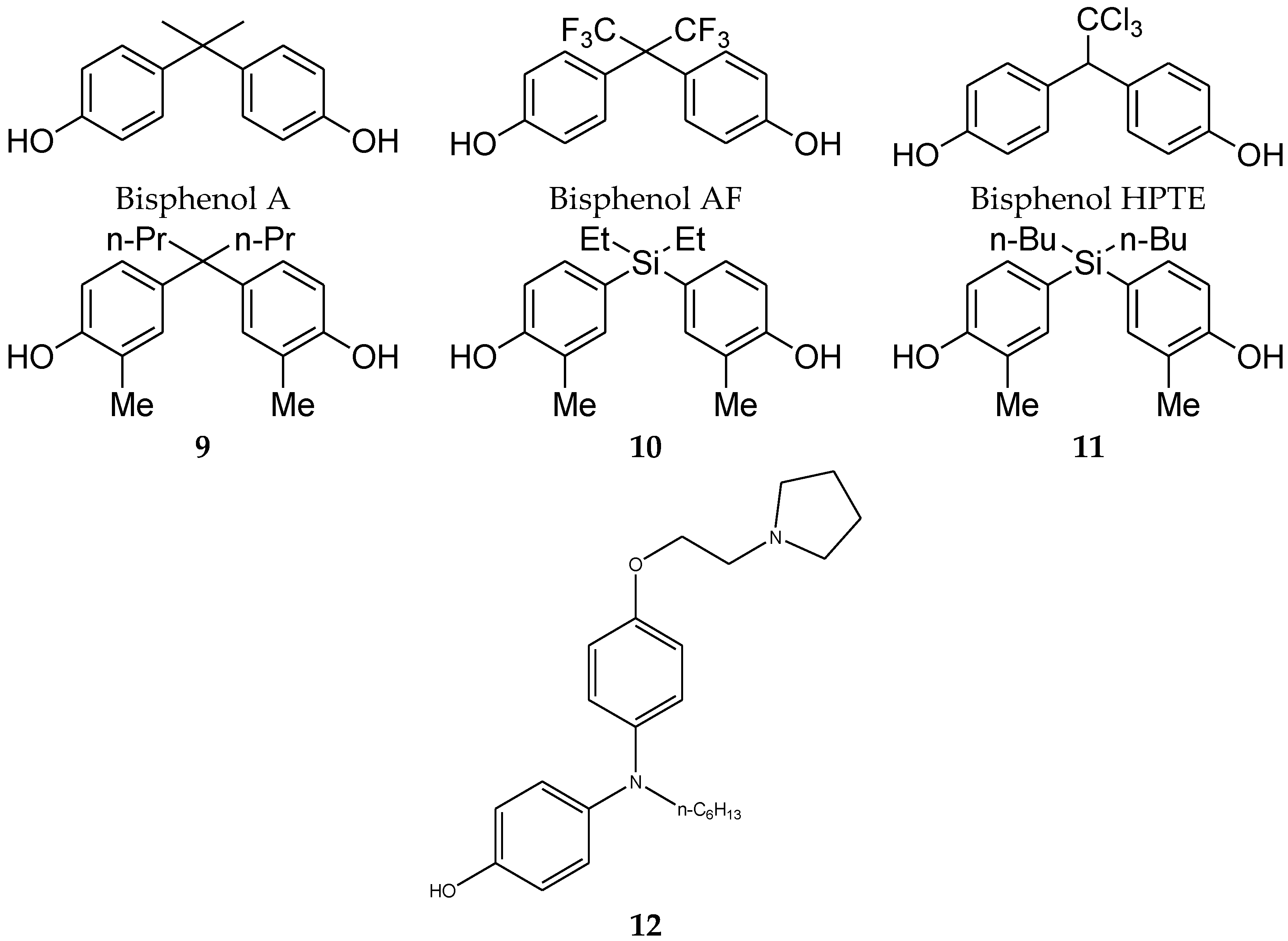
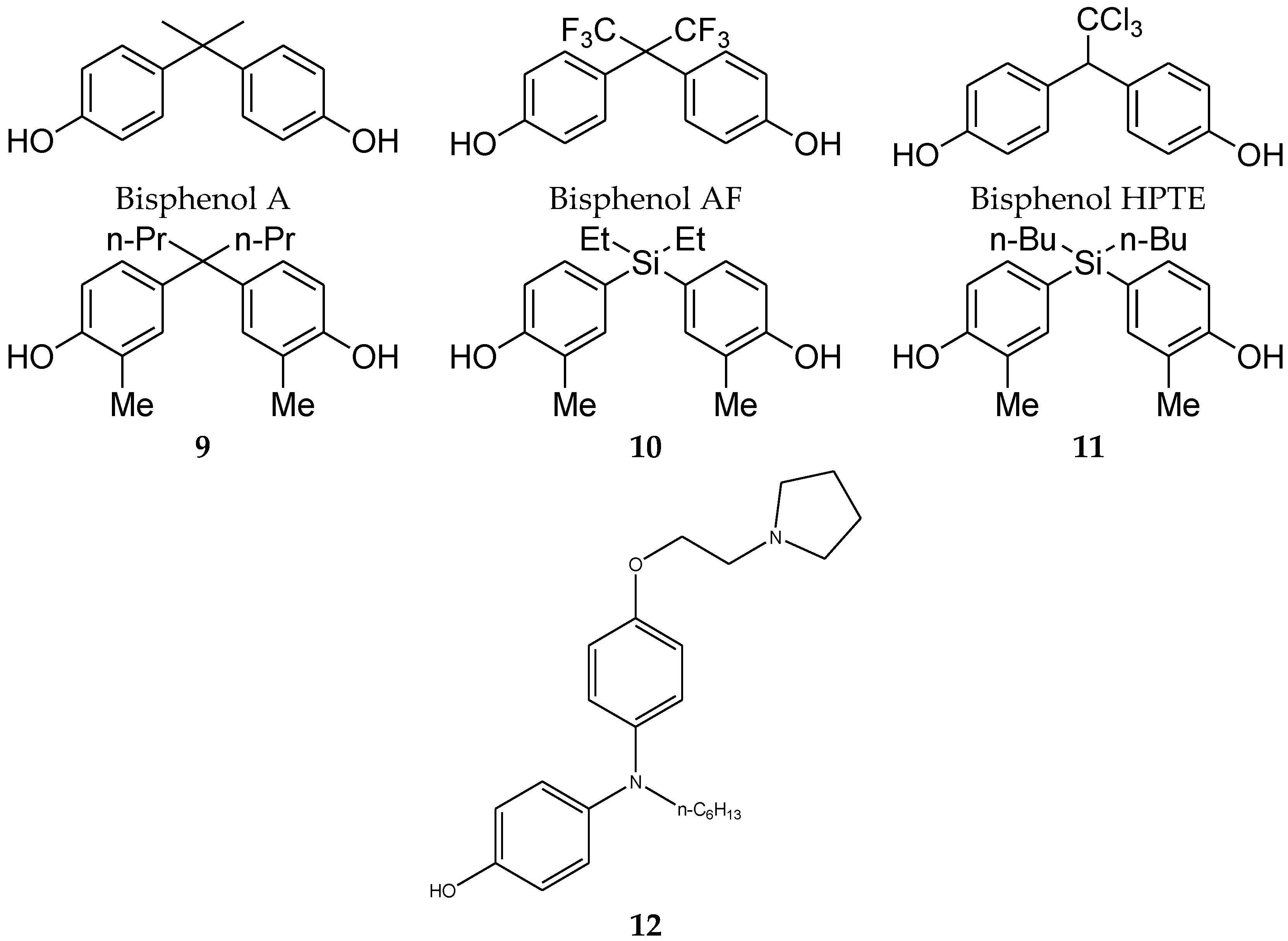
4.2. Diphenylmethane Skeleton and Related Analogues
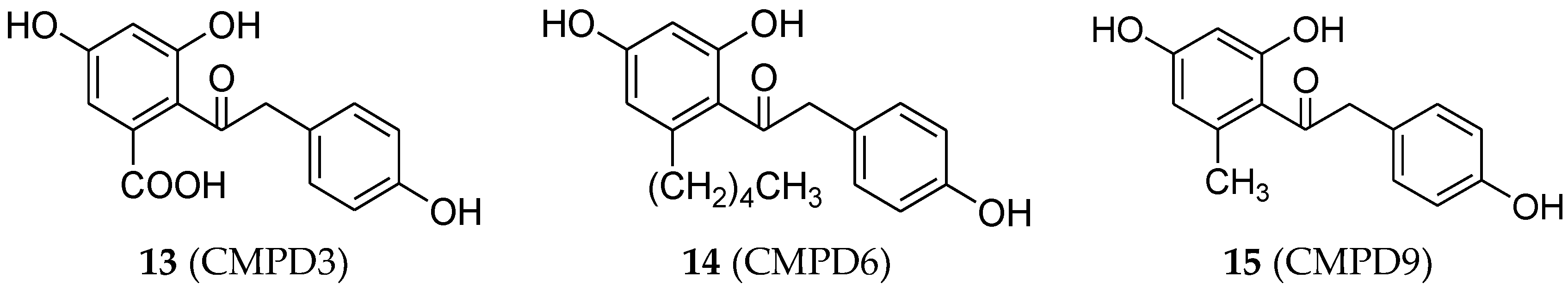
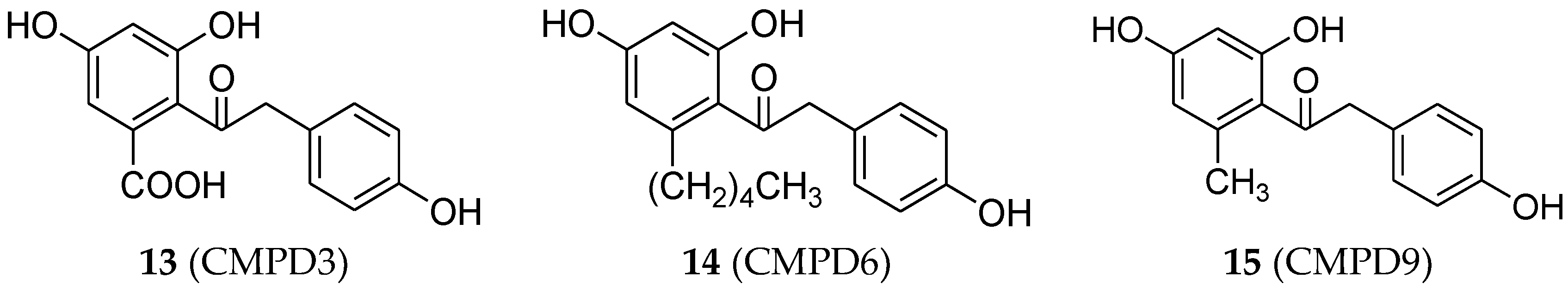
4.3. Deoxybenzoin Analogues
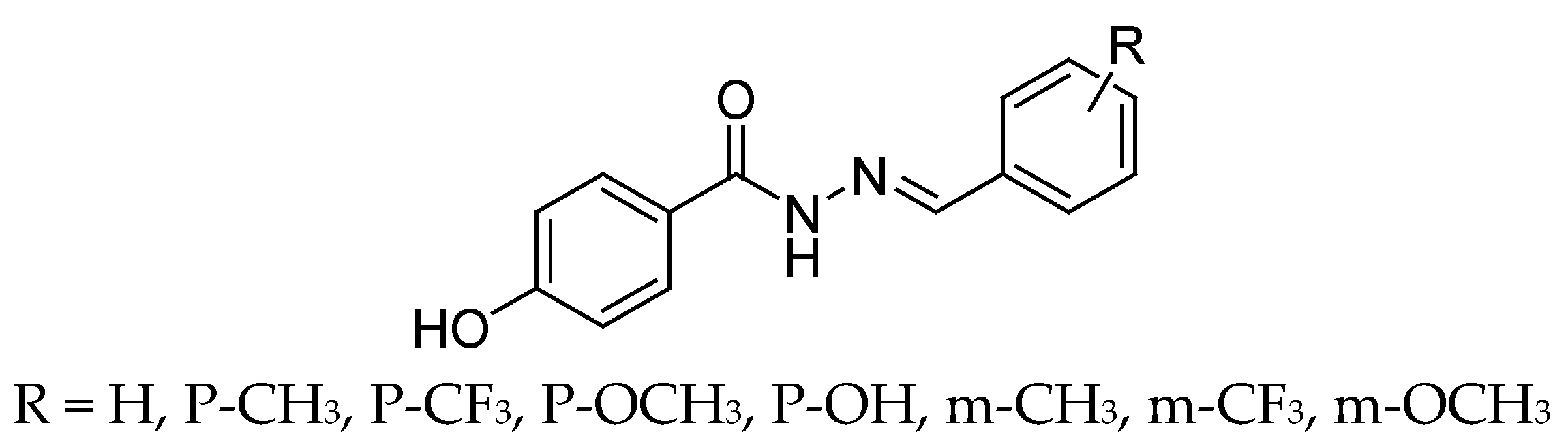
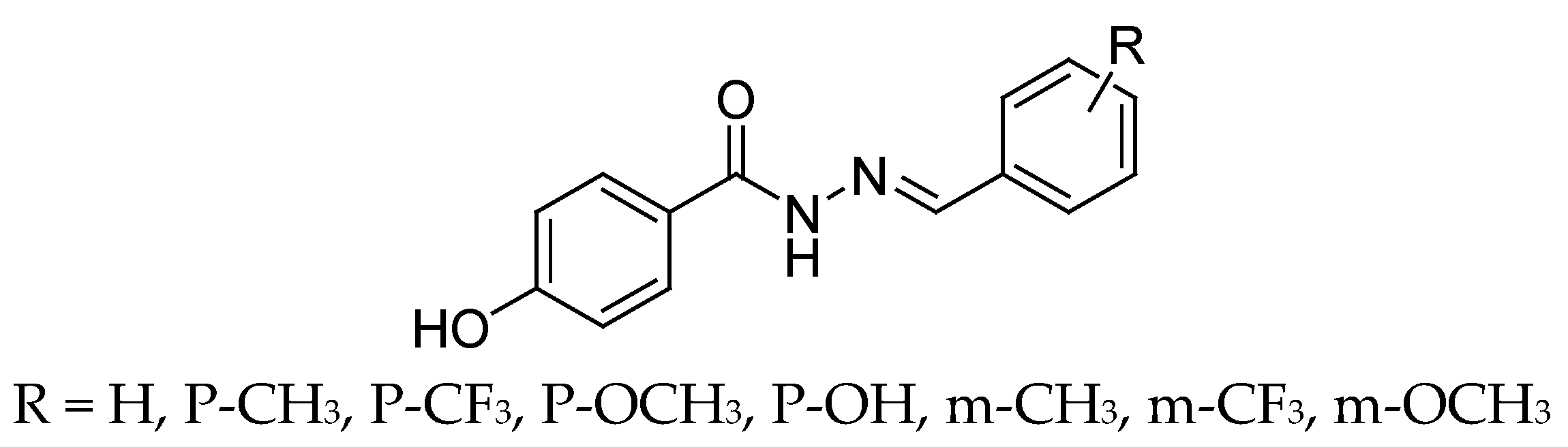
4.4. Hydrazide Derivatives
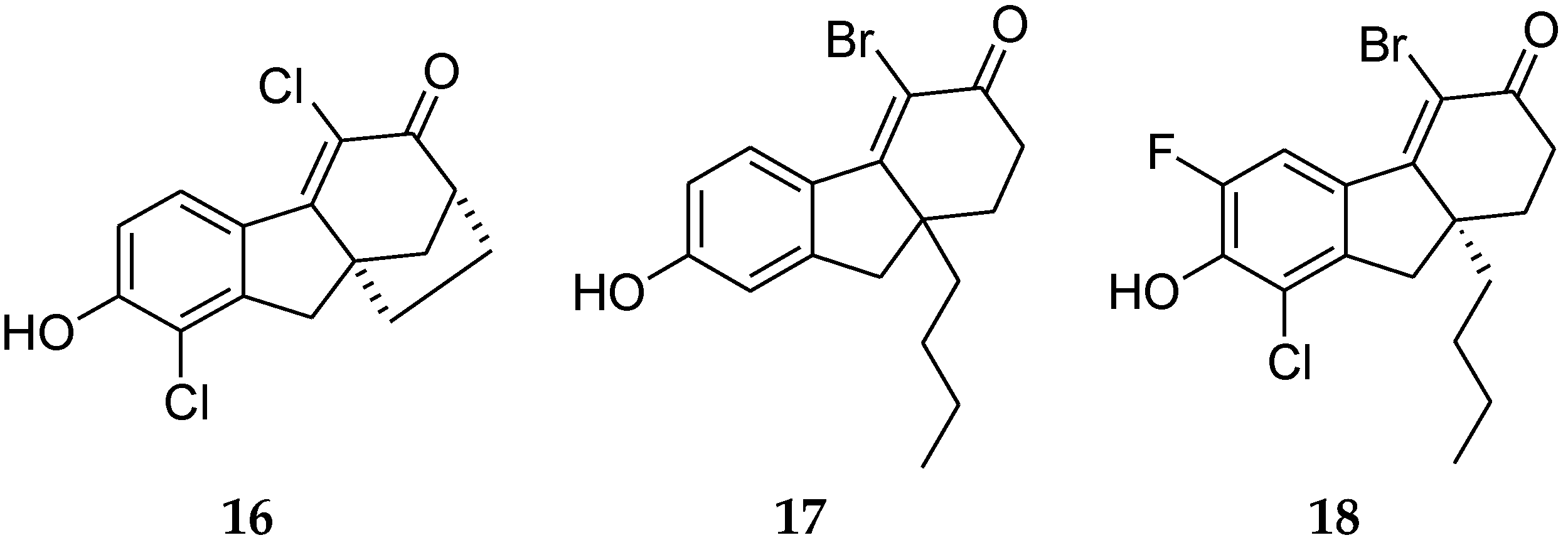
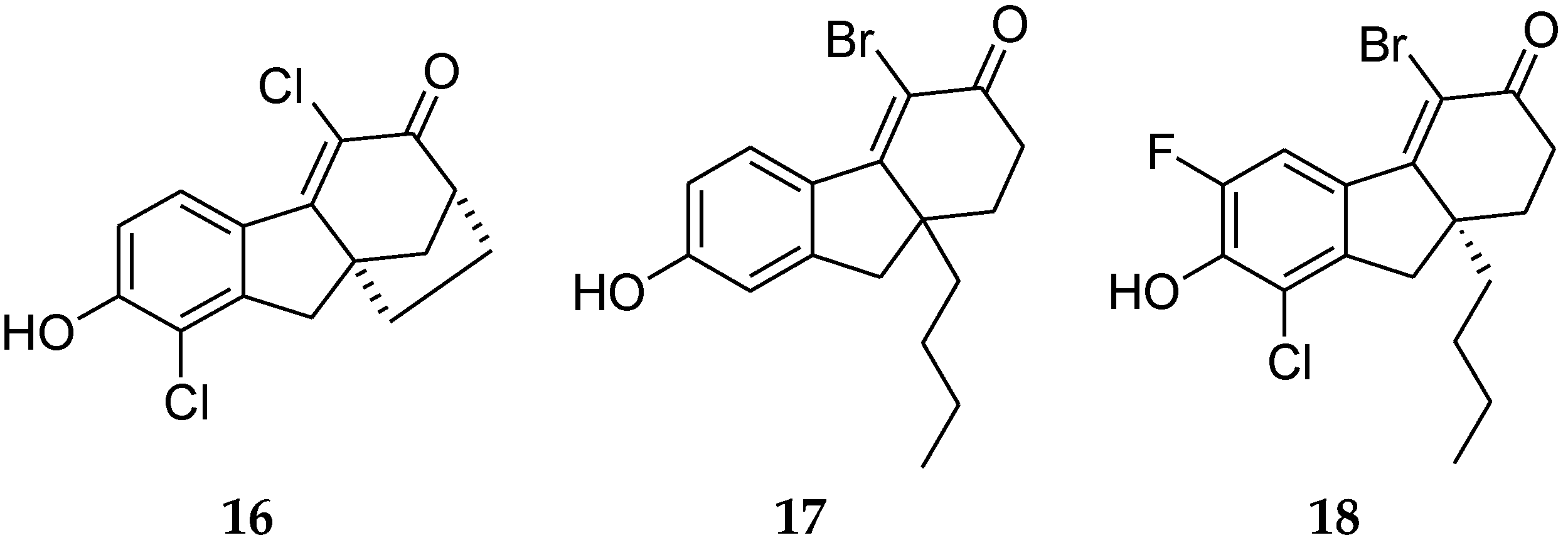
4.5. Fluoren-3-one Derivatives
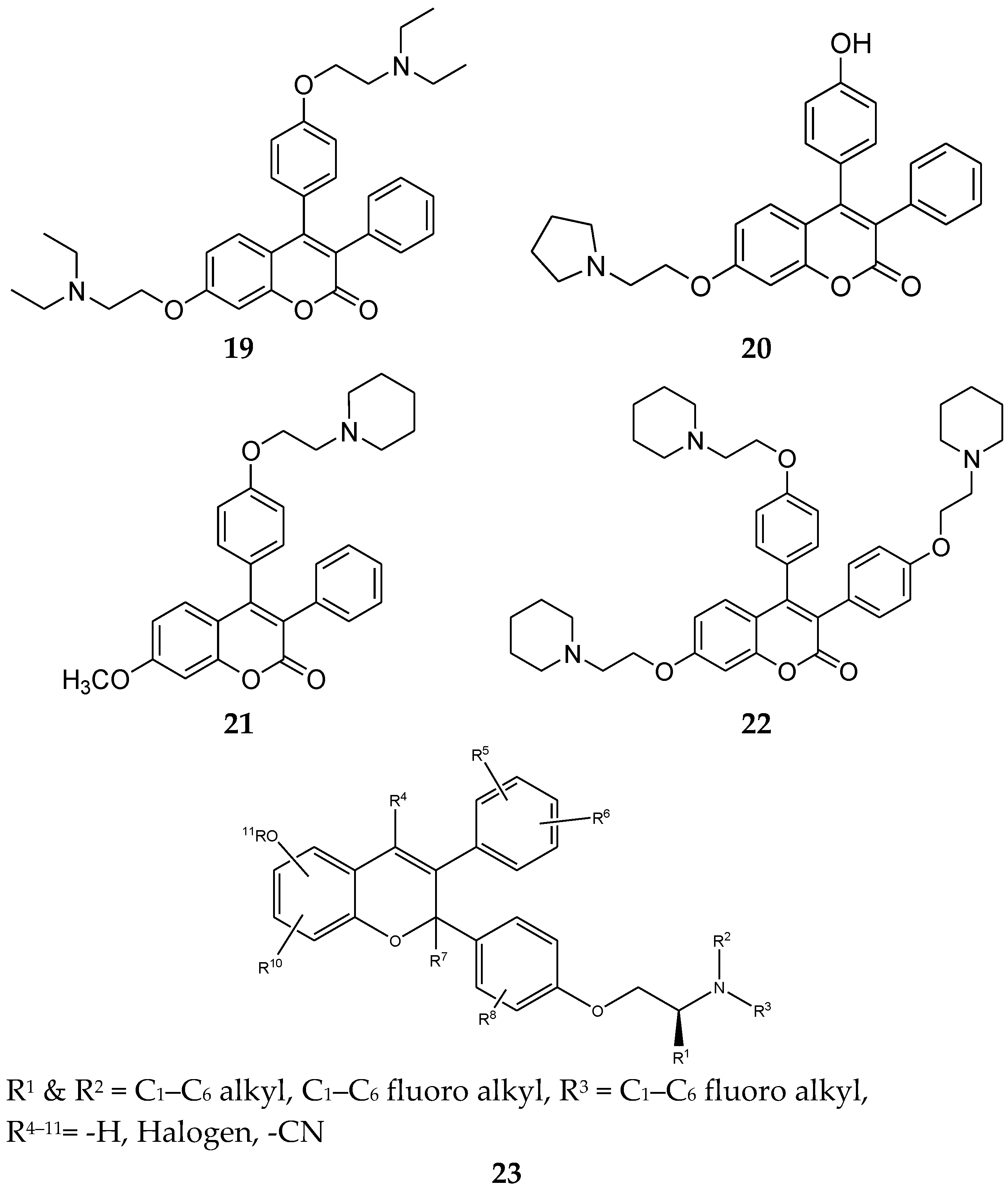
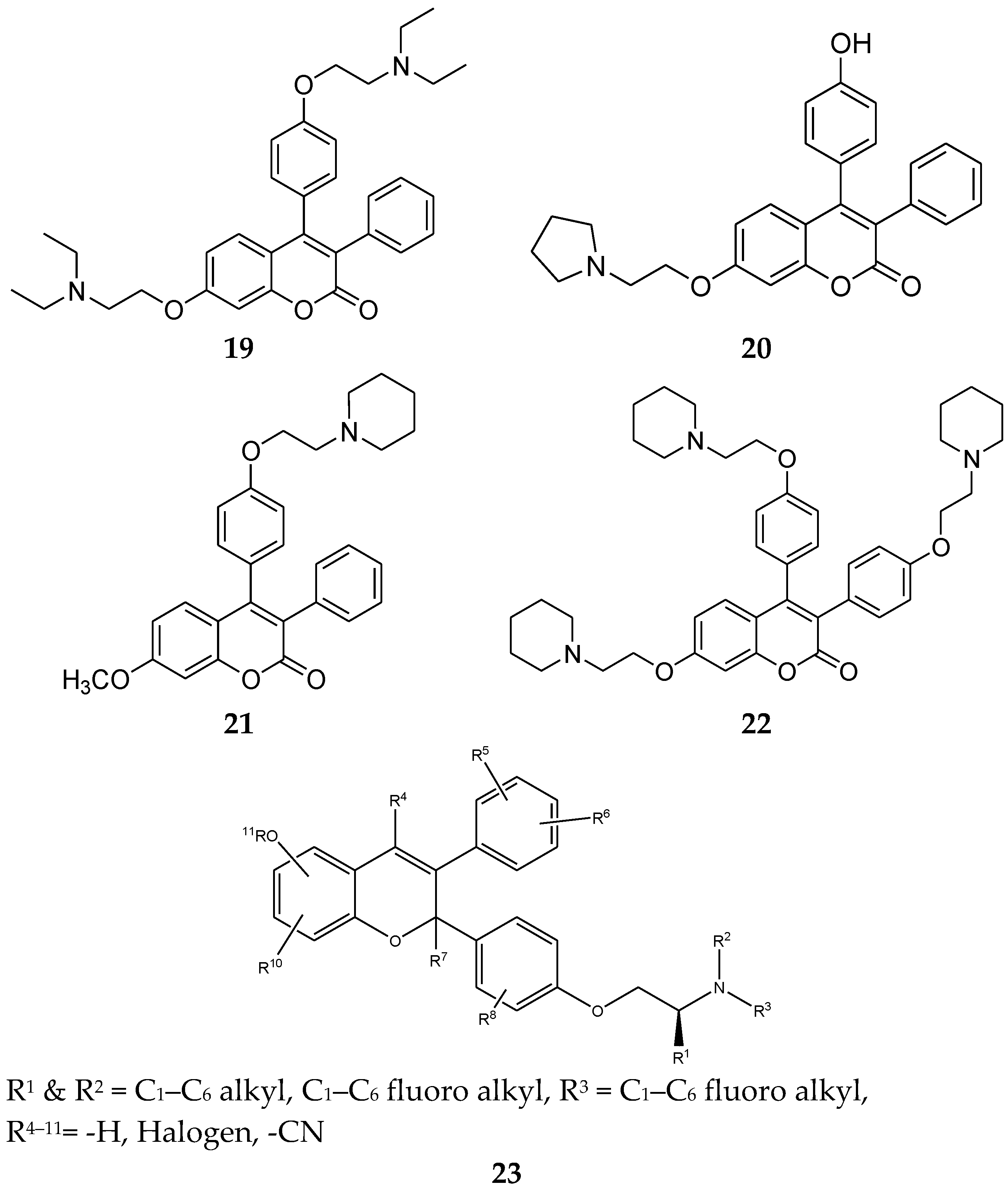
4.6. Triphenylethylene Coumarin
4.7. Combination of Anti-Estrogen and Receptor Tyrosine Kinase (RTK) Inhibitors
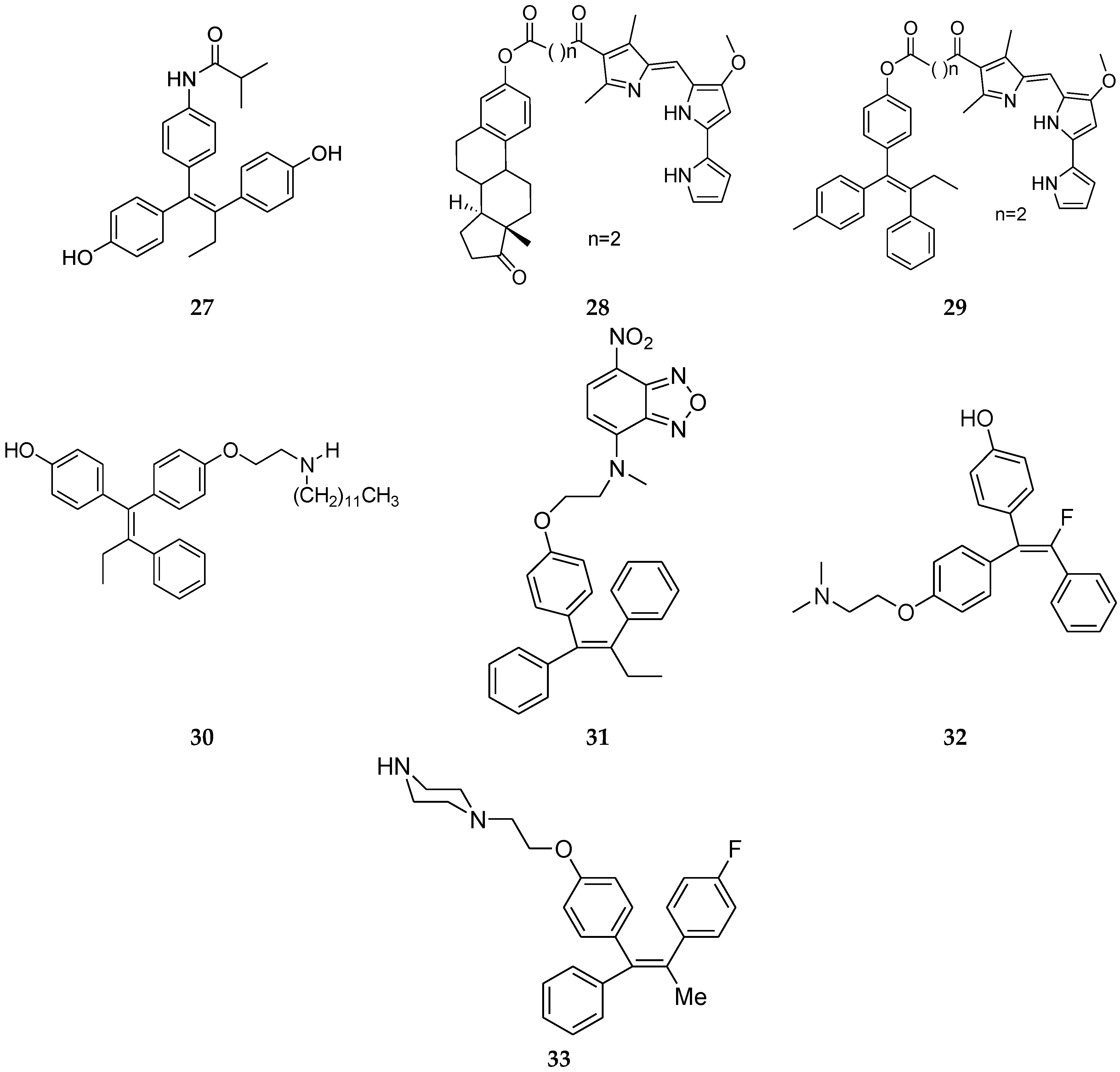
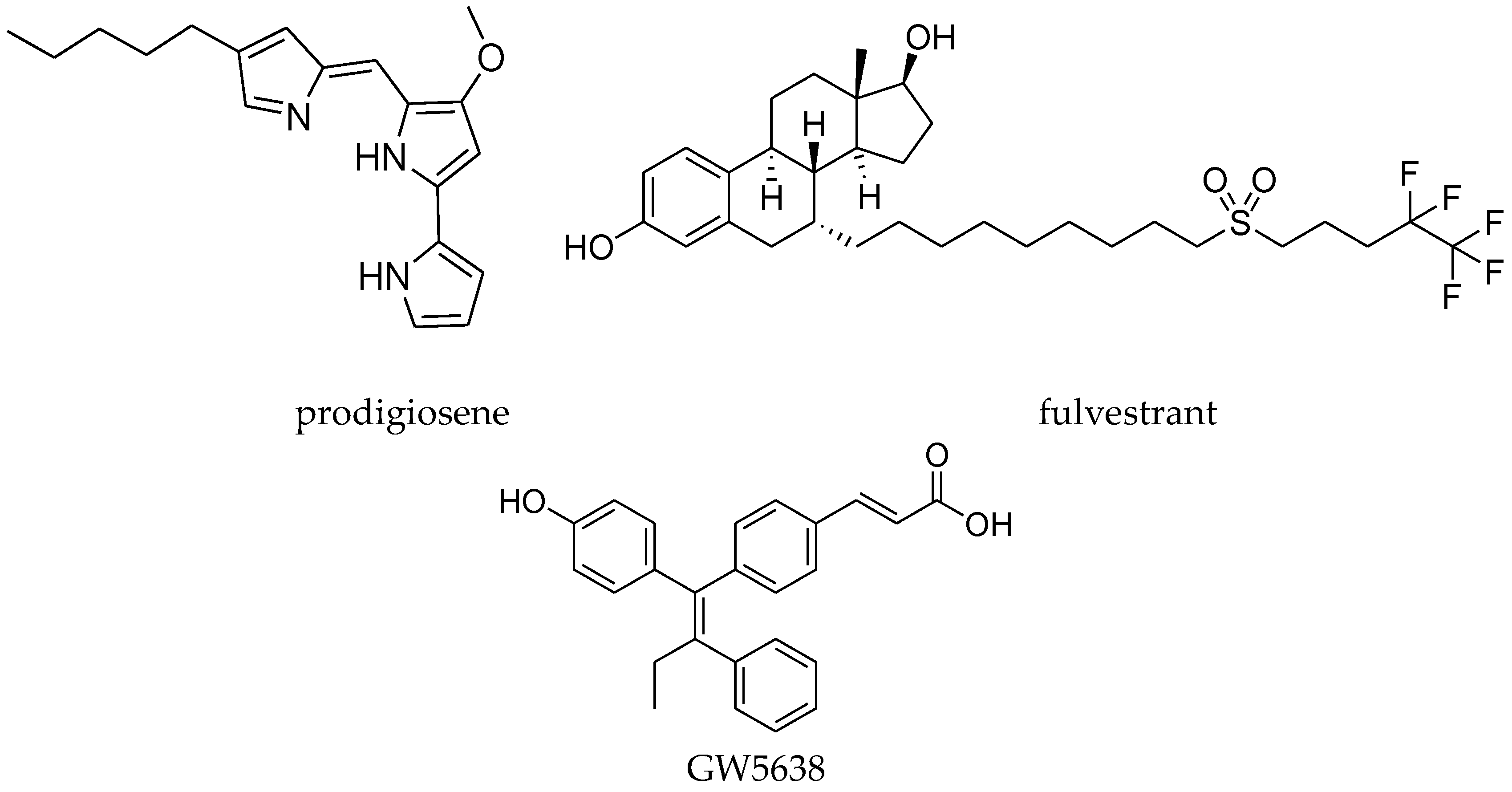
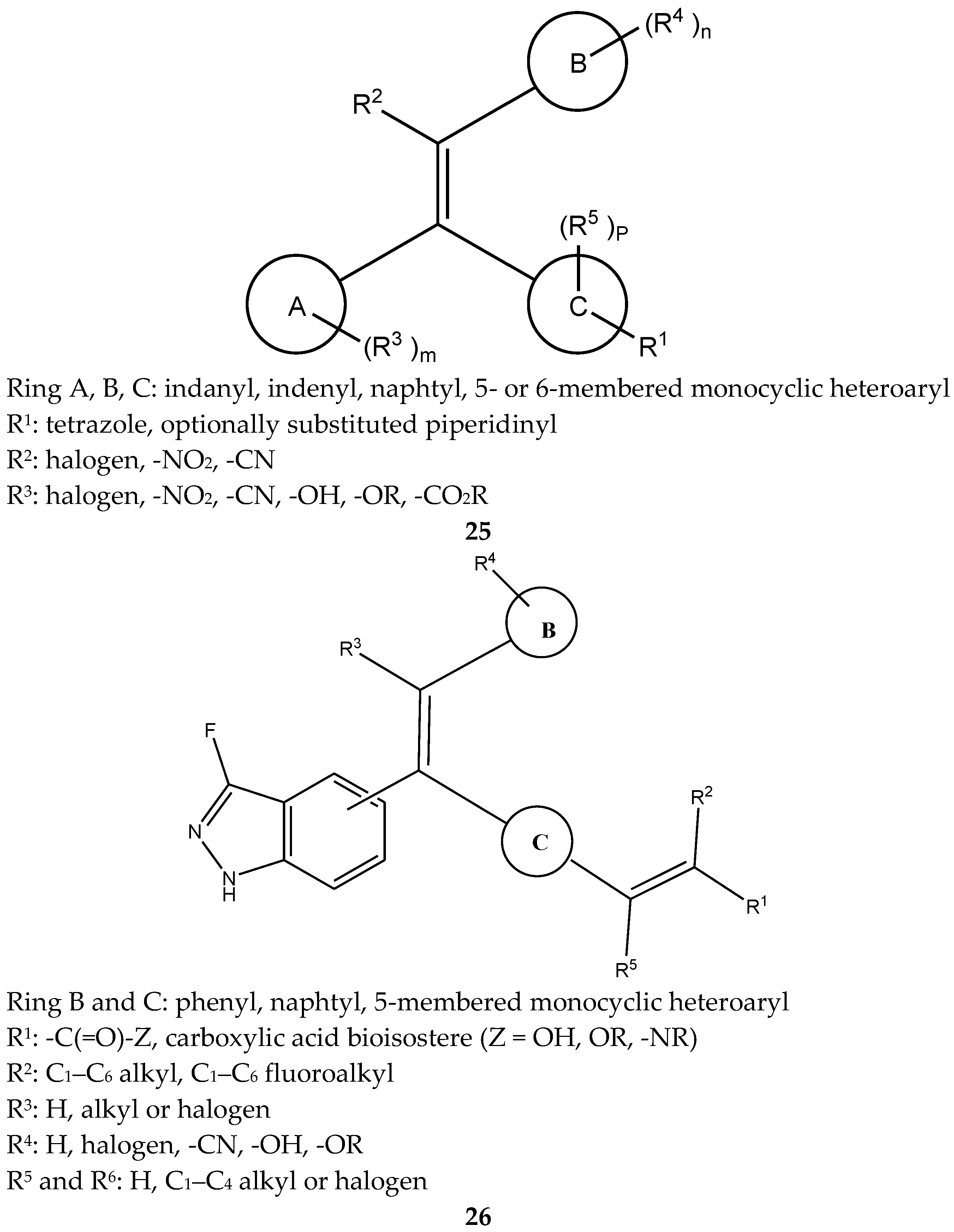
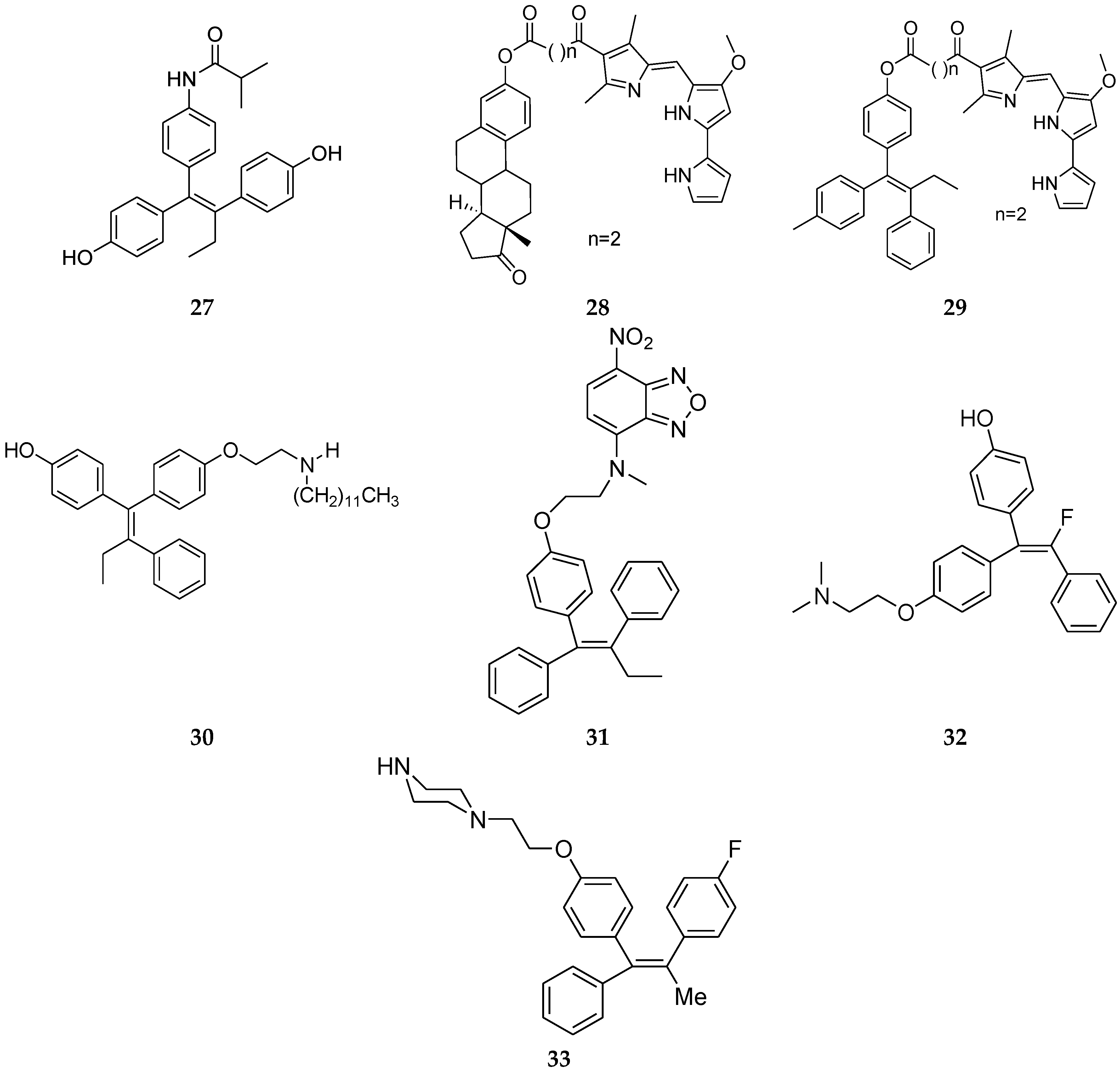
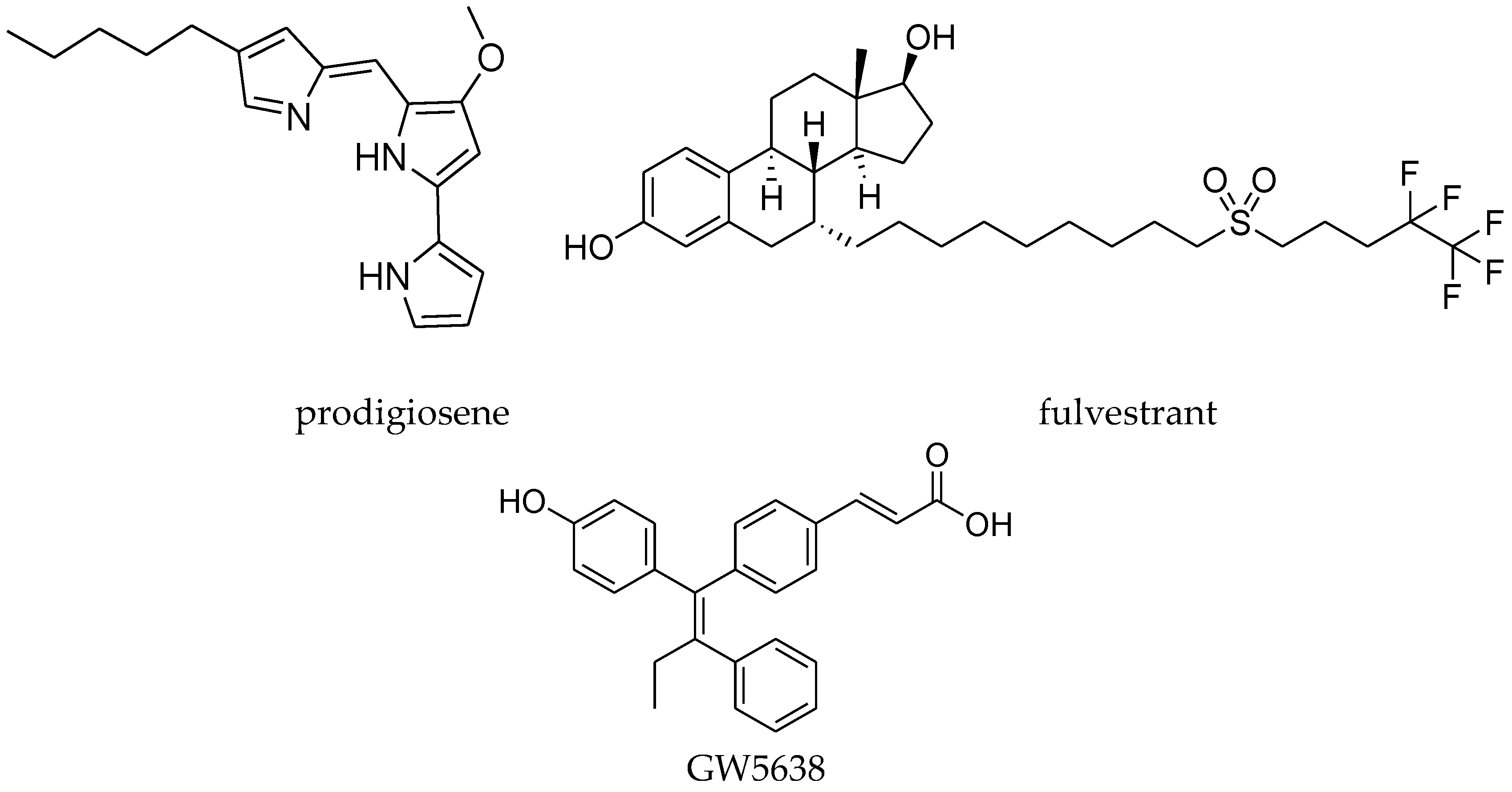
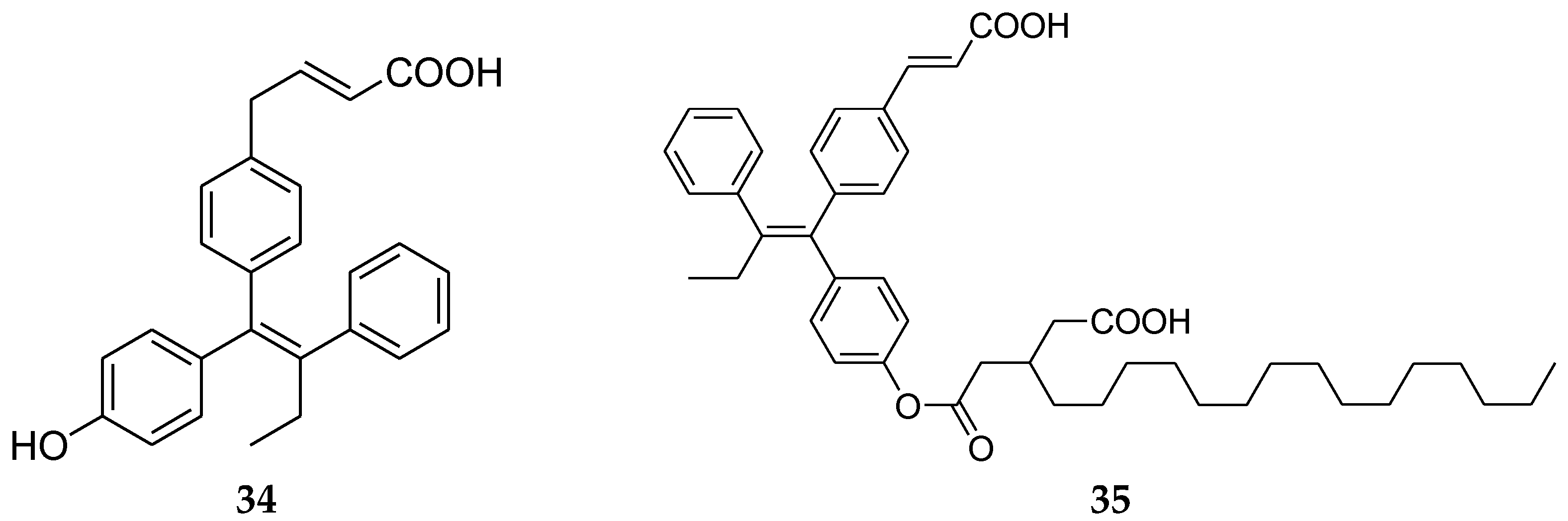
4.8. 1,1,2-Triarylolefine Derivatives
4.9. Aptamer Modulators of Estrogen Receptors
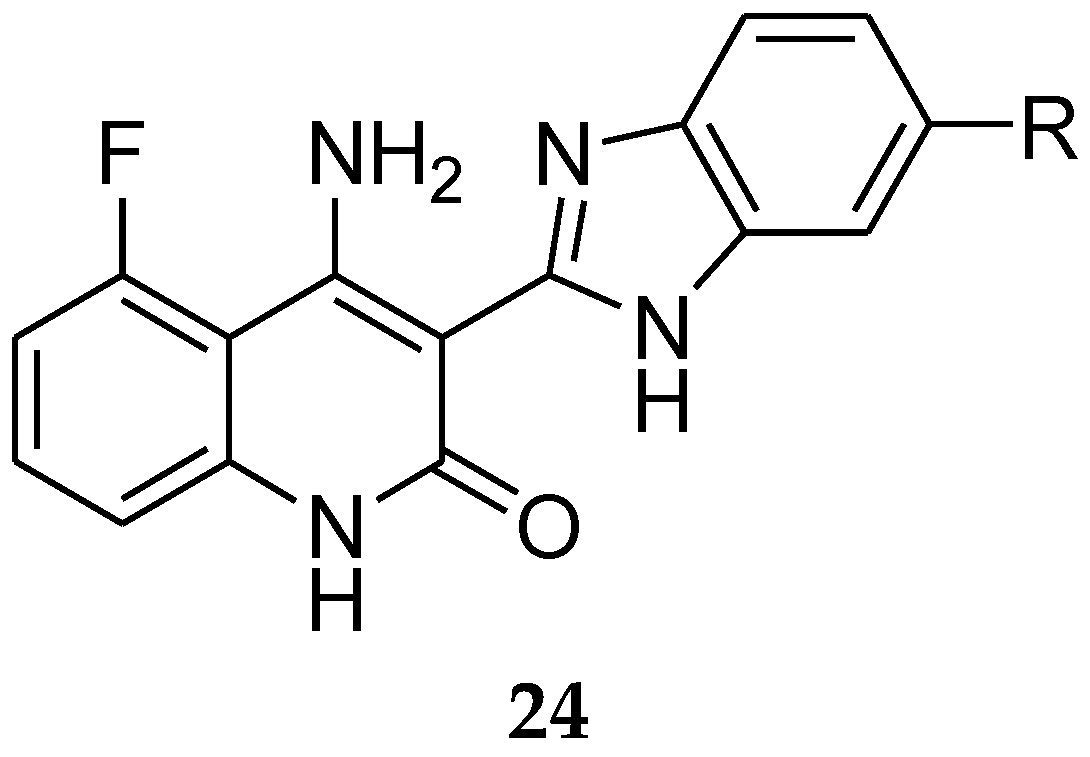
5. Tamoxifen Analogues
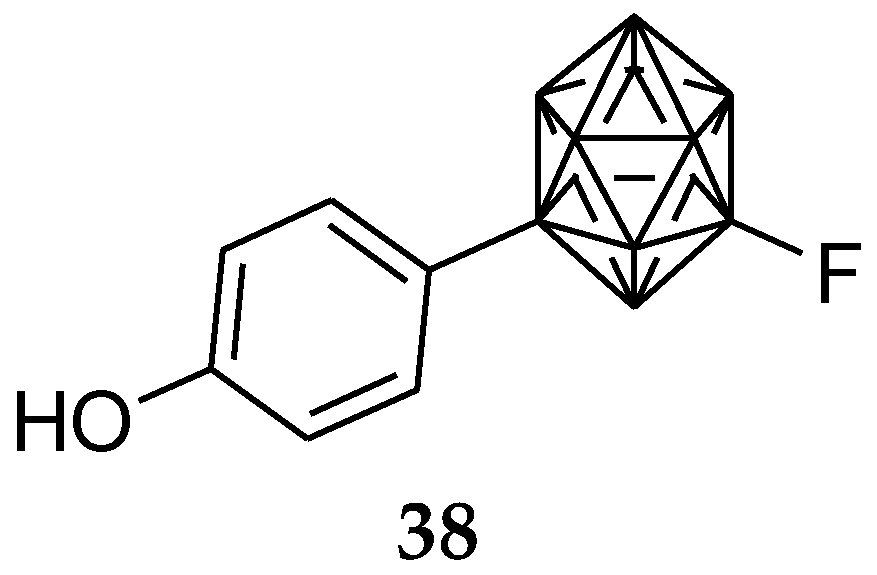
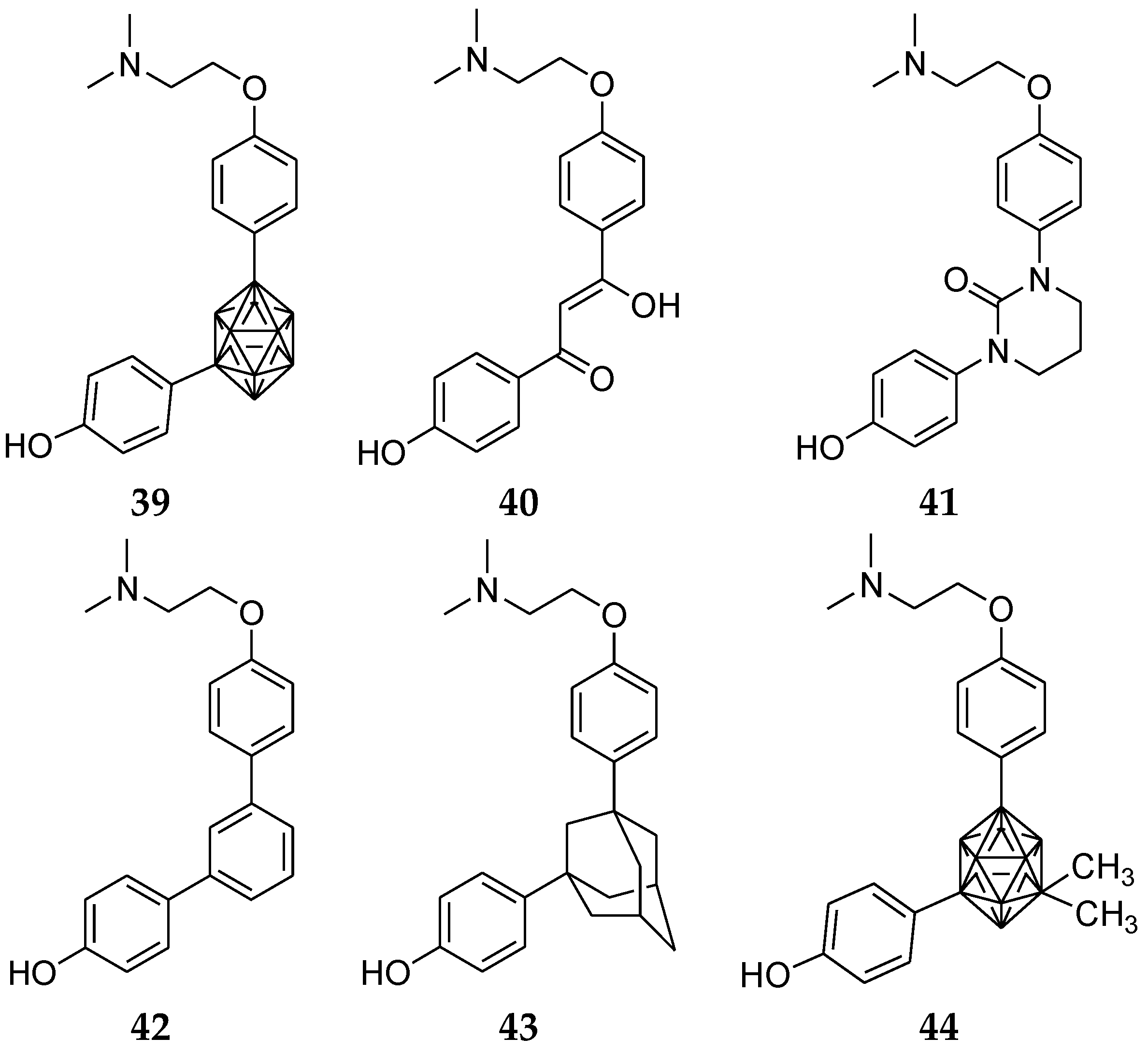
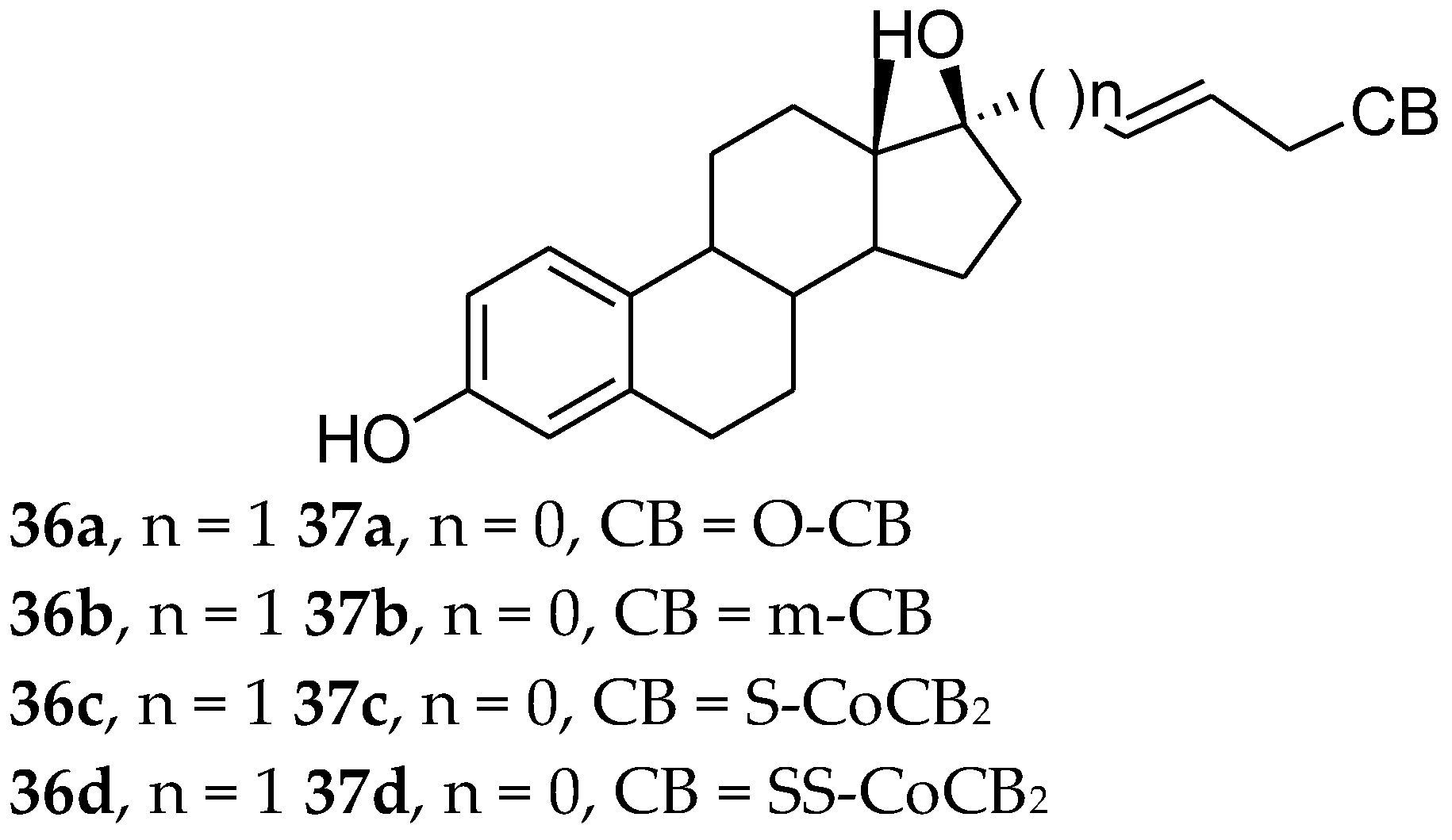
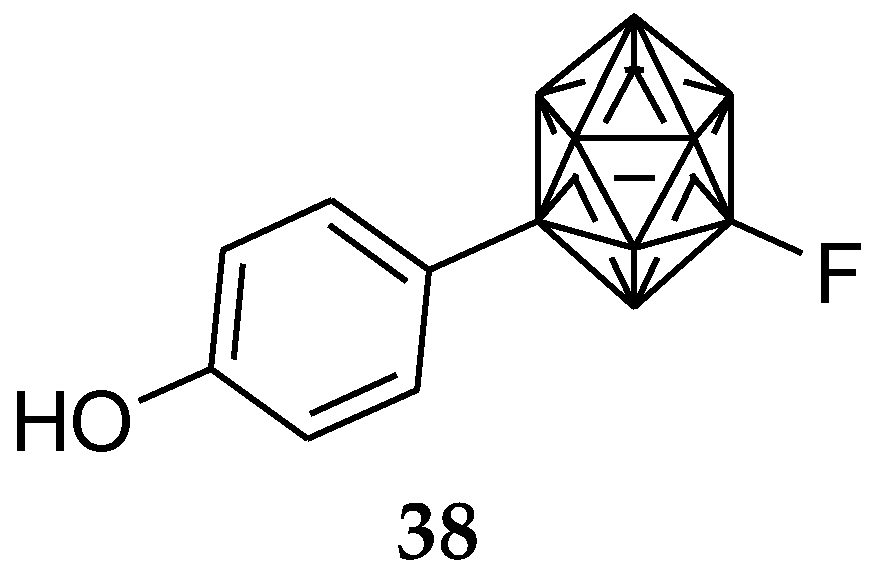
6. Carborane Analogues
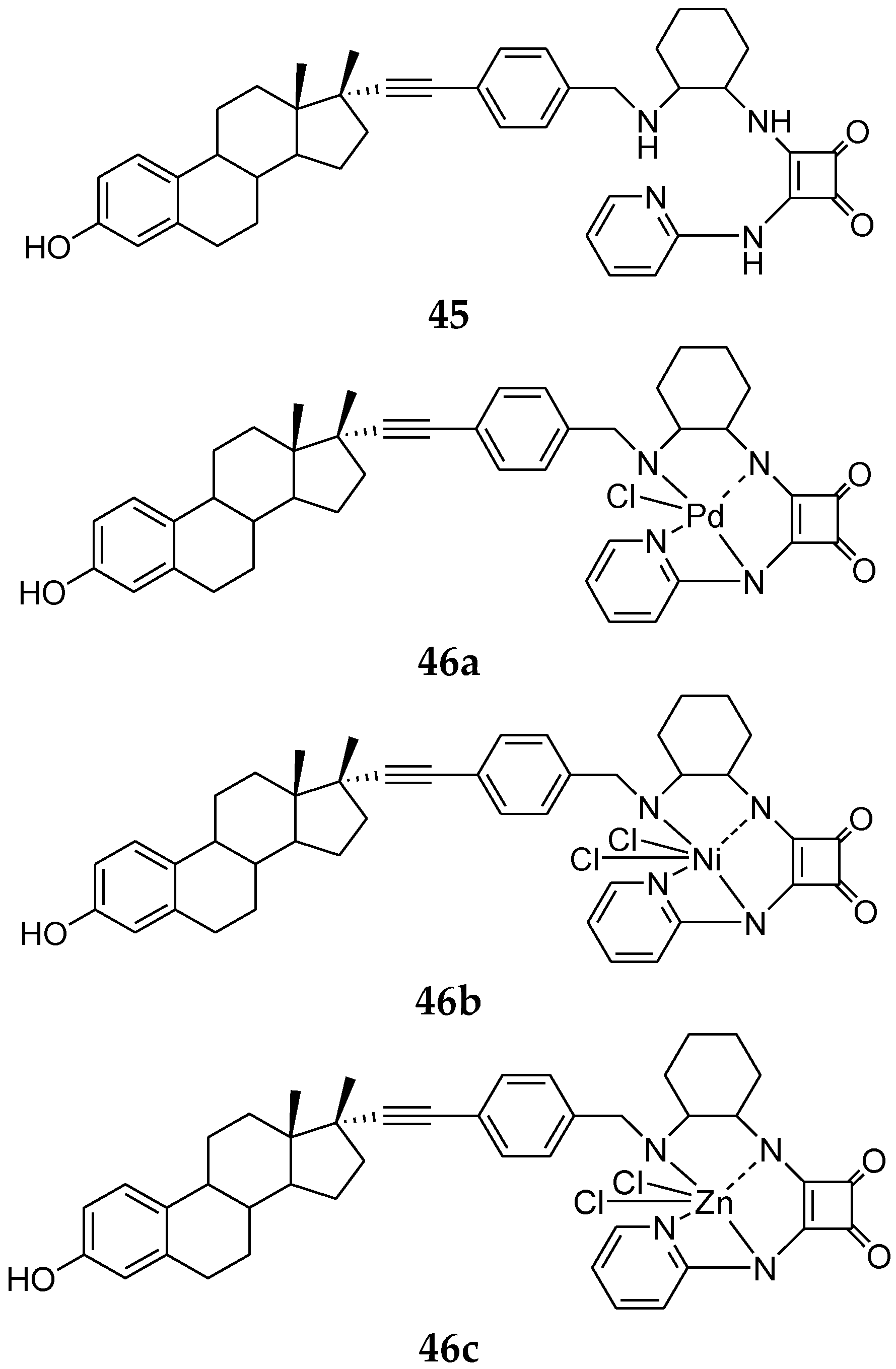
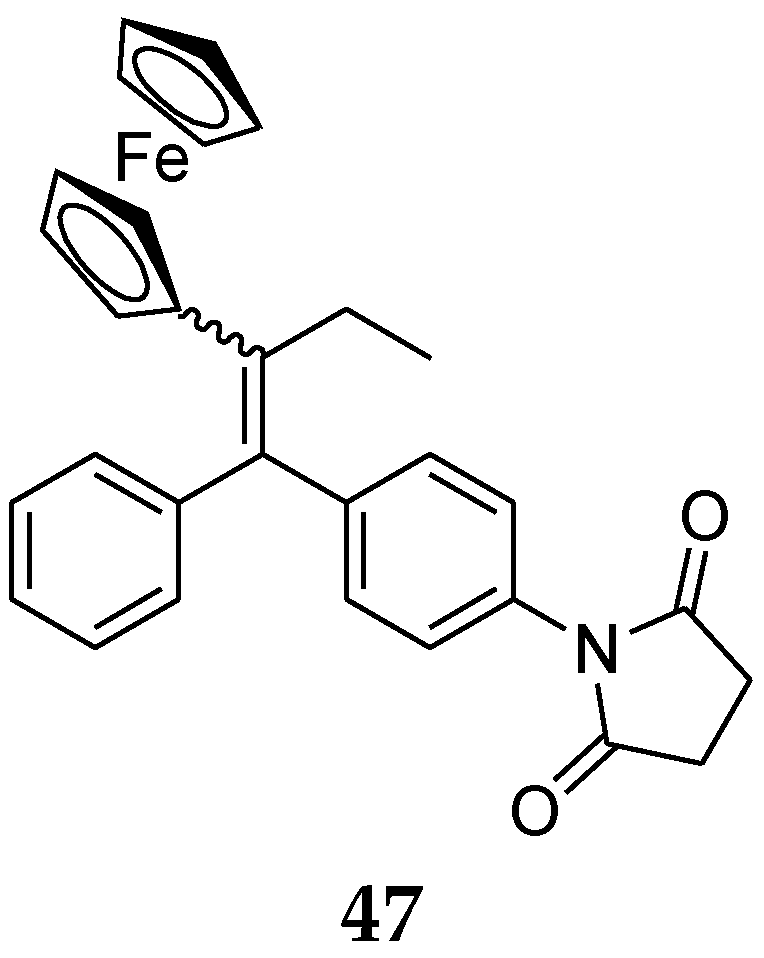
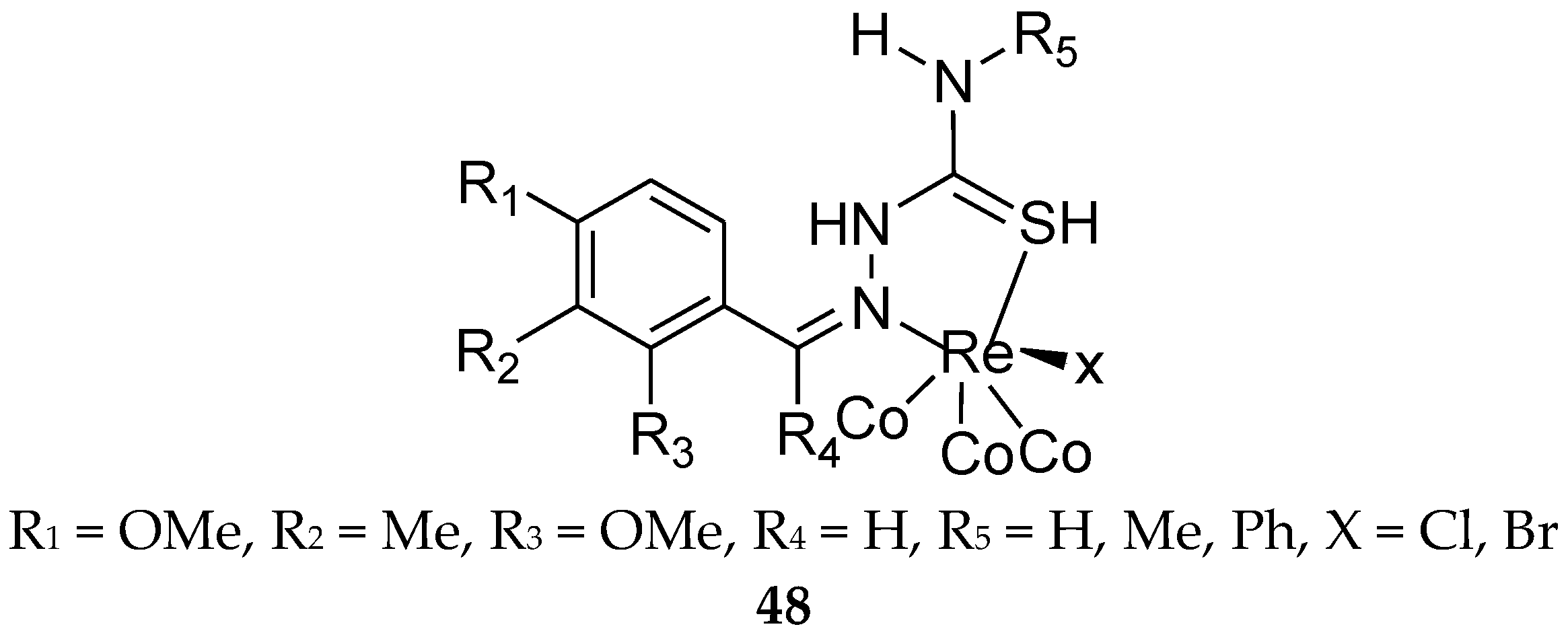
7. Metal Complexes
8. Conclusions
Author Contributions
Conflicts of Interest
References
- Maximov, P.Y.; Lee, T.M.; Jordan, V.C. The discovery and development of selective estrogen receptor modulators (SERMs) for clinical practice. Curr. Clin. Pharmacol. 2013, 8, 135. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.C. Selective estrogen receptor modulation: A personal perspective. Cancer Res. 2001, 61, 5683–5687. [Google Scholar] [CrossRef]
- Paterni, I.; Granchi, C.; Katzenellenbogen, J.A.; Minutolo, F. Estrogen receptors alpha (ERα) and beta (ERβ): Subtype-selective ligands and clinical potential. Steroids 2014, 90, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.C.; Frasor, J.; Komm, B.; Katzenellenbogen, B.S. Impact of estrogen receptor β on gene networks regulated by estrogen receptor α in breast cancer cells. Endocrinology 2006, 147, 4831–4842. [Google Scholar] [CrossRef] [PubMed]
- Archer, J. Estrogen and mood changes via CNS activity. Menopausal Med. 1999, 7, 4–8. [Google Scholar]
- Walf, A.A.; Frye, C.A. Estradiol reduces anxiety-and depression-like behavior of aged female mice. Physiol. Behav. 2010, 99, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Vitale, C.; Mendelsohn, M.E.; Rosano, G.M. Gender differences in the cardiovascular effect of sex hormones. Nat. Rev. Cardiol. 2009, 6, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Guetta, V.; Cannon, R.O. Cardiovascular effects of estrogen and lipid-lowering therapies in postmenopausal women. Circulation 1996, 93, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, S.; Gustafsson, J.Å. Estrogen receptors: Therapies targeted to receptor subtypes. Clin. Pharmacol. Ther. 2011, 89, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Lerner, L.J.; Jordan, V.C. Development of antiestrogens and their use in breast cancer: Eighth Cain memorial award lecture. Cancer Res. 1990, 50, 4177–4189. [Google Scholar] [PubMed]
