
Distribution and Evolutionary History of the Mobile Genetic Element s2m in Coronaviruses

Abstract
:1. Introduction
2. Coronavirus Phylogeny and Distribution of s2m
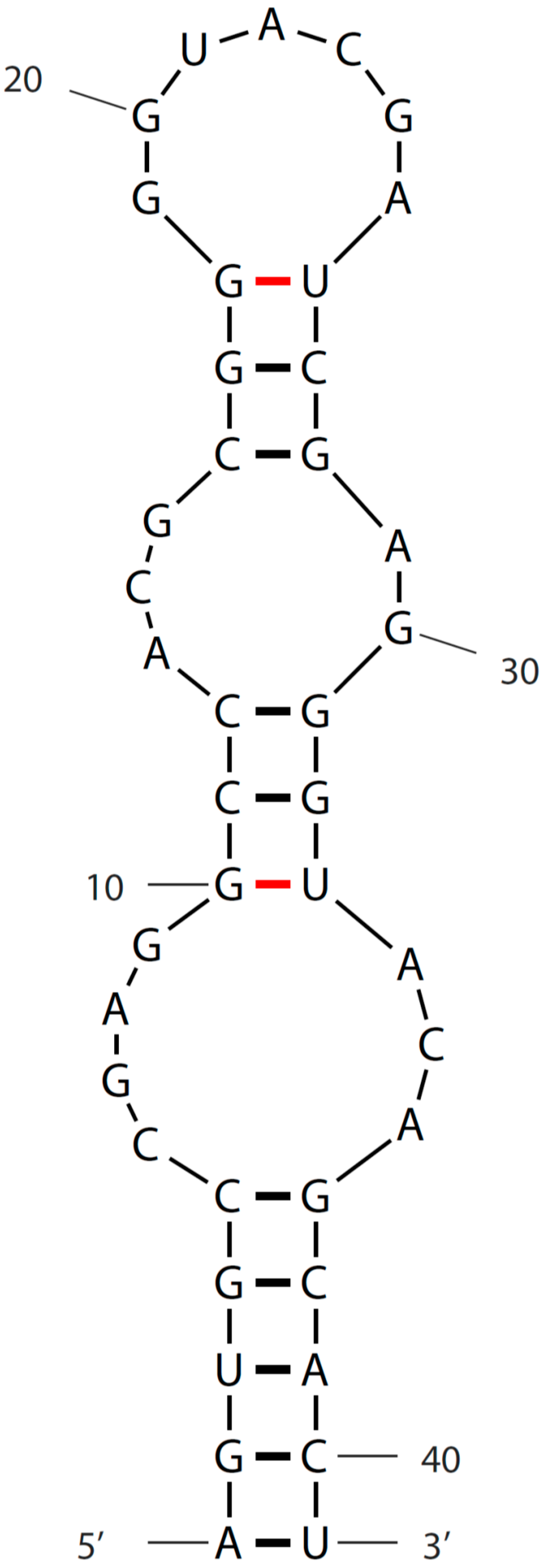
3. Sequence and Secondary Structure of s2m
4. Function of s2m
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yang, D.; Leibowitz, J.L. The structure and functions of coronavirus genomic 3′ and 5′ ends. Virus Res. 2015, 206, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, B.L.; White, K.A. Exploring the architecture of viral RNA genomes. Curr. Opin. Virol. 2015, 12, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Liu, P.; Giedroc, D.P.; Leibowitz, J. Mouse hepatitis virus stem-loop 4 functions as a spacer element required to drive subgenomic RNA synthesis. J. Virol. 2011, 85, 9199–9209. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Li, L.; Millership, J.J.; Kang, H.; Leibowitz, J.L.; Giedroc, D.P. A U-turn motif-containing stem-loop in the coronavirus 5′ untranslated region plays a functional role in replication. RNA 2007, 13, 763–780. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.D.; Chang, R.Y.; Brian, D.A. A phylogenetically conserved hairpin-type 3′ untranslated region pseudoknot functions in coronavirus RNA replication. J. Virol. 1999, 73, 8349–8355. [Google Scholar] [PubMed]
- Tengs, T.; Kristoffersen, A.B.; Bachvaroff, T.R.; Jonassen, C.M. A mobile genetic element with unknown function found in distantly related viruses. Virol. J. 2013, 10, 132. [Google Scholar] [CrossRef] [PubMed]
