
Puzzle Pieces: Neural Structure and Function in Prader-Willi Syndrome

Abstract
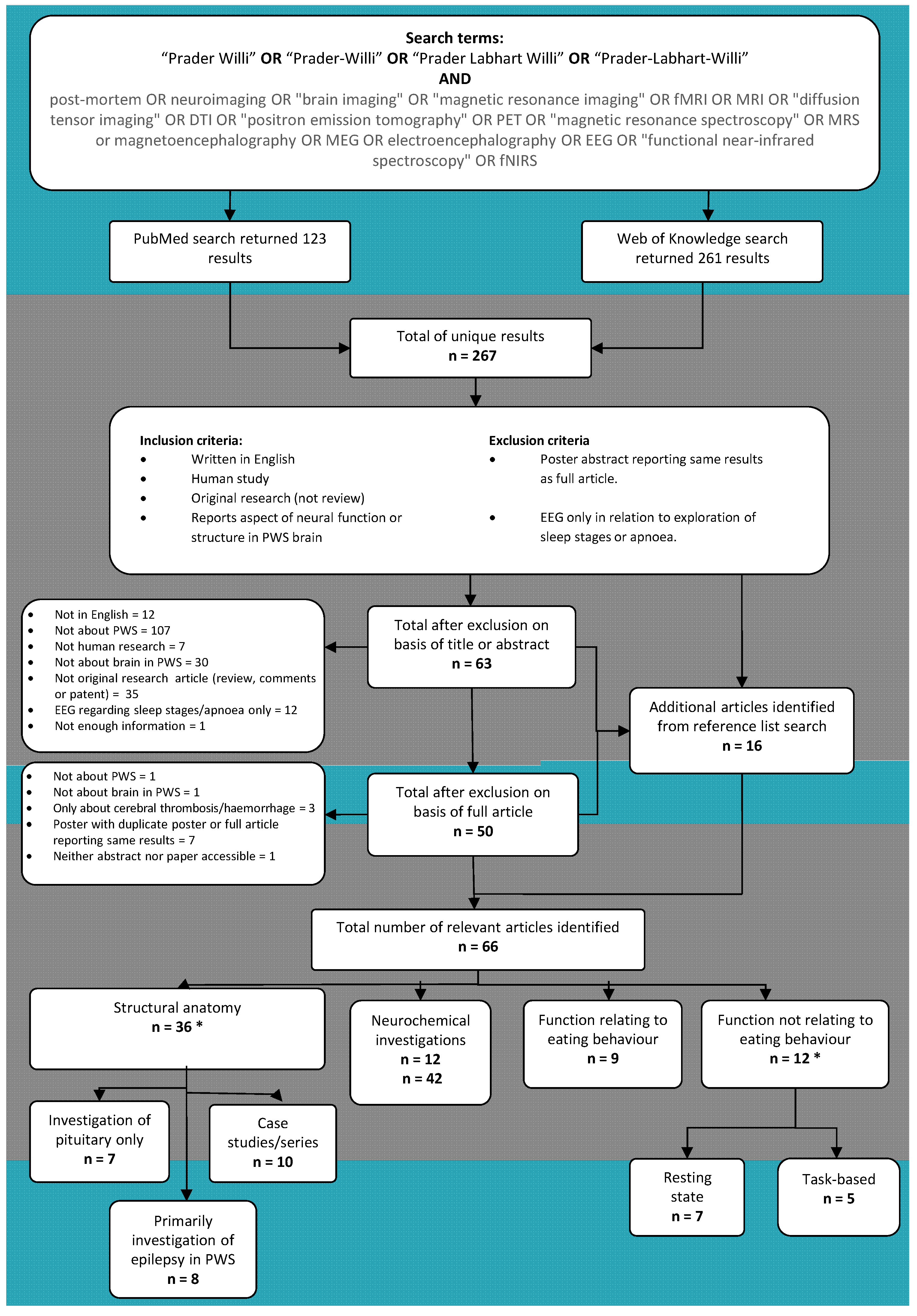
:1. Introduction
2. Literature Search
3. Anatomical Structure of the PWS Brain


{kind=link}
| Author | Methods | Sample | Key findings |
|---|---|---|---|
| Cacciari et al. 1990 [9] | MRI—pituitary evaluation only | 1 PWS patient in larger study of 101 patients (15 y) | No abnormality found. |
| Crino et al. 2008 [10] | MRI—pituitary evaluation only | 8 y old male with PWS | No abnormality found. |
| Fan et al. 2005 [15] | Retrospective case reviews of available MRI data | MRI available for 9/10 patients with seizures from total PWS sample of 56 (31 males, 1–37 y) | MRI normal for 8/9; haemorrhage in 1/9. |
| Gilboa & Gross-Tsur 2013 [16] | Retrospective case reviews of available imaging data for patients: MRI, CT, ultrasonography | Imaging data available for 59 of 125 PWS patients (50% male, 1 month–48 y): MRI for 30; CT for 17; ultrasonography for 12 | Abnormalities in 20/59, incl. 4/5 with epilepsy: including ventriculomegaly (3/59); thalamic abnormalities (3/59); partial corpus callosum agenesis (3/59); disturbed myelination (3/59); partial empty sellar (1/59). |
| Grugni et al. 2000 [14] | MRI—pituitary only. | 17 PWS patients (9 males, mean 20 y) | Pituitary hypoplasia (10/17); empty sella (3/17). |
| Hashimoto et al. 1998 [17] | MRI and 1H-MRS | 5 PWS patients (2 males, 1–14.5 y) 37 control infants and children (1–15 y) | Abnormalities in 5/5: mild ventriculomegaly (4/5); frontal cortical atrophy (3/5); small brainstem (1/5); delayed myelination (1/5). Metabolite abnormalities: Reduced NAA peak in; NAA:Choline and NAA:Creatine ratios tended to be lower in PWS group with a signification relationship between these metabolite rations and developmental level. |
| Hayashi et al. 1992 [18] | Post-mortem | 6 month old female with PWS | No pituitary abnormality; abnormalities of cortical gyrification, cerebellar white matter and dentate gyrus. |
| Honea et al. 2012 [19] | MRI | 23 participants with PWS (8 males, 10–39 y) 25 typically-developing control participants (11 males, 10–48 y) | Deletion vs. UPD: reduced grey matter volume in PFC & temporal cortex and reduced white matter volume in parietal cortex in deletion; reduced grey and white matter volume in OFC and limbic cortex in UPD. |
| Iughetti et al. 2008 [12] | Retrospective case reviews of available MRI data | 91 children with PWS (49 males, 0.7–16.8 y) | Abnormalities in 61/91: reduced pituitary height (45/91); absence of posterior pituitary bright spot (6/91); ventriculomegaly (8/91); thin corpus callosum (2/91). |
| Kumada et al. 2005 [20] | Retrospective case reviews of available MRI data | Imaging data available for 3/4 patients with PWS and epilepsy (1 males, 3–12 y) in larger study of chromosomal abnormalities and epilepsy | Cerebral atrophy in 1/3. |
| Leonard et al. 1993 [21] | MRI | 4 children with PWS (3 males, 3–10 y) 6 with AS (3 males, 2–7 y) | Abnormality in sylvian fissure of 1/4 PWS children (unilateral), but 6/6 AS (2 bilateral, 4 unilateral). |