- Jordan, V.C. Antiestrogens and selective estrogen receptor modulators as multi-functional medicines 1. Receptor interactions. J. Med. Chem. 2003, 46, 883–908. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.C. Antiestrogens and selective estrogen receptor modulators as multi-functional medicines 2. Clinical considerations and new agents. J. Med Chem. 2003, 46, 1081–1111. [Google Scholar] [CrossRef]
- Minutolo, F.; Macchia, M.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Estrogen receptor β ligands: Recent advances and biomedical applications. Med. Res. Rev. 2011, 31, 364–442. [Google Scholar] [CrossRef] [PubMed]
- Paterni, I.; Bertini, S.; Granchi, C.; Macchia, M.; Minutolo, F. Estrogen receptor ligands: A patent review update. Expert Opin. Ther. Pat. 2013, 23, 1247–1271. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, S.; Koehler, K.F.; Gustafsson, J.-Å. Development of subtype-selective oestrogen receptor-based therapeutics. Nat. Rev. Drug Discov. 2011, 10, 778–792. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-X.; Labaree, D.C.; Hochberg, R.B. Synthesis of 11β-ether-17α-ethinyl-3,17β-estradiols with strong ER antagonist activities. Chin. Chem. Lett. 2014, 25, 567–570. [Google Scholar] [CrossRef]
- Griggs, J.; Metcalfe, J.C.; Hesketh, R. Targeting tumour vasculature: The development of combretastatin A4. Lancet Oncol. 2001, 2, 82–87. [Google Scholar] [CrossRef]
- Parihar, S.; Kumar, A.; Chaturvedi, A.K.; Sachan, N.K.; Luqman, S.; Changkija, B.; Manohar, M.; Prakash, O.; Chanda, D.; Khan, F.; et al. Synthesis of combretastatin A4 analogues on steroidal framework and their anti-breast cancer activity. J. Steroid Biochem. Mol. Biol. 2013, 137, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Haines, D.R.; Tseng, C.K.; Marquez, V.E. Synthesis and biological activity of unsaturated carboacyclic purine nucleoside analogues. J. Med. Chem. 1987, 30, 943–947. [Google Scholar] [CrossRef] [PubMed]
- Huryn, D.M.; Okada, M. AIDS-driven nucleoside chemistry. Chem. Rev. 1992, 92, 1745–1768. [Google Scholar] [CrossRef]
- Tuncbilek, M.; Guven, E.B.; Onder, T.; Cetin Atalay, R. Synthesis of novel 6-(4-substituted piperazine-1-yl)-9-(b-d-ribofuranosyl) purine derivatives, which lead to senescence-induced cell death in liver cancer cells. J. Med. Chem. 2012, 55, 3058–3065. [Google Scholar] [CrossRef] [PubMed]
- Norman, T.C.; Gray, N.S.; Koh, J.T.; Schultz, P.G. A structure-based library approach to kinase inhibitors. J. Am. Chem. Soc. 1996, 118, 7430–7431. [Google Scholar] [CrossRef]
- Legraverend, M.; Grierson, D.S. The purines: Potent and versatile small molecule inhibitors and modulators of key biological targets. Bioorg. Med. Chem. 2006, 14, 3987–4006. [Google Scholar] [CrossRef] [PubMed]
- Bressi, J.C.; Choe, J.; Hough, M.T.; Buckner, F.S.; VanVoorhis, W.C.; Verlinde, C.L.; Hol, W.G.; Gelb, M.H. Adenosine analogues as inhibitors of Trypanosoma brucei phosphoglycerate kinase: Elucidation of a novel binding mode for a 2-amino-N6-substituted adenosine. J. Med. Chem. 2000, 43, 4135–4150. [Google Scholar] [CrossRef] [PubMed]
