

Pt(II)-PLGA Hybrid in a pH-Responsive Nanoparticle System Targeting Ovarian Cancer
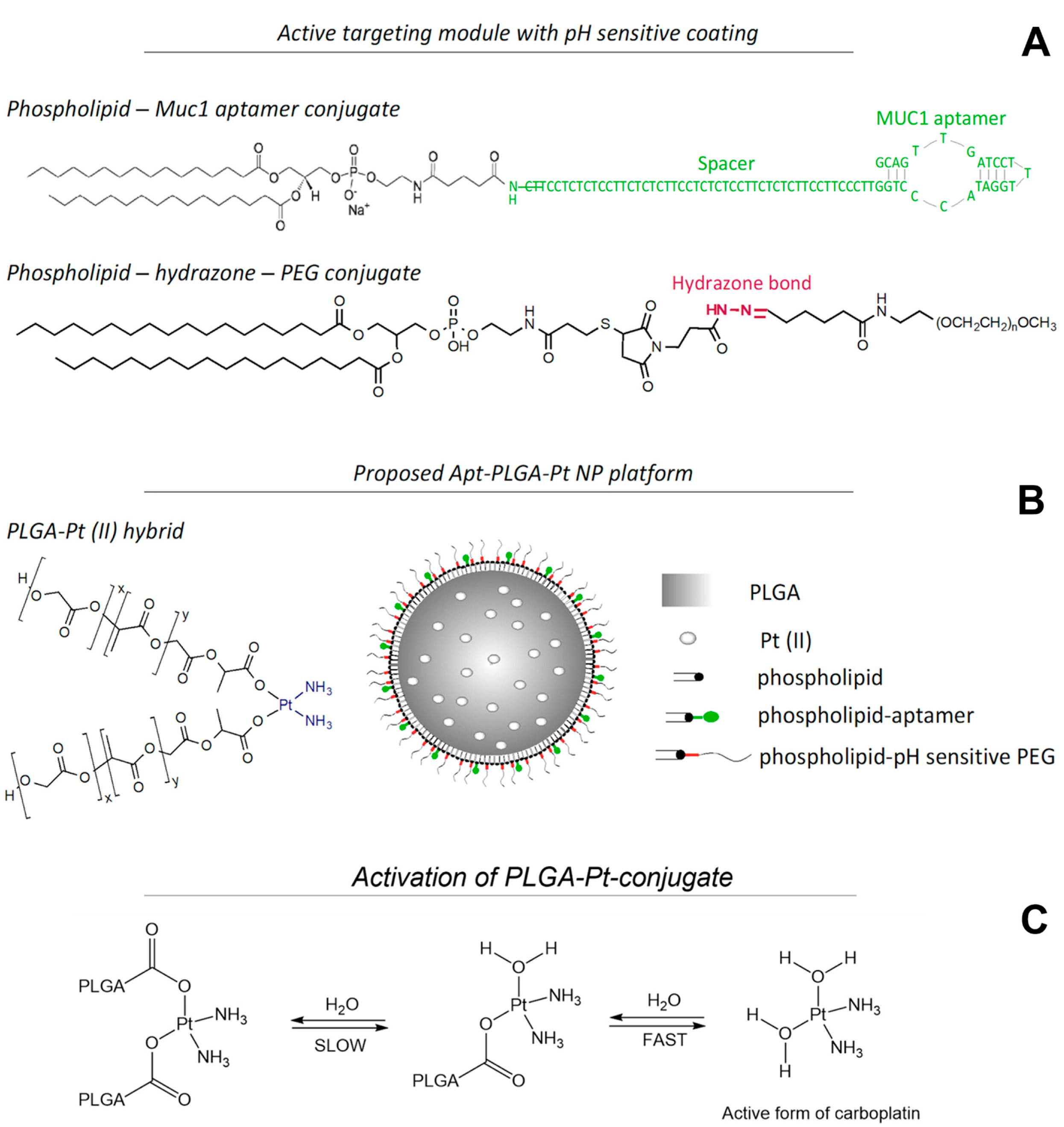{kind=link}
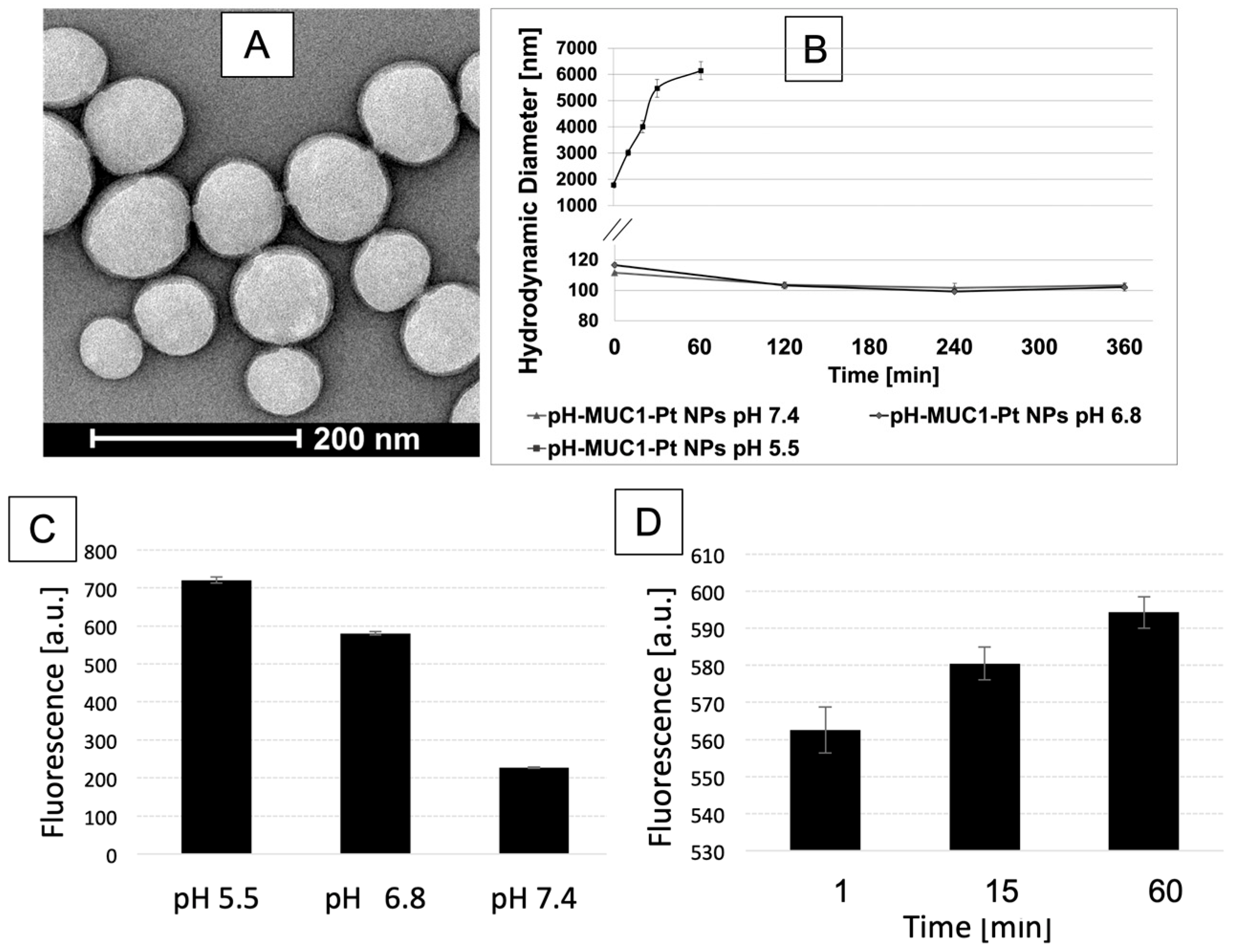{kind=link}
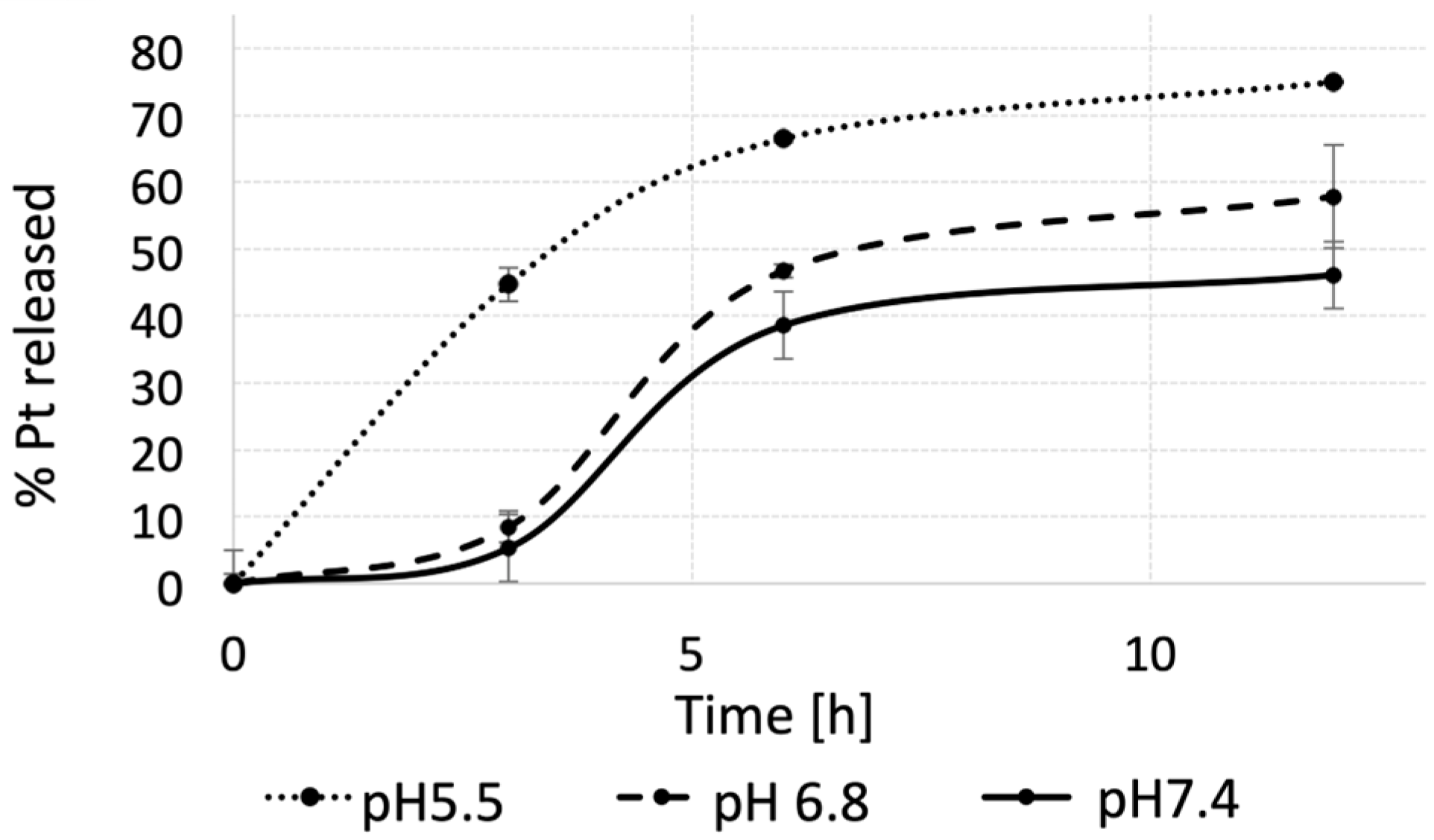{kind=link}
{kind=link}
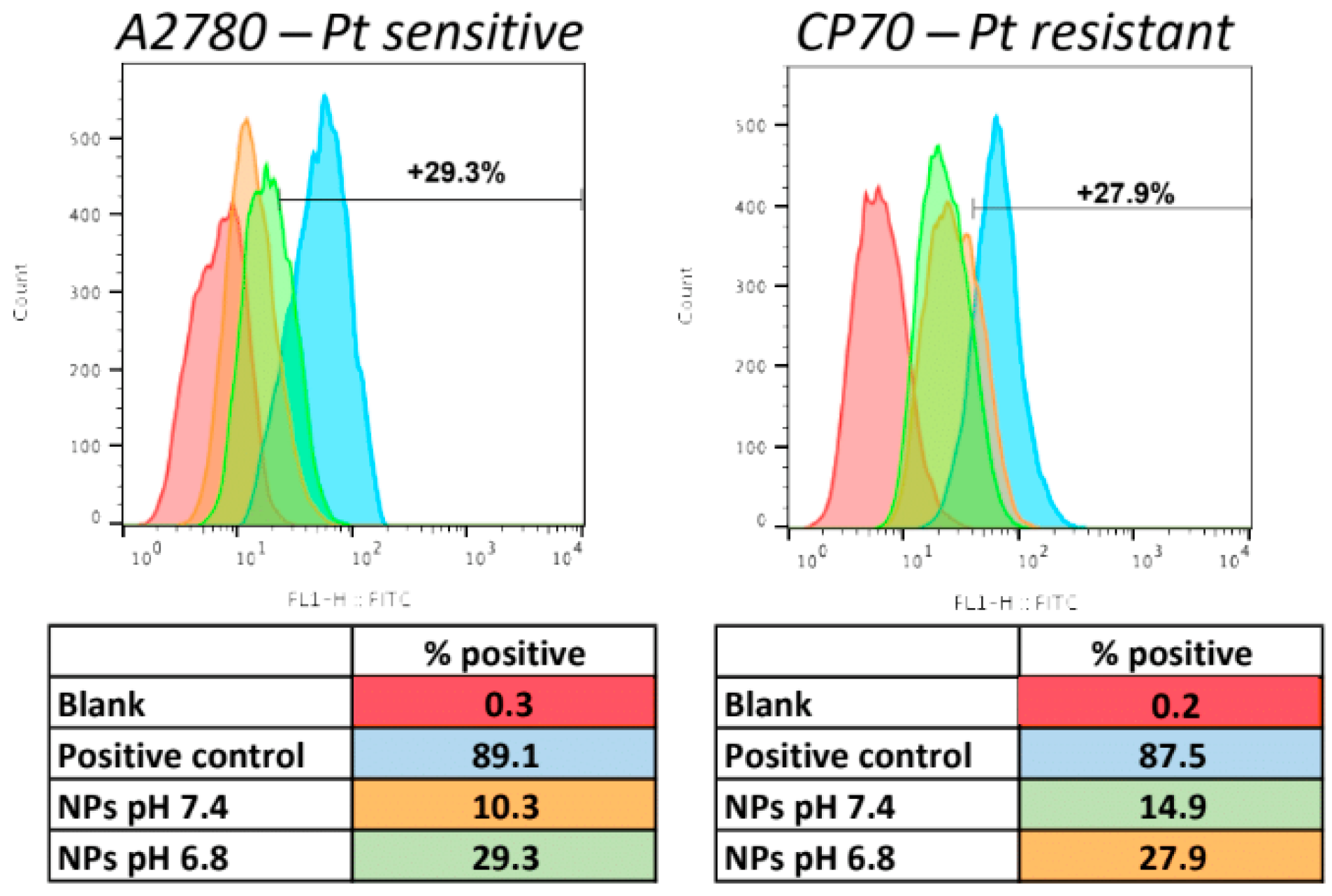{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Materials
2.2. Synthesis of pH-Sensitive DSPE-PEG
2.2.1. Synthesis of 3-(2,5-Dioxo-2,5-dihydro-1H-pyrrol-1-yl)propanoic Acid (1)
2.2.2. Synthesis of Precursor tert-Butyl 2-(3-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)propanoyl)hydrazine-1-carboxylate (2)
2.2.3. Synthesis of 3-(2,5-Dioxo-2,5-dihydro-1H-pyrrol-1-yl)propanehydrazide (3)
2.2.4. Synthesis of Precursor (2,3-Bis(stearoyloxy)propyl (2-((1-(3-hydrazineyl-3-oxopropyl)-2,5-dioxopyrrolidin-3-yl)thio)ethyl) Phosphate (4)
2.2.5. Synthesis of Lipid-hydrazone-PEG2000 (5)
2.3. The Synthesis of Phospholipid-MUC1 Conjugate
2.4. Synthesis of PLGA-Pt (II) Conjugate
2.5. Nanoparticle Synthesis
2.6. Synthesis of PLGA-Cy5.5 Conjugate
2.7. Synthesis of PLGA-FITC Conjugate
2.8. Synthesis of DSPE-Cy7 Conjugate
2.9. Characterization
2.10. In Vitro Drug Release
2.11. Fluorescamine Test
2.12. Cellular Uptake of the NPs
2.13. ELISA Test for Mucin 1 Presence in OC Cells
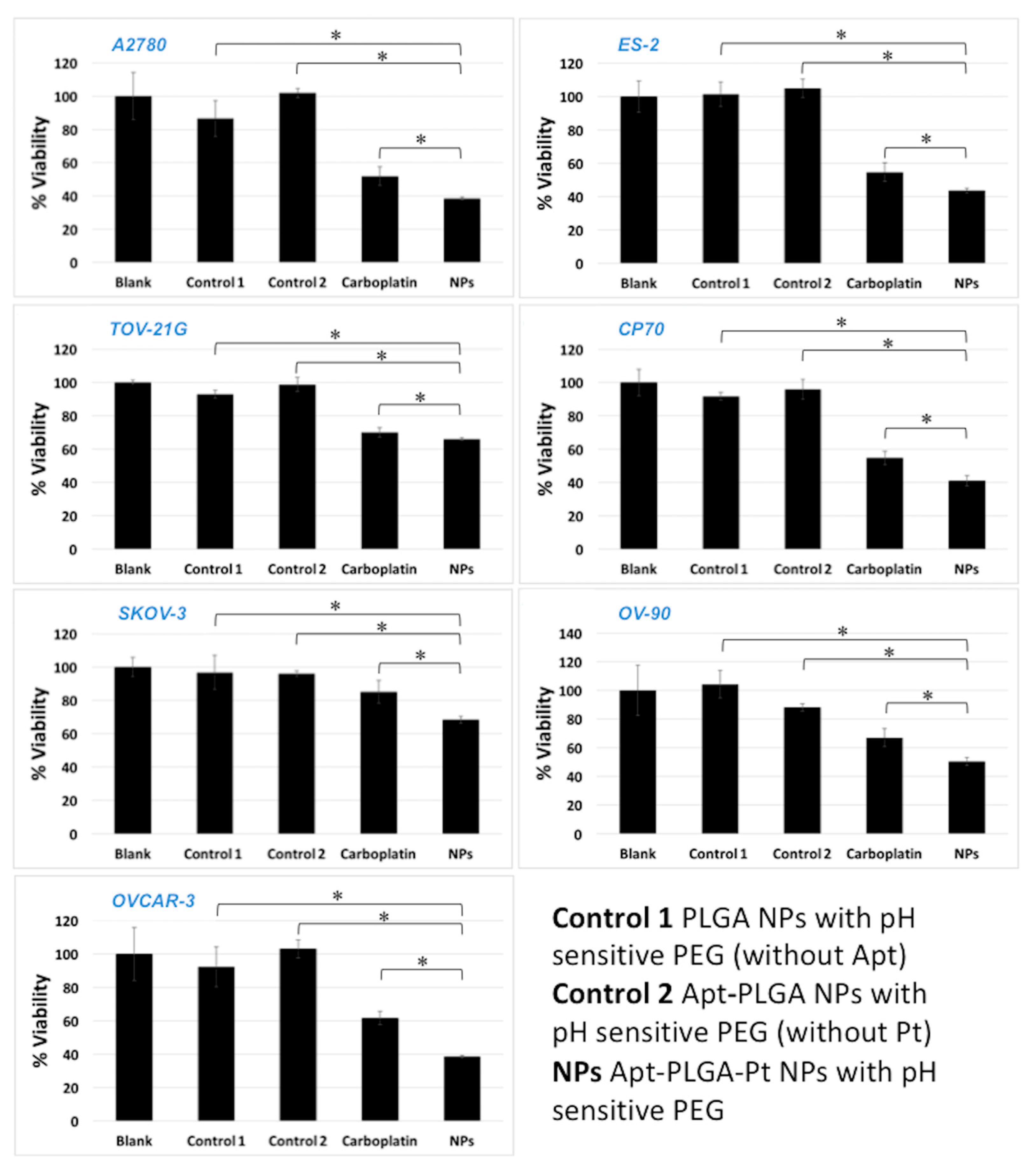
2.14. In Vitro Viability Assay
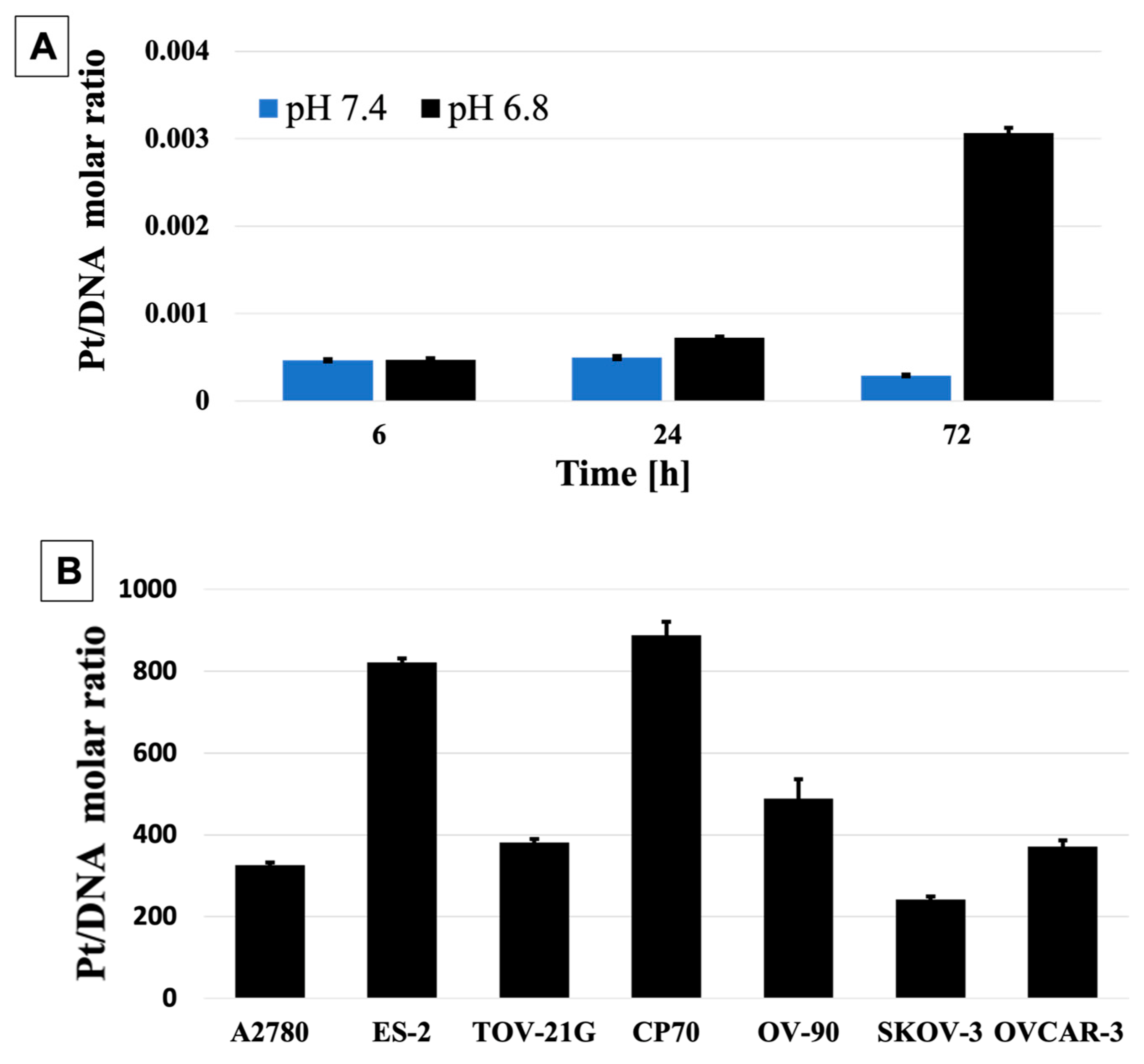
2.15. DNA:Pt Quantification
2.16. In Vivo Mouse Model Studies
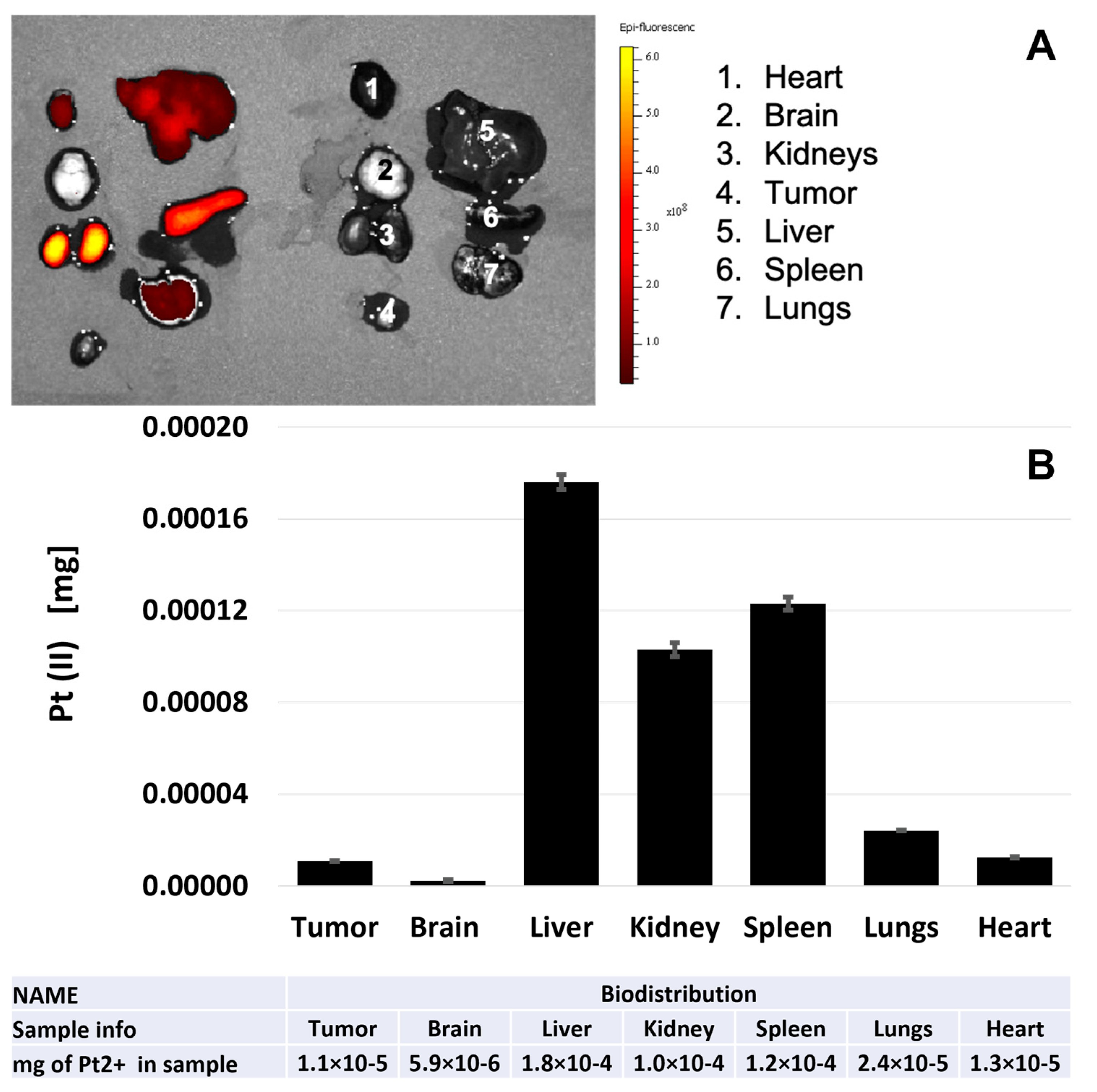
2.17. In Vivo Fluorescence Imaging
2.18. AAS Analysis of Pt (II) in Organs
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| NP | nanoparticle |
| Apt | aptamer |
| PLGA | poly(lactic)co-(glycolic) acid |
| PEG | polyethylene glycol |
| AAS | atomic absorption spectroscopy |
| TLC | thin layer chromatography |
| DLS | dynamic light scattering |
| TEM | transmission electron microscopy |
| FBS | fetal bovine serum |
References
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: Past, present and future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef] [PubMed]
- Baert, T.; Ferrero, A.; Sehouli, J.; O’Donnell, D.; González-Martín, A.; Joly, F.; van der Velden, J.; Blecharz, P.; Tan, D.; Querleu, D.; et al. The systemic treatment of recurrent ovarian cancer revisited. Ann. Oncol. 2021, 32, 710–725. [Google Scholar] [CrossRef] [PubMed]
- Ahmed-Lecheheb, D.; Joly, F. Ovarian cancer survivors’ quality of life: A systematic review. J. Cancer Surviv. 2016, 10, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Cristea, M.; Han, E.; Salmon, L.; Morgan, J.R.J. Review: Practical considerations in ovarian cancer chemotherapy. Ther. Adv. Med. Oncol. 2010, 2, 175–187. [Google Scholar] [CrossRef]
- Lheureux, S.; Braunstein, M.; Oza, A.M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef]
- Herzog, T.J.; Pothuri, B. Ovarian cancer: A focus on management of recurrent disease. Nat. Clin. Prac. Oncol. 2006, 3, 604–611. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Englander, E.W. DNA damage response in peripheral nervous system: Coping with cancer therapy-induced DNA lesions. DNA Repair 2013, 12, 685–690. [Google Scholar] [CrossRef]