| Linnemann et al. 1999 [11] | MRI case study | 6 y old male with PWS | Small flat pituitary; no further abnormalities. |
| Lukoshe et al. 2013 [22] | MRI | 20 children with PWS (9 males, 6–17 y) 11 sibling control participants (8 males, 7–15 y) | Significantly reduced brainstem volume in PWS group compared to control group; trend to reduced white matter volume and total cortical surface area in PWS. Greater ventriculomegaly and surface CSF in UPD compared to deletion subtypes. |
| Lukoshe et al. 2014 [23] | MRI | 24 children with PWS (9 males, 6–18 y) 11 sibling control participants (8 males, 7–15 y) | Decreased cortical complexity in four clusters in frontal, temporal and parietal lobes. Cortical complexity positively correlated with IQ. Greater cortical thickness and lower cortical complexity in some clusters in UPD. |
| Mantoulan et al. 2011 [24] | MRI | 9 PWS teenagers (6 males, 12.7–18.6y, mean 16.4 y) 9 typically developing young adults (6 males, mean 21.2 y) | No abnormalities found. |
| Maski et al. 2009 [25] | Retrospective case reviews of available “neuroimaging” data: unspecified modality | Imaging data available for 14/21 PWS patients with seizures | Unspecified non-focal abnormalities in 3/14. |
| Miller et al. 1996 [26] | MRI | 15 individuals with PWS (6 males, 2–28 y) 16 age- & sex-matched group with isolated growth hormone deficiency (10 males, 3–14 y) 49 age-matched typically-developing controls from previous literature (1–29 y) | No significant difference in pituitary height. Posterior pituitary bright spot absent in 3/15 PWS participants and reduced in 1/15. |
| Miller et al. 2006 [27] | MRI | 17 PWS participants (11 males, 4–39 y) 18 individuals with early-onset morbid obesity (EMO) (9 males, 4–22 y) 21 typically-developing siblings (8 males, 3.5–43 y) | White matter abnormalities in 6/8 adults with PWS and 5 participants with EMO, but 0/9 children with PWS and none in typically-developing controls participants. |
| Miller et al. 2007 [28] | MRI | 27 PWS participants (16 males, 3 m–39 y) 13 EMO participants (4 males, 4–16 y) 26 typically developing siblings (10 males, 2 m–43 y). | Incomplete insular closure in PWS, compared to both sibling and EMO control groups. |
| Miller et al. 2007 [29] | MRI | 20 PWS participants (12 males, 3 m–39 y) 16 individuals with EMO (8 males, 15 m–22 y) 21 typically-developing siblings (10 males, 2–43 y) | Abnormalities in all 20 PWS participants: ventriculomegaly (20/20); incomplete closure of the insula (13/20); sylvian fissure polymicrogyria (12/20); decreased parietal-occipital volume (10/20). None of these abnormalities in either EMO or sibling control groups. |
| Miller et al. 2008 [30] | MRI—pituitary evaluation only | 27 individuals with PWS (16 males, 3 m–39 y) 16 individuals with EMO (6 males, 4-22 y) 25 typically-developing siblings (10 males, 2–43 y) | Pituitary abnormalities in 20/27 PWS, 10/16 EMO & 2/25 controls. Abnormality not specific to PWS. |