- Bavetsias, V.; Large, J.M.; Sun, C.; Bouloc, N.; Kosmopoulou, M.; Matteucci, M.; Wilsher, N.E.; Martins, V.; Reynisson, J.; Atrash, B.; et al. Imidazo[4,5-b]pyridine derivatives as inhibitors of aurora kinases: Lead optimization studies toward the identification of an orally bioavailable preclinical development candidate. J. Med. Chem. 2010, 53, 5213–5228. [Google Scholar] [CrossRef] [PubMed]
- Boumendjel, A.; Nicolle, E.; Moraux, T.; Gerby, B.; Blanc, M.; Ronot, X.; Boutonnat, J. Piperazino-benzopyranones and phenalkylaminobenzopyranones: Potent inhibitors of breast cancer resistance protein (ABCG2). J. Med. Chem. 2005, 48, 7275–7281. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xu, H.; Yang, Z.; Zheng, Y.; Liu, H. Synthesis and anticancer activity of novel C6-piperazine substituted purine steroid-nucleosides analogues. Steroids 2014, 82, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, Y.; Xu, H.; Zheng, Y.; Liu, H. Synthesis and biological evaluation of novel C6-cyclo secondary amine substituted purine steroid-nucleosides analogues. Steroids 2014, 85, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, D.; Rivera, G.; Sanchez, J.L.; Rivera, J.; Granados, J.C.; Guerrero, A.M.; Chang, F.-M.; Dearth, R.K.; Short, J.D.; Banik, B.K. Bismuth nitrate-induced novel nitration of estradiol: An entry to new anticancer agents. Eur. J. Med. Chem. 2014, 82, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Longatto Filho, A.; Lopes, J.M.; Schmitt, F.C. Angiogenesis and breast cancer. J. Oncol. 2010, 2010, 576384. [Google Scholar] [CrossRef]
- Tang, Z.; Niu, S.; Liu, F.; Lao, K.; Miao, J.; Ji, J.; Wang, X.; Yan, M.; Zhang, L.; You, Q.; et al. Synthesis and biological evaluation of 2,3-diaryl isoquinolinone derivatives as anti-breast cancer agents targeting ERα and VEGFR-2. Bioorg. Med. Chem. Lett. 2014, 24, 2129–2133. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, A.; Liu, X.; Okada, H.; Shimohigashi, M.; Shimohigashi, Y. Bisphenol AF is a full agonist for the estrogen receptor ERalpha but a highly specific antagonist for ERbeta. Environ. Health Perspect. 2010, 118, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Sumbayev, V.V.; Jensen, J.K.; Hansen, J.A.; Andreasen, P.A. Novel modes of oestrogen receptor agonism and antagonism by hydroxylated and chlorinated biphenyls, revealed by conformation-specific peptide recognition patterns. Mol. Cell. Endocrinol. 2008, 287, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Showell, G.A.; Mills, J.S. Chemistry challenges in lead optimization: Silicon isosteres in drug discovery. Drug Discov. Today 2003, 8, 551–556. [Google Scholar] [CrossRef]
- Franz, A.K.; Wilson, S.O. Organosilicon molecules with medicinal applications. J. Med. Chem. 2012, 56, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Miyajima, Y.; Masuno, H.; Kagechika, H. Increased Hydrophobicity and Estrogenic Activity of Simple Phenols with Silicon and Germanium-Containing Substituents. J. Med. Chem. 2012, 56, 160–166. [Google Scholar] [CrossRef]