- Boeckman, H.J.; Trego, K.S.; Turchi, J.J. Cisplatin Sensitizes Cancer Cells to Ionizing Radiation via Inhibition of Nonhomologous End Joining. Mol. Cancer Res. 2005, 3, 277–285. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2011, 31, 1869–1883. [Google Scholar] [CrossRef]
- Amable, L. Cisplatin resistance and opportunities for precision medicine. Pharmacol. Res. 2016, 106, 27–36. [Google Scholar] [CrossRef]
- Wang, E.; Wang, A. Nanoparticles and their applications in cell and molecular biology. Integr. Biol. 2013, 6, 9–26. [Google Scholar] [CrossRef]
- Aghebati-Maleki, A.; Dolati, S.; Ahmadi, M.; Baghbanzhadeh, A.; Asadi, M.; Fotouhi, A.; Yousefi, M.; Aghebati-Maleki, L. Nanoparticles and cancer therapy: Perspectives for application of nanoparticles in the treatment of cancers. J. Cell. Physiol. 2020, 235, 1962–1972. [Google Scholar] [CrossRef]
- Chaturvedi, V.K.; Singh, A.; Singh, V.K.; Singh, M.P. Cancer Nanotechnology: A New Revolution for Cancer Diagnosis and Therapy. Curr. Drug Metab. 2019, 20, 416–429. [Google Scholar] [CrossRef]
- Iyer, A.K.; Khaled, G.; Fang, J.; Maeda, H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov. Today 2006, 11, 812–818. [Google Scholar] [CrossRef]
- Elkhodiry, M.A.; Momah, C.C.; Suwaidi, S.R.; Gadalla, D.; Martins, A.M.; Vitor, R.F.; Husseini, G.A. Synergistic Nanomedicine: Passive, Active, and Ultrasound-Triggered Drug Delivery in Cancer Treatment. J. Nanosci. Nanotechnol. 2016, 16, 1–18. [Google Scholar] [CrossRef]
- Alavi, M.; Hamidi, M. Passive and active targeting in cancer therapy by liposomes and lipid nanoparticles. Drug Metab. Pers. Ther. 2019, 34. [Google Scholar] [CrossRef]
- Browning, R.J.; Reardon, P.J.T.; Parhizkar, M.; Pedley, R.B.; Edirisinghe, M.; Knowles, J.C.; Stride, E. Drug Delivery Strategies for Platinum-Based Chemotherapy. ACS Nano 2017, 11, 8560–8578. [Google Scholar] [CrossRef]
- Boztepe, T.; Castro, G.R.; León, I.E. Lipid, polymeric, inorganic-based drug delivery applications for platinum-based anticancer drugs. Int. J. Pharm. 2021, 605, 120788. [Google Scholar] [CrossRef]
- Kim, E.S.; Lu, C.; Khuri, F.R.; Tonda, M.; Glisson, B.S.; Liu, D.; Jung, M.; Hong, W.K.; Herbst, R.S. A phase II study of STEALTH cisplatin (SPI-77) in patients with advanced non-small cell lung cancer. Lung Cancer 2001, 34, 427–432. [Google Scholar] [CrossRef]
- Alavi, M.; Webster, T.J. Nano liposomal and cubosomal formulations with platinum-based anticancer agents: Therapeutic advances and challenges. Nanomedicine 2020, 15, 2399–2410. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.; Gu, F.X.; Langer, R.; Farokhzad, O.C.; Lippard, S.J. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA–PEG nanoparticles. Proc. Natl. Acad. Sci. USA 2008, 105, 17356–17361. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Jiang, W.; Shen, Y.; Li, H.; Sun, C.; Ouahab, A.; Tu, J. A Poly(γ, l-glutamic acid)-citric acid based nanoconjugate for cisplatin delivery. Biomaterials 2012, 33, 7182–7193. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhang, D.Y.; Peng, Z.; Guan, S.; Zhai, J. Delivery of Platinum(IV) Prodrugs via Bi2Te3 Nanoparticles for Photothermal-Chemotherapy and Photothermal/Photoacoustic Imaging. Mol. Pharm. 2020, 17, 3403–3411. [Google Scholar] [CrossRef]
- Babu, A.; Wang, Q.; Muralidharan, R.; Shanker, M.; Munshi, A.; Ramesh, R. Chitosan Coated Polylactic Acid Nanoparticle-Mediated Combinatorial Delivery of Cisplatin and siRNA/Plasmid DNA Chemosensitizes Cisplatin-Resistant Human Ovarian Cancer Cells. Mol. Pharm. 2014, 11, 2720–2733. [Google Scholar] [CrossRef]
- Eriksson, M.; Hassan, S.; Larsson, R.; Linder, S.; Ramqvist, T.; Lövborg, H.; Vikinge, T.; Figgemeier, E.; Müller, J.; Stetefeld, J.; et al. Utilization of a right-handed coiled-coil protein from archaebacterium Staphylothermus marinus as a carrier for cisplatin. Anticancer Res. 2009, 29, 11–18. [Google Scholar]
- Kumar, A.; Huo, S.; Zhang, X.; Liu, J.; Tan, A.; Li, S.; Jin, S.; Xue, X.; Zhao, Y.; Ji, T.; et al. Neuropilin-1-Targeted Gold Nanoparticles Enhance Therapeutic Efficacy of Platinum(IV) Drug for Prostate Cancer Treatment. ACS Nano 2014, 8, 4205–4220. [Google Scholar] [CrossRef]
- Dhar, S.; Liu, Z.; Thomale, J.; Dai, H.; Lippard, S.J. Targeted Single-Wall Carbon Nanotube-Mediated Pt(IV) Prodrug Delivery Using Folate as a Homing Device. J. Am. Chem. Soc. 2008, 130, 11467–11476. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic. Bioeng. Transl. Med. Mar. 2016, 1, 10–29. [Google Scholar] [CrossRef]
- Jeevanandam, J.; Chan, Y.S.; Danquah, M.K. Nano-formulations of drugs: Recent developments, impact and challenges. Biochimie 2016, 128, 99–112. [Google Scholar] [CrossRef]
- Upreti, M.; Jyoti, A.; Sethi, P. Tumor microenvironment and nanotherapeutics. Transl. Cancer Res. 2013, 2, 309–319. [Google Scholar] [CrossRef]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Lai, H.; Ding, X.; Ye, J.; Deng, J.; Cui, S. pH-responsive hyaluronic acid-based nanoparticles for targeted curcumin delivery and enhanced cancer therapy. Colloids Surf. B Biointerfaces 2021, 198, 111455. [Google Scholar] [CrossRef]
- Quazi, M.Z.; Lee, U.; Park, S.; Shin, S.; Sim, E.; Son, H.; Park, N. Cancer Cell-Specific Enhanced Raman Imaging and Photothermal Therapeutic Effect Based on Reversibly pH-Responsive Gold Nanoparticles. ACS Appl. Bio Mater. 2021, 4, 8377–8385. [Google Scholar] [CrossRef]
- Jeong, Y.-H.; Shin, H.-W.; Kwon, J.-Y.; Lee, S.-M. Cisplatin-Encapsulated Polymeric Nanoparticles with Molecular Geometry-Regulated Colloidal Properties and Controlled Drug Release. ACS Appl. Mater. Interfaces 2018, 10, 23617–23629. [Google Scholar] [CrossRef]