- Monceyron, C.; Grinde, B.; Jonassen, T.O. Molecular characterisation of the 3′-end of the astrovirus genome. Arch. Virol. 1997, 142, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Jonassen, T.O.; Monceyron, C.; Lee, T.W.; Kurtz, J.B.; Grinde, B. Detection of all serotypes of human astrovirus by the polymerase chain reaction. J. Virol. Methods 1995, 52, 327–334. [Google Scholar] [CrossRef]
- Robertson, M.P.; Igel, H.; Baertsch, R.; Haussler, D.; Ares, M., Jr.; Scott, W.G. The structure of a rigorously conserved RNA element within the SARS virus genome. PLoS Biol. 2005, 3, e5. [Google Scholar] [CrossRef] [PubMed]
- Jonassen, C.M. Detection and sequence characterization of the 3′-end of coronavirus genomes harboring the highly conserved RNA motif s2m. Methods Mol. Biol. 2008, 454, 27–34. [Google Scholar] [PubMed]
- Jonassen, C.M.; Jonassen, T.O.; Grinde, B. A common RNA motif in the 3′ end of the genomes of astroviruses, avian infectious bronchitis virus and an equine rhinovirus. J. Gen. Virol. 1998, 79 Pt 4, 715–718. [Google Scholar] [CrossRef] [PubMed]
- Schutze, H.; Ulferts, R.; Schelle, B.; Bayer, S.; Granzow, H.; Hoffmann, B.; Mettenleiter, T.C.; Ziebuhr, J. Characterization of white bream virus reveals a novel genetic cluster of nidoviruses. J. Virol. 2006, 80, 11598–11609. [Google Scholar] [CrossRef] [PubMed]
- RAPID RISK ASSESSMENT. Severe Respiratory Disease Associated with a Novel Coronavirus. Available online: http://ecdc.europa.eu/en/publications/Publications/novel-coronavirus-rapid-risk-assessment-update.pdf (accessed on 26 July 2016).