- Nakamura, M.; Makishima, M.; Hashimoto, Y. Development of silicon-containing bis-phenol derivatives as androgen receptor antagonists: Selectivity switching by C/Si exchange. Bioorg. Med. Chem. 2013, 21, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; Nakamura, M.; Tomoshige, S.; Sugita, K.; Makishima, M.; Hashimoto, Y.; Ishikawa, M. Structure–activity relationships of bisphenol A analogs at estrogen receptors (ERs): Discovery of an ERα-selective antagonist. Bioorg. Med. Chem. Lett. 2013, 23, 4031–4036. [Google Scholar] [CrossRef] [PubMed]
- Ohta, K.; Chiba, Y.; Kaise, A.; Endo, Y. Structure–activity relationship study of diphenylamine-based estrogen receptor (ER) antagonists. Bioorg. Med. Chem. 2015, 23, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekharan, S.; Bhaskar, B.; Muthiah, R.; Chandrasekharan, A.K.; Ramamurthy, V. Estrogenic effect of three substituted deoxybenzoins. Steroids 2013, 78, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Forman, B.; Yu, D. Ligands for Estrogen Related Receptors and Methods for Synthesis of Said Ligands. U.S. Patent 20,130,131,340, 23 May 2013. [Google Scholar]
- Wallace, D.J.; Reamer, R.A. New synthesis of a selective estrogen receptor modulator using an enatioselective phosphine-mediated 2 + 3 cycloaddition. Tetrahedron Lett. 2013, 54, 4425–4428. [Google Scholar] [CrossRef]
- Clark, J.A.; Alves, S.; Gundlah, C.; Rocha, B.; Birzin, E.T.; Cai, S.J.; Flick, R.; hayes, E.; Ho, K.; Warrier, S.; et al. Selective estrogen receptor-beta (SERM-beta) compounds modulate raphe nuclei tryptophan hydroxylase-1 (TPH-1) mRNA expression and cause antidepressant-like effects in the forced swim test. Neuropharmacology 2012, 63, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, S.; Yao, Y.; Zhou, L.; Zhao, J.; Gu, Y.; Wang, K.; Li, X. Design, synthesis, and anti-tumor activities of novel triphenylethylene–coumarin hybrids, and their interactions with Ct-DNA. Bioorg. Med. Chem. Lett. 2013, 23, 4785–4789. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.D.; Govek, S.P.; Kahraman, M.; Nagasawa, J.Y.; Lai, A.G.; Bonnefous, C. Fluorinated Estrogen Receptor Modulators and Uses Thereof. U.S. Patent 20,150,005,286, 1 January 2015. [Google Scholar]
- Yovine, A.; Squires, M.; Reddick, C.; Zhang, Y. Combination of a RTK Inhibitor with an Anti-Estrogen and Use Thereof for the Treatment of Cancer. Patent WO2,013,116,293, 8 August 2013. [Google Scholar]
- Smith, N.D.; Kahraman, M.; Govek, S.P.; Nagasawa, J.Y.; Lai, A.G.; Julien, J.D.; Herbert, M.R.; Bonnefous, C.; Douglas, K.L. Estrogen Receptor Modulators and Uses Thereof. U.S. Patent 20,130,231,333, 5 September 2013. [Google Scholar]
- Smith, N.D.; Govek, S.P.; Kahraman, M.; Julien, J.D.; Nagasawa, J.Y.; Douglas, K.L.; Bonnefous, C.; Lai, A.G. Estrogen Receptor Modulators and Uses Thereof. U.S. Patent 20,150,105,403, 28 January 2015. [Google Scholar]
- Hager, J.H.; Chow maneval, E.; Smith, N.D.; Friedman, L.; Sampath, D. Therapeutic Combinations with Estrogen Receptor Modulators. Patent WO2,015,136,016, 10 December 2015. [Google Scholar]