- Xu, H.L.; Fan, Z.L.; ZhuGe, D.L.; Tong, M.Q.; Shen, B.X.; Lin, M.T.; Zhu, Q.Y.; Jin, B.H.; Sohawon, Y.; Yao, Q.; et al. Ratiometric delivery of two therapeutic candidates with inherently dissimilar physicochemical property through pH-sensitive core-shell nanoparticles targeting the heterogeneous tumor cells of glioma. Drug Deliv. 2018, 25, 1302–1318. [Google Scholar] [CrossRef]
- Li, X.X.; Chen, J.; Shen, J.M.; Zhuang, R.; Zhang, S.Q.; Zhu, Z.Y.; Ma, J.B. pH-Sensitive nanoparticles as smart carriers for selective intracellular drug delivery to tumor. Int. J. Pharm. 2018, 545, 274–285. [Google Scholar] [CrossRef]
- Pan, C.; Liu, Y.; Zhou, M.; Wang, W.; Shi, M.; Xing, M.; Liao, W. Theranostic pH-sensitive nanoparticles for highly efficient targeted delivery of doxorubicin for breast tumor treatment. Int. J. Nanomed. 2018, 13, 1119–1137. [Google Scholar] [CrossRef]
- Tang, B.; Zaro, J.L.; Shen, Y.; Chen, Q.; Yu, Y.; Sun, P.; Wang, Y.; Shen, W.-C.; Tu, J.; Sun, C. Acid-sensitive hybrid polymeric micelles containing a reversibly activatable cell-penetrating peptide for tumor-specific cytoplasm targeting. J. Control. Release 2018, 279, 147–156. [Google Scholar] [CrossRef]
- Tang, S.; Meng, Q.; Sun, H.; Su, J.; Yin, Q.; Zhang, Z.; Yu, H.; Chen, L.; Gu, W.; Li, Y. Dual pH-sensitive micelles with charge-switch for controlling cellular uptake and drug release to treat metastatic breast cancer. Biomaterials 2017, 114, 44–53. [Google Scholar] [CrossRef]
- Li, Y.; Zhi, X.; Lin, J.; You, X.; Yuan, J. Preparation and characterization of DOX loaded keratin nanoparticles for pH/GSH dual responsive release. Mater. Sci. Eng. C 2017, 73, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Qiu, Y.; Yang, W.; Guo, Y.; Hou, H.; Liu, Z.; Zhao, X. Charge reversible and biodegradable nanocarriers showing dual pH-/reduction-sensitive disintegration for rapid site-specific drug delivery. Colloids Surf. B Biointerfaces 2018, 169, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Song, X.; Lu, J.; Liu, S.; Sha, X.; Wang, Q.; Cao, X.; Xu, K.; Li, J. DNA aptamer-based dual-responsive nanoplatform for targeted MRI and combination therapy for cancer. RSC Adv. 2022, 12, 3871–3882. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, X.; Zhong, W.; Ren, X.; Sha, X.; Fang, X. Matrix metalloproteinases-2/9-sensitive peptide-conjugated polymer micelles for site-specific release of drugs and enhancing tumor accumulation: Preparation and in vitro and in vivo evaluation. Int. J. Nanomed. 2016, 11, 1643–1661. [Google Scholar]
- Huang, C.; Sun, Y.; Shen, M.; Zhang, X.; Gao, P.; Duan, Y. Altered Cell Cycle Arrest by Multifunctional Drug-Loaded Enzymatically-Triggered Nanoparticles. ACS Appl. Mater. Interfaces 2016, 8, 1360–1370. [Google Scholar] [CrossRef]
- Nazli, C.; Demirer, G.S.; Yar, Y.; Acar, H.Y.; Kizilel, S. Targeted delivery of doxorubicin into tumor cells via MMP-sensitive PEG hydrogel-coated magnetic iron oxide nanoparticles (MIONPs). Colloids Surf. B Biointerfaces 2014, 122, 674–683. [Google Scholar] [CrossRef]