| Miller et al. 2009 [31] | MRI | 16 PWS participants (10 males, mean 16.53 y) 12 EMO participants (4 males, mean 9.25 y) 15 typically-developing siblings (5 males, mean 12.08 y) | Reduced cerebellar volume and cerebellar/cerebral ratio in PWS and EMO compared to sibling group, but no difference between PWS and EMO. |
| Ogura et al. 2011 [32] | MRI | 12 adults with PWS (6 males, 19–31 y) 13 age- & sex-matched controls (6 males, 19–29 y) | Reduced total brain, grey matter and white matter volume, and focally reduced OFC and somatomotor area volume in PWS. |
| Stevenson et al. 2004 [33] | MRI and post-mortem | Post-mortem: 5 month, 9 month, & 3.5 y old with PWS (2 males) MRI: 9 month old female with PWS | Cortical grey & white matter abnormalities in 3/4; midbrain, hindbrain & cerebellar abnormalities in 1/4; normal in 1/4. |
| Takeshita et al. 2013 [34] | Retrospective case reviews of available imaging data for patients: MRI, CT | MRI available for 9/31 PWS patients with seizures (6 males, neonatal–3 y) | Diffuse atrophy in 1/9, but appears to have followed some injury to the brain of undisclosed nature. 8/9 within normal limits. |
| Tauber et al. 2000 [13] | Retrospective case reviews of available MRI data—pituitary only | MRI for 16/28 with PWS (mean 11.8 y) | Pituitary hypoplasia in 10/16. |
| van Nieuwpoort et al. 2011 [35] | MRI—pituitary only | 15 adults with PWS (4 males, 19.2–42.9 y) 14 typically-developing siblings (7 males; 17.5–41.3 y) | Reduced anterior pituitary size in 12/15 adults with PWS compared to sibling group. |
| Vendrame et al. 2010 [36] | Retrospective case reviews of available MRI data | Imaging available for 20/30 PWS patients with seizures (6 males, 4–21 y) | Abnormalities in 5/20: ventriculomegaly (4/20); diffuse cortical atrophy (1/20). |
| Verrotti et al. 2015 [37] | Retrospective case reviews of available imaging data for patients: MRI, CT | Imaging data available for 35/28 PWS patients with seizures (22 males, seizure onset 2 days–11 y, age at imaging evaluation unspecified): MRI for 25; CT for 10 | Abnormalities in 11/35: ventriculomegaly (5/35), cortical atrophy (4/35), corpus callosum hypoplasia (1/35), periventricular leukomalacia (1/35). |
| Yamada et al. 2006 [38] | DTI | 8 participants with PWS (6 males, 8–29 y) 8 age- & sex-matched typically-developing controls (6 males, 8–29 y) | Atypical diffusivity indicating abnormalities of frontal white matter, posterior limb of internal capsule, and splenium of corpus callosum. |
| Yoshii et al. 2002 [39] | MRI case study | 40 week old female with PWS | Abnormal gyrification and cortical grey-white matter boundaries; partially uncovered right insula. |
| Study | Methods | Sample | Mai findings |
|---|---|---|---|
| Akefeldt et al. 1997 [40] | EEG: Auditory brainstem response. | 7 participants with PWS (6 males, 4–25 y) 7 control participants with intellectual disability (5 males, 5–26 y) | Atypicalities of auditory brainstem response compared to control group and laboratory reference values. |