- Shi, H.; Xu, D.; Chatakonda, V.K. Aptamer Modulators of Estrogen Receptors. U.S. Patent 20,150,031,754, 29 January 2015. [Google Scholar]
- Christodoulou, M.S.; Fokialakis, N.; Passarella, D.; García-Argáez, A.N.; Gia, O.M.; Pongratz, I.; Dalla Via, L.; Haroutounian, S.A. Synthesis and biological evaluation of novel tamoxifen analogues. Bioorg. Med. Chem. 2013, 21, 4120–4131. [Google Scholar] [CrossRef] [PubMed]
- Fürstner, A. Chemistry and biology of roseophilin and the prodigiosin alkaloids: A survey of the last 2500 years. Angew. Chem. Int. Ed. 2003, 42, 3582–3603. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.; Chander, R.; Sainis, K. Prodigiosins as anti cancer agents: Living upto their name. Curr. Pharm. Des. 2009, 15, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Mazza, M.; Uchegbu, I.; Schätzlein, A. Cancer and the blood–brain barrier: ‘Trojan horses’ for courses? Br. J. Pharmacol. 2008, 155, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Spradau, T.W.; Katzenellenbogen, J.A. Ligands for the estrogen receptor, containing cyclopentadienyltricarbonylrhenium units. Bioorg. Med. Chem. Lett. 1998, 8, 3235–3240. [Google Scholar] [CrossRef]
- Perron, V.; Rabouin, D.; Asselin, É.; Parent, S.; C-Gaudreault, R.; Bérubé, G. Synthesis of 17β-estradiol-linked platinum (II) complexes and their cytocidal activity on estrogen-dependent and-independent breast tumor cells. Bioorg. Chem. 2005, 33, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Van Themsche, C.; Parent, S.; Leblanc, V.; Descôteaux, C.; Simard, A.-M.; Bérubé, G.; Asselin, E. VP-128, a novel oestradiol-platinum (II) hybrid with selective anti-tumour activity towards hormone-dependent breast cancer cells in vivo. Endocr.-Relat. Cancer 2009, 16, 1185–1195. [Google Scholar] [CrossRef] [PubMed]
- Provencher-Mandeville, J.; Debnath, C.; Mandal, S.K.; Leblanc, V.; Parent, S.; Asselin, É.; Bérubé, G. Design, synthesis and biological evaluation of estradiol-PEG-linked platinum (II) hybrid molecules: Comparative molecular modeling study of three distinct families of hybrids. Steroids 2011, 76, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Cyrus, K.; Wehenkel, M.; Choi, E.Y.; Lee, H.; Swanson, H.; Kim, K.B. Jostling for Position: Optimizing Linker Location in the Design of Estrogen Receptor-Targeting PROTACs. Chem. Med. Chem. 2010, 5, 979–985. [Google Scholar] [CrossRef]
- Gacio, A.F.; Fernandez-Marcos, C.; Swamy, N.; Dunn, D.; Ray, R. Photodynamic cell-kill analysis of breast tumor cells with a tamoxifen-pyropheophorbide conjugate. J. Cell. Biochem. 2006, 99, 665–670. [Google Scholar] [CrossRef] [PubMed]
- El Amouri, H.; Vessieres, A.; Vichard, D.; Top, S.; Gruselle, M.; Jaouen, G. Syntheses and affinities of novel organometallic-labeled estradiol derivatives: A structure-affinity relationship. J. Med. Chem. 1992, 35, 3130–3135. [Google Scholar] [CrossRef]
- Top, S.; El Hafa, H.; Vessieres, A.; Quivy, J.; Vaissermann, J.; Hughes, D.W.; McGlinchey, M.J.; Mornon, J.-P.; Thoreau, E.; Jaouen, G. Rhenium Carbonyl Complexes of β-Estradiol Derivatives with High Affinity for the Estradiol Receptor: An Approach to Selective OrganometallicRadiopharmaceuticals. J. Am. Chem. Soc. 1995, 117, 8372–8380. [Google Scholar] [CrossRef]