- Hu, Q.; Sun, W.; Lu, Y.; Bomba, H.N.; Ye, Y.; Jiang, T.; Isaacson, A.J.; Gu, Z. Tumor Microenvironment-Mediated Construction and Deconstruction of Extracellular Drug-Delivery Depots. Nano Lett. 2016, 16, 1118–1126. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, K.; Wang, H.; Liu, Y.; Bao, B.; Fang, Y.; Zhang, X.; Lu, W. Design, Synthesis, and Biological Evaluation of New Cathepsin B-Sensitive Camptothecin Nanoparticles Equipped with a Novel Multifuctional Linker. Bioconjug. Chem. 2016, 27, 1267–1275. [Google Scholar] [CrossRef]
- Farokhzad, O.C.; Karp, J.M.; Langer, R. Nanoparticle–aptamer bioconjugates for cancer targeting. Expert Opin. Drug Deliv. 2006, 3, 311–324. [Google Scholar] [CrossRef]
- Morita, Y.; Leslie, M.; Kameyama, H.; Volk, D.; Tanaka, T. Aptamer Therapeutics in Cancer: Current and Future. Cancers 2018, 10, 80. [Google Scholar] [CrossRef]
- Musumeci, D.; Platella, C.; Riccardi, C.; Moccia, F.; Montesarchio, D. Fluorescence Sensing Using DNA Aptamers in Cancer Research and Clinical Diagnostics. Cancers 2017, 9, 174. [Google Scholar] [CrossRef]
- Röthlisberger, P.; Gasse, C.; Hollenstein, M. Nucleic Acid Aptamers: Emerging Applications in Medical Imaging, Nanotechnology, Neurosciences, and Drug Delivery. Int. J. Mol. Sci. 2017, 18, 2430. [Google Scholar] [CrossRef]
- Filippi, L.; Bagni, O.; Nervi, C. Aptamer-based technology for radionuclide targeted imaging and therapy: A promising weapon against cancer. Expert Rev. Med. Devices 2020, 17, 751–758. [Google Scholar] [CrossRef]
- He, F.; Wen, N.; Xiao, D.; Yan, J.; Xiong, H.; Cai, S.; Liu, Z.; Liu, Y. Aptamer-Based Targeted Drug Delivery Systems: Current Potential and Challenges. Curr. Med. Chem. 2020, 27, 2189–2219. [Google Scholar] [CrossRef]
- Zhu, G.; Chen, X. Aptamer-based targeted therapy. Adv. Drug Deliv. Rev. 2018, 134, 65–78. [Google Scholar] [CrossRef]
- D’Souza, A.A.; Shegokar, R. Polyethylene glycol (PEG): A versatile polymer for pharmaceutical applications. Expert Opin. Drug Deliv. 2016, 13, 1257–1275. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef]
- Cho, E.C.; Zhang, Q.; Xia, Y. The effect of sedimentation and diffusion on cellular uptake of gold nanoparticles. Nat. Nanotechnol. 2011, 6, 385–391. [Google Scholar] [CrossRef]
- Li, Y.; Kröger, M.; Liu, W.K. Endocytosis of PEGylated nanoparticles accompanied by structural and free energy changes of the grafted polyethylene glycol. Biomaterials 2014, 35, 8467–8478. [Google Scholar] [CrossRef]
- Hatakeyama, H.; Akita, H.; Harashima, H. The Polyethyleneglycol Dilemma: Advantage and Disadvantage of PEGylation of Liposomes for Systemic Genes and Nucleic Acids Delivery to Tumors. Biol. Pharm. Bull. 2013, 36, 892–899. [Google Scholar] [CrossRef]
- Gianella, A.; Mieszawska, A.J.; Hoeben, F.J.M.; Janssen, H.M.; Jarzyna, P.A.; Cormode, D.P.; Costa, K.D.; Rao, S.; Farokhzad, O.C.; Langer, R.; et al. Synthesis and in vitro evaluation of a multifunctional and surface-switchable nanoemulsion platform. Chem. Commun. 2013, 49, 9392–9394. [Google Scholar] [CrossRef] [PubMed]