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Varani, G.; McClain, W.H. The G × U wobble base pair. A fundamental building block of RNA structure crucial to RNA function in diverse biological systems. EMBO Rep. 2000, 1, 18–23. [Google Scholar] [CrossRef] [PubMed]
- The mfold Web Server. Available online: http://unafold.rna.albany.edu/?q=mfold (accessed on 26 July 2016).
- Goebel, S.J.; Taylor, J.; Masters, P.S. The 3′ cis-acting genomic replication element of the severe acute respiratory syndrome coronavirus can function in the murine coronavirus genome. J. Virology 2004, 78, 7846–7851. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; State Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, Harbin, China. Personal communication, 2012.
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Shapiro, J.S.; Varble, A.; Pham, A.M.; Tenoever, B.R. Noncanonical cytoplasmic processing of viral microRNAs. RNA 2010, 16, 2068–2074. [Google Scholar] [CrossRef] [PubMed]
- Boss, I.W.; Renne, R. Viral miRNAs and immune evasion. Biochim. Biophys. Acta 2011, 1809, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Hackenberg, M.; Rodriguez-Ezpeleta, N.; Aransay, A.M. Miranalyzer: An update on the detection and analysis of microRNAs in high-throughput sequencing experiments. Nucleic Acids Res. 2011, 39, W132–W138. [Google Scholar] [CrossRef] [PubMed]