- Tang, J.; Top, S.; Vessières, A.; Sellier, N.; Vaissermann, J.; Jaouen, G. Synthesis of 17α-ruthenocenyl-17β-oestradiol and its potential as a radiopharmaceutical agent. Appl. Organomet. Chem. 1997, 11, 771–781. [Google Scholar] [CrossRef]
- Hawco, C.L.A.; Marchal, E.; Uddin, M.I.; Baker, A.E.G.; Corkery, D.P.; Dellaire, G.; Thompson, A. Synthesis and biological evaluation of prodigiosene conjugates of porphyrin, estrone and 4-hydroxytamoxifen. Bioorg. Med. Chem. 2013, 21, 5995–6002. [Google Scholar] [CrossRef] [PubMed]
- Howell, S.J.; Johnston, S.R.D.; Howell, A. The use of selective estrogen receptor modulators and selective estrogen receptor down-regulators in breast cancer. Best Pract. Res. Clin. Endocrinol. Metab. 2004, 18, 47–66. [Google Scholar] [CrossRef] [PubMed]
- Baumann, C.K.; Castiglione-Gertsch, M. Estrogen Receptor Modulators and Down Regulators. Drugs 2007, 67, 2335–2353. [Google Scholar] [CrossRef] [PubMed]
- Buzdar, A.U.; Robertson, J.F. Fulvestrant: Pharmacologic profile versus existing endocrine agents for the treatment of breast cancer. Ann. Pharmacother. 2006, 40, 1572–1583. [Google Scholar] [CrossRef]
- Bross, P.F.; Baird, A.; Chen, G.; Jee, J.M.; Lostritto, R.T.; Morse, D.E.; Rosario, L.A.; Williams, G.M.; Yang, P.; Rahman, A.; et al. Fulvestrant in postmenopausal women with advanced breast cancer. Clin. Cancer Res. 2003, 9, 4309–4317. [Google Scholar] [PubMed]
- Howell, A.; Abram, P. Clinical development of fulvestrant (“Faslodex”). Cancer Treat. Rev. 2005, 31 (Suppl. S2), S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.F.; Howell, A.; Gorbunova, V.; Watanabe, T.; Pienkowski, T.; Lichinitser, M. Sensitivity to further endocrine therapy is retained following progression on first-line fulvestrant. Breast Cancer Res. Treat. 2005, 92, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Willson, T.M.; Henke, B.R.; Momtahen, T.M.; Charifson, P.S.; Batchelor, K.W.; Lubahn, D.B.; Moore, L.B.; Oliver, B.B.; Sauls, H.R. 3-[4-(1,2-Diphenylbut-1-enyl)phenyl]acrylic acid: A non-steroidal estrogen with functional selectivity for bone over uterus in rats. J. Med. Chem. 1994, 37, 1550–1552. [Google Scholar] [CrossRef] [PubMed]
- Shoda, T.; Okuhira, K.; Kato, M.; Demizu, Y.; Inoue, H.; Naito, M.; Kurihara, M. Design and synthesis of tamoxifen derivatives as a selective estrogen receptor down-regulator. Bioorg. Med. Chem. Lett. 2014, 24, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Marrero-Alonso, J.; Morales, A.; García Marrero, B.; Boto, A.; Marín, R.; Cury, D.; Gómez, T.; Fernández-Pérez, L.; Lahoz, F.; Díaz, M. Unique SERM-like properties of the novel fluorescent tamoxifen derivative FLTX1. Eur. J. Pharm. Biopharm. 2013, 85, 898–910. [Google Scholar] [CrossRef] [PubMed]
- Malo-Forest, B.; Landelle, G.; Roy, J.-A.; Lacroix, J.; Gaudreault, R.C.; Paquin, J.-F. Synthesis and growth inhibition activity of fluorinated derivatives of tamoxifen. Bioorg. Med. Chem. Lett. 2013, 23, 1712–1715. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, K.R.A.; Belal, A.; Omar, H.A. Design, synthesis and biological evaluation of novel triaryl (Z)-olefins as tamoxifen analogues. Bioorg. Med. Chem. Lett. 2013, 23, 4960–4963. [Google Scholar] [CrossRef]