- Juang, V.; Chang, C.H.; Wang, C.S.; Wang, H.E.; Lo, Y.L. pH-Responsive PEG-Shedding and Targeting Peptide-Modified Nanoparticles for Dual-Delivery of Irinotecan and microRNA to Enhance Tumor-Specific Therapy. Small 2019, 15, e1903296. [Google Scholar] [CrossRef] [PubMed]
- Ting, C.-W.; Chou, Y.-H.; Huang, S.-Y.; Chiang, W.-H. Indocyanine green-carrying polymeric nanoparticles with acid-triggered detachable PEG coating and drug release for boosting cancer photothermal therapy. Colloids Surf. B Biointerfaces 2021, 208, 112048. [Google Scholar] [CrossRef] [PubMed]
- Oishi, M.; Nagasaki, Y.; Itaka, K.; Nishiyama, N.; Kataoka, K. Lactosylated poly(ethylene glycol)-siRNA conjugate through acid-labile beta-thiopropionate linkage to construct pH-sensitive polyion complex micelles achieving enhanced gene silencing in hepatoma cells. J. Am. Chem. Soc. 2005, 127, 1624–1625. [Google Scholar] [CrossRef] [PubMed]
- Quan, C.Y.; Chen, J.X.; Wang, H.Y.; Li, C.; Chang, C.; Zhang, X.Z.; Zhuo, R.X. Core-shell nanosized assemblies mediated by the alpha-beta cyclodextrin dimer with a tumor-triggered targeting property. ACS Nano 2010, 4, 4211–4219. [Google Scholar] [CrossRef] [PubMed]
- Sawant, R.M.; Hurley, J.P.; Salmaso, S.; Kale, A.; Tolcheva, E.; Levchenko, T.S.; Torchilin, V.P. “SMART” drug delivery systems: Double-targeted pH-responsive pharmaceutical nanocarriers. Bioconjug. Chem. 2006, 17, 943–949. [Google Scholar] [CrossRef]
- Engelstaedter, V.; Heublein, S.; Schumacher, A.L.; Lenhard, M.; Engelstaedter, H.; Andergassen, U.; Guenthner-Biller, M.; Kuhn, C.; Rack, B.; Kupka, M.; et al. Mucin-1 and its relation to grade, stage and survival in ovarian carcinoma patients. BMC Cancer 2012, 12, 600. [Google Scholar] [CrossRef]
- Zhang, L.; Chan, J.M.; Gu, F.X.; Rhee, J.-W.; Wang, A.Z.; Radovic-Moreno, A.F.; Alexis, F.; Langer, R.; Farokhzad, O.C. Self-Assembled Lipid−Polymer Hybrid Nanoparticles: A Robust Drug Delivery Platform. ACS Nano 2008, 2, 1696–1702. [Google Scholar] [CrossRef]
- Jagur-Grodzinski, J. Biomedical application of functional polymers. React. Funct. Polym. 1999, 39, 99–138. [Google Scholar] [CrossRef]
- Cryan, S.-A. Carrier-based strategies for targeting protein and peptide drugs to the lungs. AAPS J. 2005, 7, E20–E41. [Google Scholar] [CrossRef]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef]
- Cappellano, G.; Comi, C.; Chiocchetti, A.; Dianzani, U. Exploiting PLGA-Based Biocompatible Nanoparticles for Next-Generation Tolerogenic Vaccines against Autoimmune Disease. Int. J. Mol. Sci. 2019, 20, 204. [Google Scholar] [CrossRef]
- Markel, J.E.; Lacinski, R.A.; Lindsey, B.A. Nanocapsule Delivery of IL-12. Curr. Adv. Osteosarcoma Clin. Perspect. Past Present Future 2020, 1257, 155–168. [Google Scholar] [CrossRef]
- Nenna, A.; Nappi, F.; Larobina, D.; Verghi, E.; Chello, M.; Ambrosio, L. Polymers and Nanoparticles for Statin Delivery: Current Use and Future Perspectives in Cardiovascular Disease. Polymers 2021, 13, 711. [Google Scholar] [CrossRef]