- Bredenbeek, P.J.; Pachuk, C.J.; Noten, A.F.; Charite, J.; Luytjes, W.; Weiss, S.R.; Spaan, W.J. The primary structure and expression of the second open reading frame of the polymerase gene of the coronavirus MHV-A59; a highly conserved polymerase is expressed by an efficient ribosomal frameshifting mechanism. Nucleic Acids Res. 1990, 18, 1825–1832. [Google Scholar] [CrossRef] [PubMed]
- Sawicki, S.G.; Sawicki, D.L. A new model for coronavirus transcription. Adv. Exp. Med. Biol. 1998, 440, 215–219. [Google Scholar] [PubMed]
- Silva, P.A.G.C.; Pereira, C.F.; Dalebout, T.J.; Spaan, W.J.M.; Bredenbeek, P.J. An RNA pseudoknot is required for production of yellow fever virus subgenomic RNA by the host nuclease XRN1. J. Virol. 2010, 84, 11395–11406. [Google Scholar] [CrossRef] [PubMed]
- Pijlman, G.P.; Funk, A.; Kondratieva, N.; Leung, J.; Torres, S.; van der Aa, L.; Liu, W.J.; Palmenberg, A.C.; Shi, P.Y.; Hall, R.A.; et al. A highly structured, nuclease-resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell Host Microbe 2008, 4, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Chlebowski, A.; Lubas, M.; Jensen, T.H.; Dziembowski, A. RNA decay machines: The exosome. Biochim. Biophys. Acta 2013, 1829, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Villordo, S.M.; Gamarnik, A.V. Genome cyclization as strategy for flavivirus RNA replication. Virus Res. 2009, 139, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Fricke, M.; Dunnes, N.; Zayas, M.; Bartenschlager, R.; Niepmann, M.; Marz, M. Conserved RNA secondary structures and long-range interactions in hepatitis C viruses. RNA 2015, 21, 1219–1232. [Google Scholar] [CrossRef] [PubMed]