- Cho, M.; Hackett, M.; Mossinghoff, G.J. Selective Estrogen Receptor Degraders for Treatment of Tamoxifen Resistant Tumors. Patent WO2,014,008,159, 9 January 2014. [Google Scholar]
- Boivin, R.P.; Luu-The, V.; Lachance, R.; Labrie, F.; Poirier, D. Structure-Activity Relationships of 17α-Derivatives of Estradiol as Inhibitors of Steroid Sulfatase. J. Med. Chem. 2000, 43, 4465–4478. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.; Reddy, B.R.; Delgado, P.; Stotter, P.L.; Fulcher, L.C.; Chamness, G.C. 17 alpha-substituted analogs of estradiol for the development of fluorescent estrogen receptor ligands. Steroids 1991, 56, 375–387. [Google Scholar] [CrossRef]
- Peters, R.H.; Crowe, D.F.; Avery, M.A.; Chong, W.K.; Tanabe, M. 17-Desoxy estrogen analogs. J. Med. Chem. 1989, 32, 1642–1652. [Google Scholar] [CrossRef] [PubMed]
- Vichard, D.; Gruselle, M.; El Amouri, H.; Jaouen, G.; Vaissermann, J. Controlled and selective introduction of a Cp*Ru+ (Cp* = C5Me5) fragment to the 17α-substituent of 17α-phenylestradiol. Organometallics 1992, 11, 2952–2956. [Google Scholar] [CrossRef]
- Foy, N.; Stéphan, E.; Jaouen, G. Synthesis of 17α-4-amino-and 4-iodophenylestradiols. Tetrahedron Lett. 2000, 41, 8089–8092. [Google Scholar] [CrossRef]
- Foy, N.; Stéphan, E.; Vessieres, A.; Salomon, E.; Heldt, J.M.; Huché, M.; Jaouen, M. Synthesis, receptor binding, molecular modeling, and proliferative assays of a series of 17α-arylestradiols. Chem. Biol. Chem. 2003, 4, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, E.; Fiaschi, R.; Carlson, K.E.; Katzenellenbogen, J.A. 11. beta.-Substituted Estradiol Derivatives, Potential High-Affinity Carbon-11-Labeled Probes for the Estrogen Receptor: A Structure-Affinity Relationship Study. J. Med. Chem. 1995, 38, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Sedlak, D.; Eignerova, B.; Dračínský, M.; Janoušek, Z.; Bartůněk, P.; Kotora, M. Synthesis and evaluation of 17α-(carboranylalkyl) estradiols as ligands for estrogen receptors α and β. J. Organomet. Chem. 2013, 747, 178–183. [Google Scholar] [CrossRef]
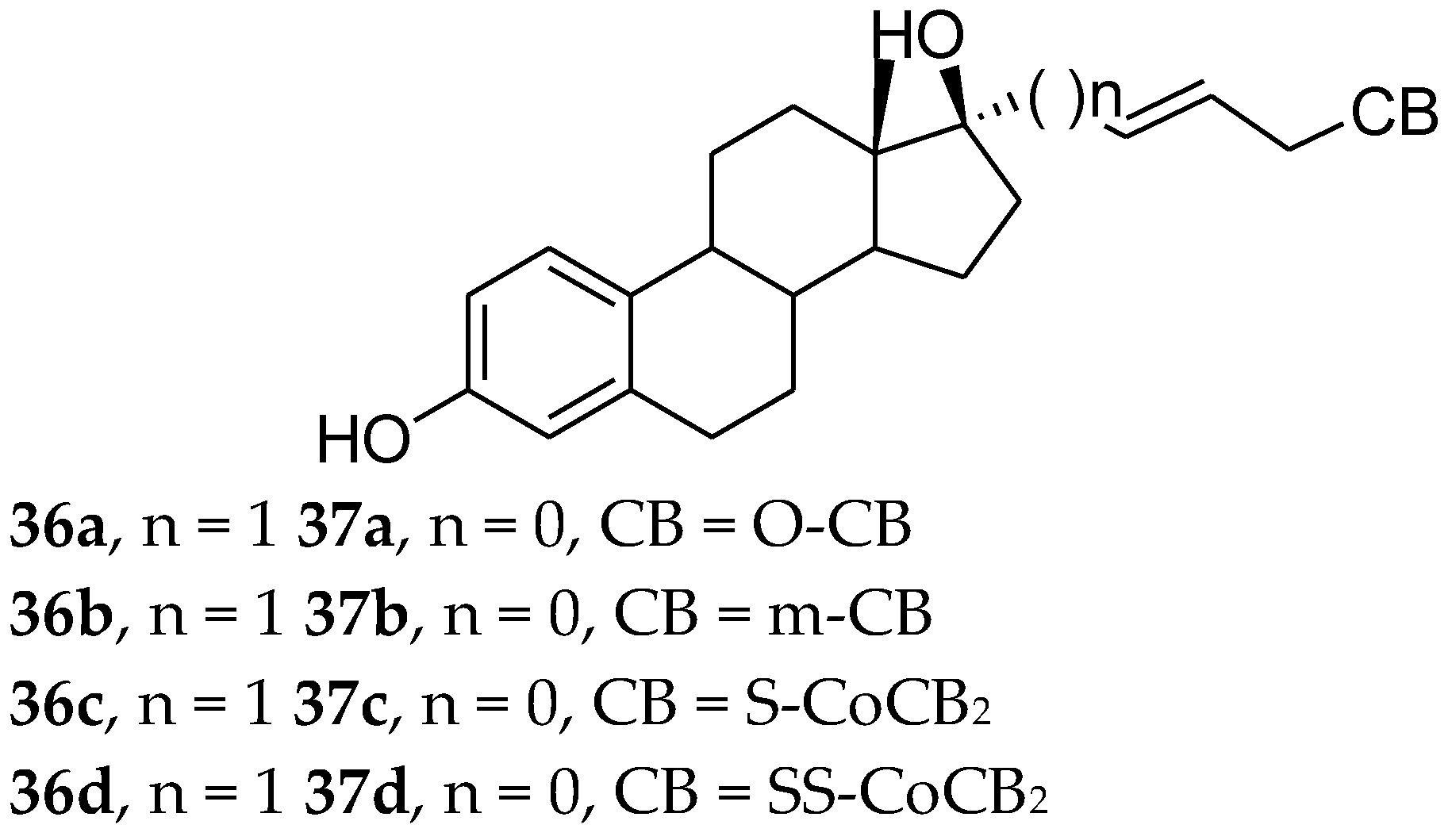
- Ohta, K.; Ogawa, T.; Kaise, A.; Endo, Y. Enhanced estrogen receptor beta (ERβ) selectivity of fluorinated carborane-containing ER modulators. Bioorg. Med. Chem. Lett. 2013, 23, 6555–6558. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Ohta, K.; Yoshimi, T.; Yamazaki, H.; Suzuki, T.; Ohta, S.; Endo, Y. m-Carborane bisphenol structure as a pharmacophore for selective estrogen receptor modulators. Bioorg. Med. Chem. Lett. 2006, 16, 3943–3946. [Google Scholar] [CrossRef]
- Ohta, K.; Ogawa, T.; Kaise, A.; Endo, Y. Novel estrogen receptor (ER) modulators containing various hydrophobic bent-core structures. Bioorg. Med. Chem. 2014, 22, 3508–3514. [Google Scholar] [CrossRef] [PubMed]
- Ohta, K.; Ogawa, T.; Kaise, A.; Endo, Y. Synthesis and biological evaluation of novel m-carborane-containing estrogen receptor partial agonists as SERM candidates. Bioorg. Med. Chem. Lett. 2015, 25, 3213–3216. [Google Scholar] [CrossRef] [PubMed]