- Kuo, Y.-C.; Chung, J.-F. Physicochemical properties of nevirapine-loaded solid lipid nanoparticles and nanostructured lipid carriers. Colloids Surf. B Biointerfaces 2011, 83, 299–306. [Google Scholar] [CrossRef]
- Rivas, C.J.M.; Tarhini, M.; Badri, W.; Miladi, K.; Greige-Gerges, H.; Nazari, Q.A.; Rodríguez, S.A.G.; Román, R.Á.; Fessi, H.; Elaissari, A. Nanoprecipitation process: From encapsulation to drug delivery. Int. J. Pharm. 2017, 532, 66–81. [Google Scholar] [CrossRef]
- Wlodarczyk, M.T. Enhanced Platinum (II) Drug Delivery for Anti-Cancer Therapy. Ph.D. Thesis, City University of New York, New York, NY, USA, 2021. [Google Scholar]
- Dragulska, S.A.; Chen, Y.; Wlodarczyk, M.T.; Poursharifi, M.; Dottino, P.; Ulijn, R.V.; Martignetti, J.A.; Mieszawska, A.J. Tripeptide-Stabilized Oil-in-Water Nanoemulsion of an Oleic Acids–Platinum(II) Conjugate as an Anticancer Nanomedicine. Bioconjug. Chem. 2018, 29, 2514–2519. [Google Scholar] [CrossRef]
- Mabey, W.; Mill, T. Critical review of hydrolysis of organic compounds in water under environmental conditions. J. Phys. Chem. Ref. Data 1978, 7, 383–415. [Google Scholar] [CrossRef]
- Canovese, L.; Cattalini, L.; Chessa, G.; Tobe, M.L. Kinetics of the displacement of cyclobutane-1,1-dicarboxylate from diammine(cyclobutane-1,1-dicarboxylato)platinum(II) in aqueous solution. J. Chem. Soc. Dalton Trans. 1988, 2135–2140. [Google Scholar] [CrossRef]
- Hay, R.W.; Miller, S. Reactions of platinum(II) anticancer drugs. Kinetics of acid hydrolysis of cis-diammine(cyclobutane-1,1-dicarboxylato)platinum(II) “Carboplatin”. Polyhedron 1998, 17, 2337–2343. [Google Scholar] [CrossRef]
- Yu, W.; Liu, R.; Zhou, Y.; Gao, H. Size-Tunable Strategies for a Tumor Targeted Drug Delivery System. ACS Cent. Sci. 2020, 6, 100–116. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, S.K.; Monsky, W.L.; Yuan, F.; Roberts, W.G.; Griffith, L.; Torchilin, V.P.; Jain, R.K. Regulation of transport pathways in tumor vessels: Role of tumor type and microenvironment. Proc. Natl. Acad. Sci. USA 1998, 95, 4607–4612. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Gao, S.; Yang, D.; Fang, Y.; Lin, X.; Jin, X.; Liu, Y.; Liu, X.; Su, K.; Shi, K. Influencing factors and strategies of enhancing nanoparticles into tumors in vivo. Acta Pharm. Sin. B 2021, 11, 2265–2285. [Google Scholar] [CrossRef]
- Miyauchi, K.; Ogawa, M.; Shibata, T.; Matsuda, K.; Mori, T.; Ito, K.; Minamiura, N.; Yamamoto, T. Development of a radioimmunoassay for human deoxyribonuclease I. Clin. Chim. Acta 1986, 154, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, T. Deoxyribonuclease II (DNase II) activity in mouse tissues and body fluids. Exp. Anim. 1995, 44, 169–171. [Google Scholar] [CrossRef]
- Hahn, J.; Wickham, S.F.J.; Shih, W.M.; Perrault, S.D. Addressing the Instability of DNA Nanostructures in Tissue Culture. ACS Nano 2014, 8, 8765–8775. [Google Scholar] [CrossRef]
- Vaupel, P.; Kallinowski, F.; Okunieff, P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: A review. Cancer Res. 1989, 49, 6449–6465. [Google Scholar]