- Geigenmuller, U.; Ginzton, N.H.; Matsui, S.M. Construction of a genome-length cDNA clone for human astrovirus serotype 1 and synthesis of infectious RNA transcripts. J. Virol. 1997, 71, 1713–1717. [Google Scholar] [PubMed]
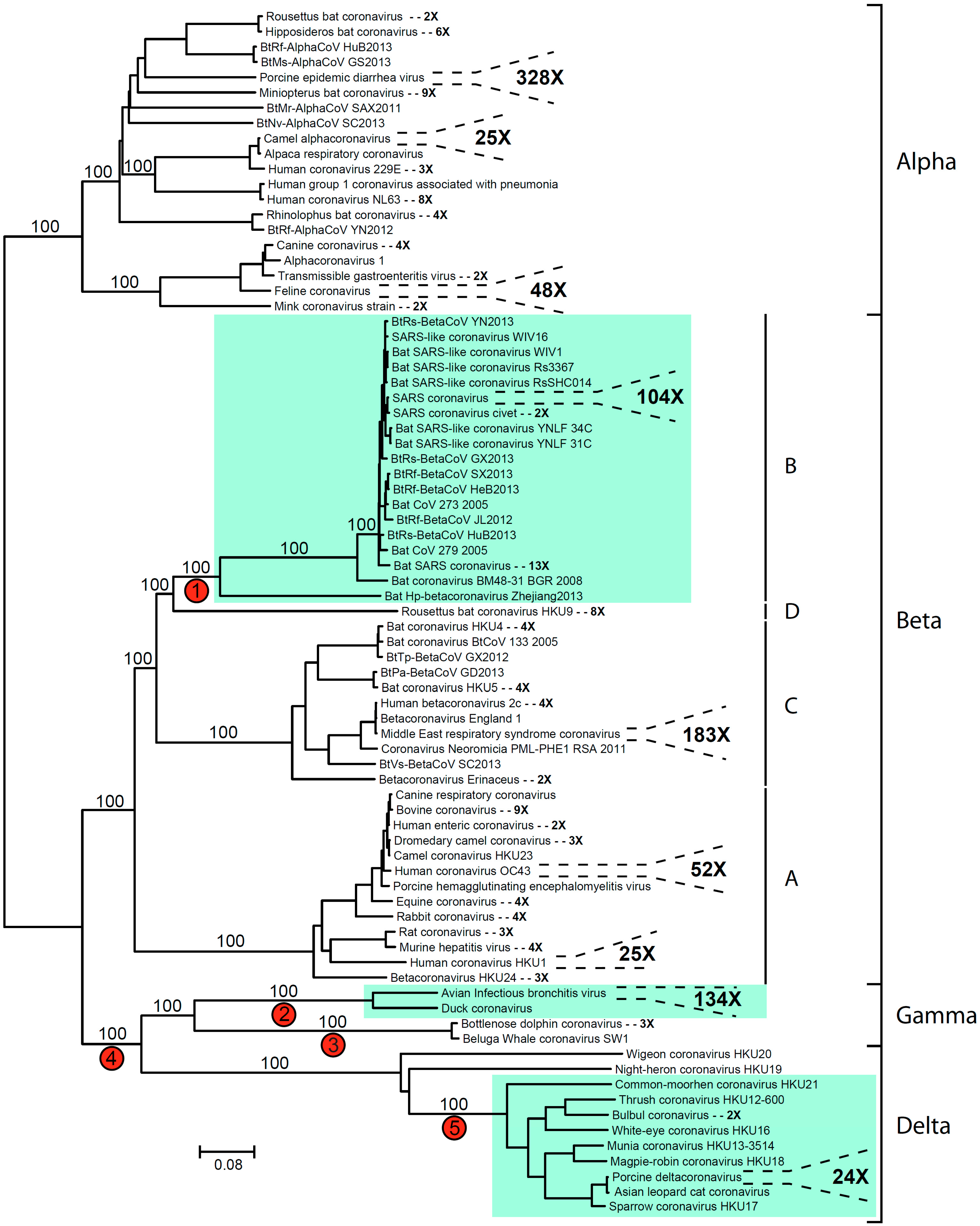


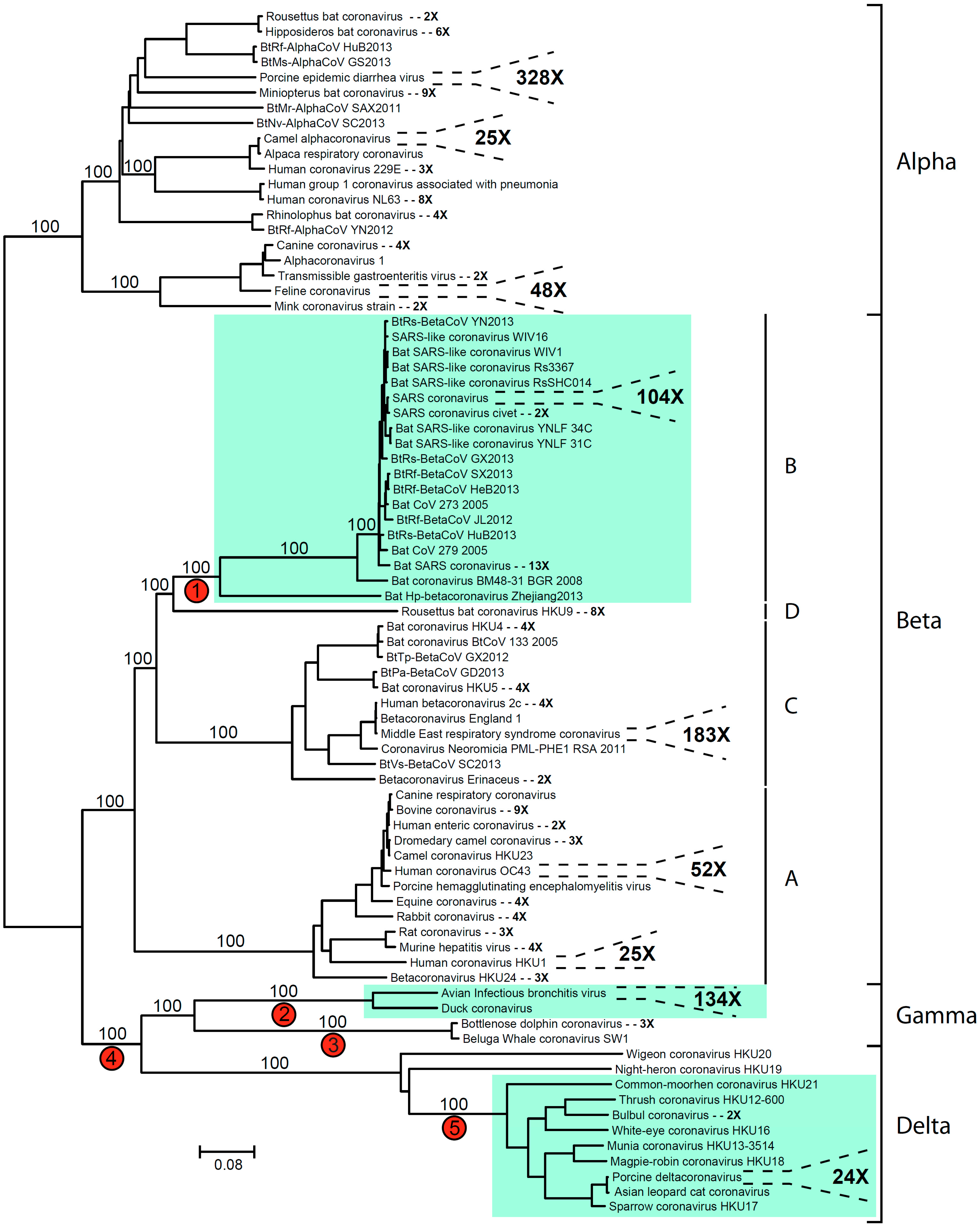{kind=link}
{kind=link}
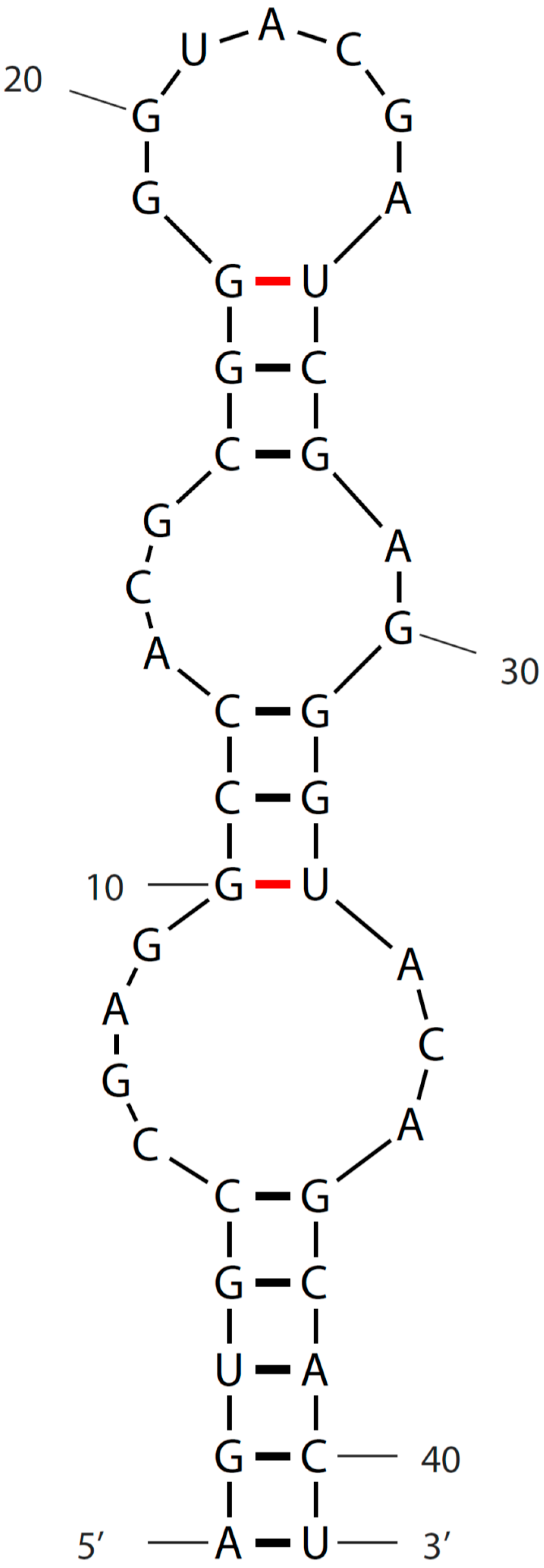{kind=link}
| Total | Containing s2m | |
|---|---|---|
| Coronavirus sequences | 20,068 | 706 (3.5%) |
| Alpha coronavirus * | 7190 | 0 |
| Beta coronavirus * | 4947 | 342 (6.9%) |
| Delta coronavirus * | 141 | 60 (42.6%) |
| Gamma coronavirus * | 6360 | 281 (4.4%) |
| Bafinivirus * | 12 | 0 |
| Torovirus * | 307 | 0 |
| Complete genomes | 1507 | 523 (34.7%) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tengs, T.; Jonassen, C.M. Distribution and Evolutionary History of the Mobile Genetic Element s2m in Coronaviruses. Diseases 2016, 4, 27. https://doi.org/10.3390/diseases4030027
Tengs T, Jonassen CM. Distribution and Evolutionary History of the Mobile Genetic Element s2m in Coronaviruses. Diseases. 2016; 4(3):27. https://doi.org/10.3390/diseases4030027
Chicago/Turabian StyleTengs, Torstein, and Christine M. Jonassen. 2016. "Distribution and Evolutionary History of the Mobile Genetic Element s2m in Coronaviruses" Diseases 4, no. 3: 27. https://doi.org/10.3390/diseases4030027
APA StyleTengs, T., & Jonassen, C. M. (2016). Distribution and Evolutionary History of the Mobile Genetic Element s2m in Coronaviruses. Diseases, 4(3), 27. https://doi.org/10.3390/diseases4030027