- Lo, K.K.W.; Zhang, K.Y.; Chung, C.K.; Kwok, K.Y. Synthesis, Photophysical and Electrochemical Properties, and Protein-Binding Studies of Luminescent Cyclometalated Iridium (III) Bipyridine Estradiol Conjugates. Chem. Eur. J. 2007, 13, 7110–7120. [Google Scholar] [CrossRef] [PubMed]
- Lo, K.K.-W.; Lee, T.K.-M.; Lau, J.S.-Y.; Poon, W.-L.; Cheng, S.-H. Luminescent biological probes derived from ruthenium (II) estradiol polypyridine complexes. Inorg. Chem. 2008, 47, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Zhu, H.; Zhang, Y.; Xu, X.; Cui, W.; Yang, G.; Shen, Y.-M. Synthesis and binding affinities of Re(I) and 99mTc(I)-containing 16α-substituted estradiol complexes: Models for potential breast cancer imaging agents. Steroids 2010, 75, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Hanson, R.N.; Kirss, R.; McCaskill, E.; Hua, E.; Tongcharoensirikul, P.; Olmsted, S.L.; Labaree, D.; Hochberg, R.B. Targeting the estrogen receptor with metal-carbonyl derivatives of estradiol. Bioorg. Med. Chem. Lett. 2012, 22, 1670–1673. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zuo, Z.; Tang, J.; Wang, K.; Wang, C.; Chen, W.; Li, C.; Xu, W.; Xiong, X.; Yuntai, K.; et al. Design, synthesis and biological evaluation of novel estrogen-derived steroid metal complexes. Bioorg. Med. Chem. Lett. 2013, 23, 3793–3797. [Google Scholar] [CrossRef] [PubMed]
- Cázares-Marinero, J.; Top, S.; Jaouen, G. Synthesis and characterization of new ferrocenyl compounds with different alkyl chain lengths and functional groups to target breast cancer cells. J. Organomet. Chem. 2014, 751, 610–619. [Google Scholar] [CrossRef]
- Nunez-Montenegro, A.; Carballo, R.; Vazquez-Lopez, E.M. Synthesis, characterization and binding affinities of rhenium(I) thiosemicarbazone complexes for the estrogen receptor (alpha/beta). J. Inorg. Biochem. 2014, 140c, 53–63. [Google Scholar] [CrossRef] [PubMed]


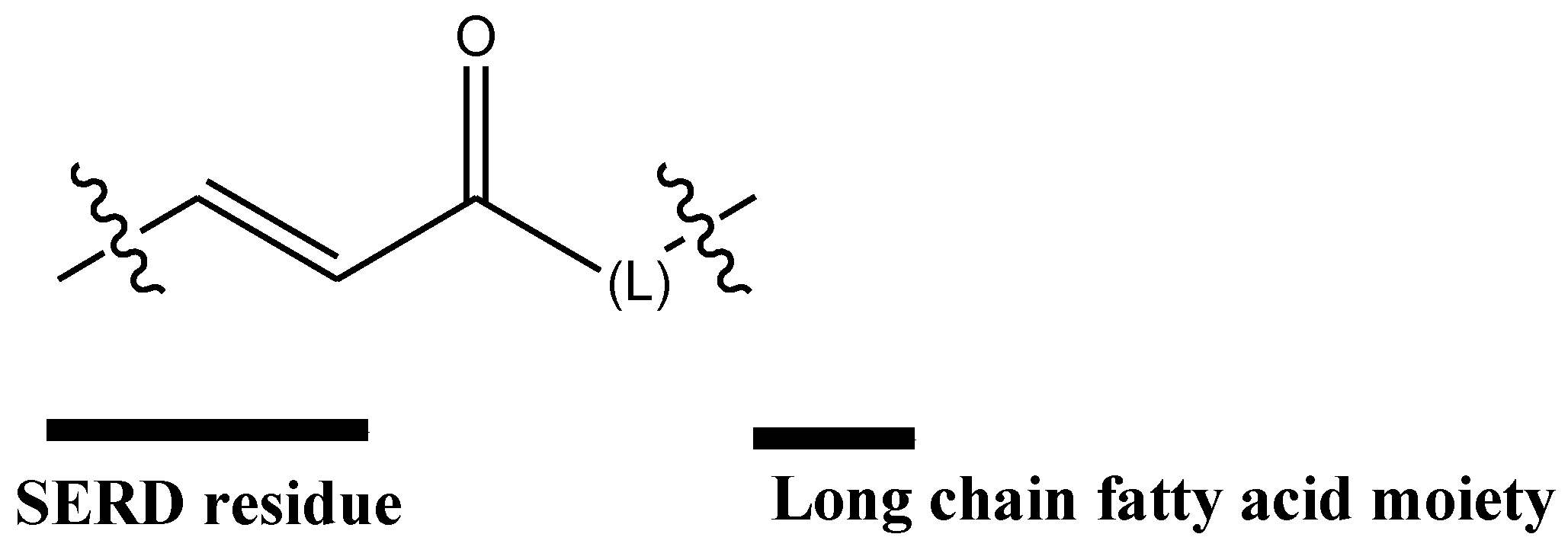

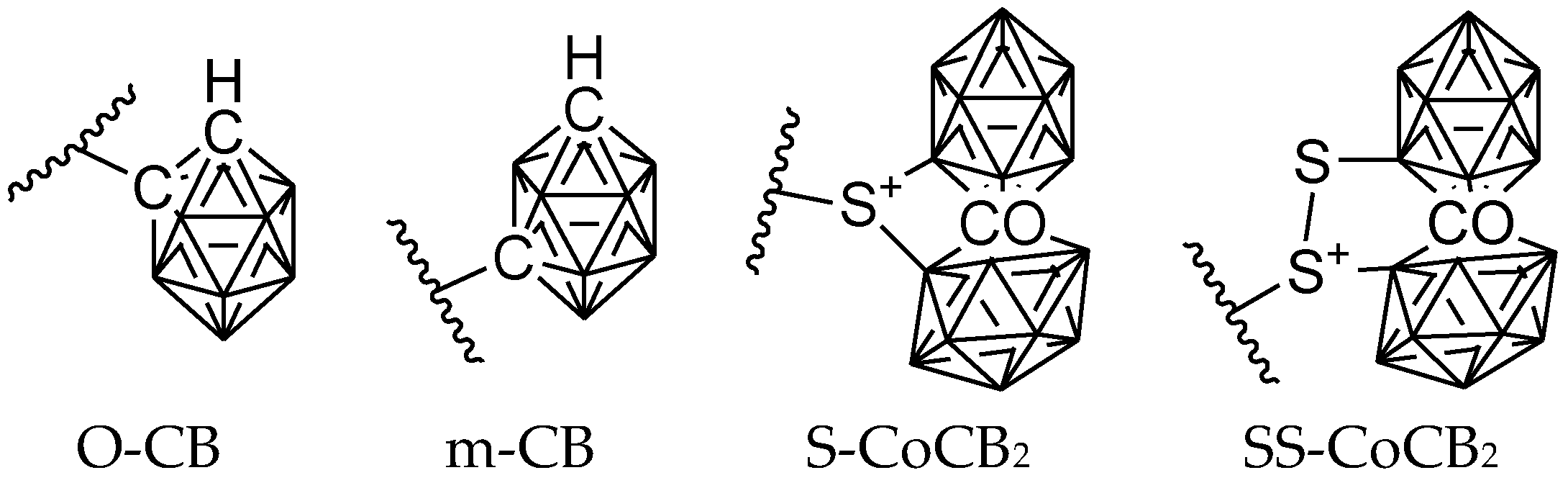




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
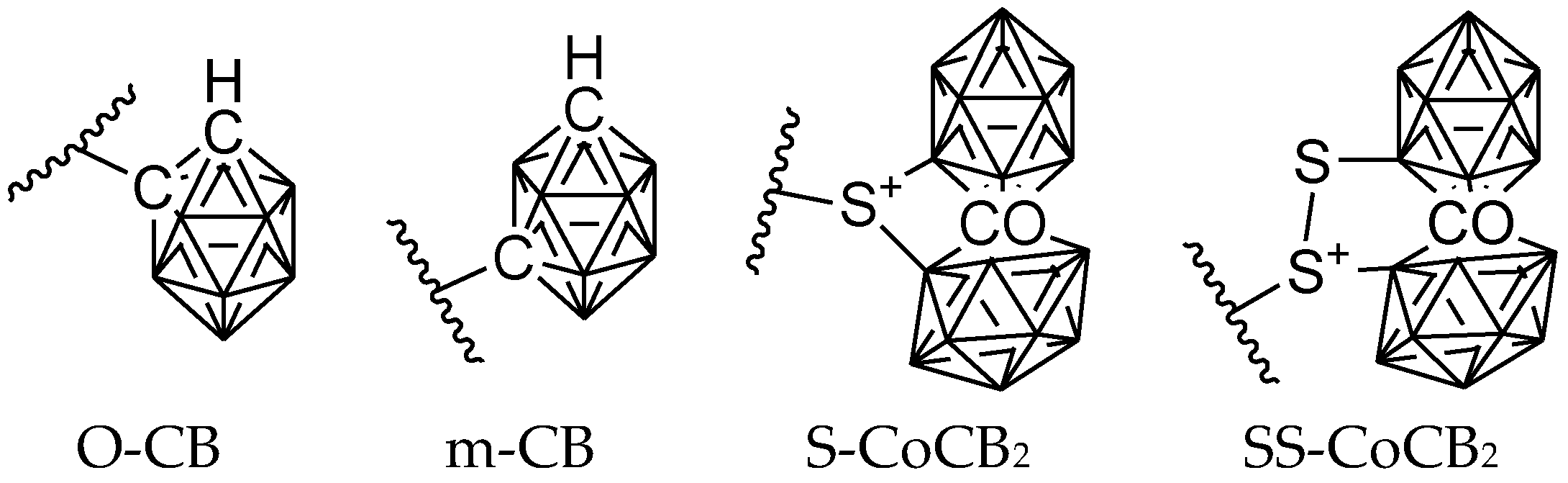{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | RTA(%) ERα | RTA(%) ERβ |
|---|---|---|
| Agonist | 100 | 100 |
| 36a | 24 | 26 |
| 36b | 52 | 35 |
| 36c | 20 | 16 |
| 36d | 53 | 43 |
| 37a | 58 | 40 |
| 37b | 71 | 63 |
| 37c | 16 | 14 |
| 37d | 31 | 27 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farzaneh, S.; Zarghi, A. Estrogen Receptor Ligands: A Review (2013–2015). Sci. Pharm. 2016, 84, 409-427. https://doi.org/10.3390/scipharm84030409
Farzaneh S, Zarghi A. Estrogen Receptor Ligands: A Review (2013–2015). Scientia Pharmaceutica. 2016; 84(3):409-427. https://doi.org/10.3390/scipharm84030409
Chicago/Turabian StyleFarzaneh, Shabnam, and Afshin Zarghi. 2016. "Estrogen Receptor Ligands: A Review (2013–2015)" Scientia Pharmaceutica 84, no. 3: 409-427. https://doi.org/10.3390/scipharm84030409
APA StyleFarzaneh, S., & Zarghi, A. (2016). Estrogen Receptor Ligands: A Review (2013–2015). Scientia Pharmaceutica, 84(3), 409-427. https://doi.org/10.3390/scipharm84030409