- Griffiths, J.R.; McIntyre, D.J.; Howe, F.A.; Stubbs, M. Why are cancers acidic? A carrier-mediated diffusion model for H+ transport in the interstitial fluid. Novartis Found Symp. 2001, 240, 46–62; discussion 62–47, 152–153. [Google Scholar]
- Attia, M.F.; Anton, N.; Wallyn, J.; Omran, Z.; Vandamme, T.F. An overview of active and passive targeting strategies to improve the nanocarriers efficiency to tumour sites. J. Pharm. Pharmacol. 2019, 71, 1185–1198. [Google Scholar] [CrossRef]
- Li, S. Hydrolytic degradation characteristics of aliphatic polyesters derived from lactic and glycolic acids. J. Biomed. Mater. Res. 1999, 48, 342–353. [Google Scholar] [CrossRef]
- Deng, J.; Wang, L.; Chen, H.; Li, L.; Ma, Y.; Ni, J.; Li, Y. The role of tumour-associated MUC1 in epithelial ovarian cancer metastasis and progression. Cancer Metastasis Rev. 2013, 32, 535–551. [Google Scholar] [CrossRef]
- Lee, D.-H.; Choi, S.; Park, Y.; Jin, H.-S. Mucin1 and Mucin16: Therapeutic Targets for Cancer Therapy. Pharmaceuticals 2021, 14, 1053. [Google Scholar] [CrossRef]
- Parker, R.J.; Eastman, A.; Bostick-Bruton, F.; Reed, E. Acquired cisplatin resistance in human ovarian cancer cells is associated with enhanced repair of cisplatin-DNA lesions and reduced drug accumulation. J. Clin. Investig. 1991, 87, 772–777. [Google Scholar] [CrossRef]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Wlodarczyk, M.T.; Dragulska, S.A.; Camacho-Vanegas, O.; Dottino, P.R.; Jarzęcki, A.A.; Martignetti, J.A.; Mieszawska, A.J. Platinum(II) Complex-Nuclear Localization Sequence Peptide Hybrid for Overcoming Platinum Resistance in Cancer Therapy. ACS Biomater. Sci. Eng. 2018, 4, 463–467. [Google Scholar] [CrossRef]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wlodarczyk, M.T.; Dragulska, S.A.; Chen, Y.; Poursharifi, M.; Acosta Santiago, M.; Martignetti, J.A.; Mieszawska, A.J. Pt(II)-PLGA Hybrid in a pH-Responsive Nanoparticle System Targeting Ovarian Cancer. Pharmaceutics 2023, 15, 607. https://doi.org/10.3390/pharmaceutics15020607
Wlodarczyk MT, Dragulska SA, Chen Y, Poursharifi M, Acosta Santiago M, Martignetti JA, Mieszawska AJ. Pt(II)-PLGA Hybrid in a pH-Responsive Nanoparticle System Targeting Ovarian Cancer. Pharmaceutics. 2023; 15(2):607. https://doi.org/10.3390/pharmaceutics15020607
Chicago/Turabian StyleWlodarczyk, Marek T., Sylwia A. Dragulska, Ying Chen, Mina Poursharifi, Maxier Acosta Santiago, John A. Martignetti, and Aneta J. Mieszawska. 2023. "Pt(II)-PLGA Hybrid in a pH-Responsive Nanoparticle System Targeting Ovarian Cancer" Pharmaceutics 15, no. 2: 607. https://doi.org/10.3390/pharmaceutics15020607
APA StyleWlodarczyk, M. T., Dragulska, S. A., Chen, Y., Poursharifi, M., Acosta Santiago, M., Martignetti, J. A., & Mieszawska, A. J. (2023). Pt(II)-PLGA Hybrid in a pH-Responsive Nanoparticle System Targeting Ovarian Cancer. Pharmaceutics, 15(2), 607. https://doi.org/10.3390/pharmaceutics15020607