| Dimitropoulos & Schultz 2008 [41] | fMRI: response to high vs. low calorie food images. | 9 participants with PWS (3 males, 8–38 y) 10 IQ and BMI matched control participants (4 males, 19–29 y) | Increased amygdala, hypothalamus, insula and OFC activity when fasted in response to images of high vs. low calorie foods. |
| Halit et al. 2008 [42] | EEG: face and gaze perception. | 8 adults with deletion subtype of PWS (20–53 y) 8 with UPD subtype of PWS (19–52 y) | Behavioural performance impaired for both genotypes, but ERP showed increased impairment in the deletion group: N170 amplitude larger for averted vs. direct gaze and inverted vs. upright faces in UPD, but not deletion group. |
| Hinton et al. 2006 [43] | PET: pre- and post-meal (400 & 1200 kcal) response to food images. | 13 participants with PWS (22–42 y) | Increased activity in medial OFC, temporal cortex and PFC usually found not seen in PWS following meals when viewing food images. |
| Hinton et al. 2006 [44] | PET: response to high and low incentive food images. | 13 participants with PWS (22–42 y) | Increased activation of amygdala and medial OFC typically seen in response to high vs. low incentive foods not found. |
| Holsen et al. 2006 [45] | fMRI: pre- and post-meal (500 kcal) response to food images and control animal images | 9 participants with PWS (1 male, mean 14.7 y) 9 age matched typically-developing controls (3 males, mean 14.4 y) | PWS group showed reduced activity when viewing food vs. animal images in medial PFC and OFC in pre-meal condition than control group and greater medial PFC, insula, parahippocampal gyrus and amygdala activity in post-meal condition. |
| Holsen et al. 2009 [46] | fMRI: pre- and post-meal (500 kcal) responses to food images and control animal images. | 9 participants with PWS deletion subtype (2 males; mean 24.4 y) 9 participants with PWS UPD subtype (3 males; mean 20.8 y) 9 healthy weight controls (3 males; mean 23.6 y) | Both PWS subtypes showed atypical response in both pre- and post-meal conditions compared to control group. Deletion vs. UPD: greater activity in deletion group in frontal/limbic areas, especially medial PFC and amygdala, in both pre- and post-meal conditions; greater activity in dorsolateral PFC and parahippocampal gyrus in UPD group in post-meal condition only. |
| Holsen et al. 2012 [47] | fMRI: pre- and post-meal 500 kcal) responses to food images and control animal images. | 14 participants with PWS (2 males, mean 23.3 y) 14 BMI & age-matched control participants (5 males; mean 25.0 y) 15 healthy weight age-matched controls (6 males; mean 23.1 y) | Pre-meal: increased activity when viewing food vs. non-food images in nucleus accumbens and amygdala in PWS than either control group control group, and lower in hypothalamus and hippocampus. Post-meal: increased activity in hypothalamus, amygdala and hippocampus in PWS than either control group, but higher dorsolateral PFC and OFC in obese group not seen in PWS or healthy weight control group. |
| Key & Dykens 2008 [48] | EEG: N1 & P3 ERP response to food images according to categorisation/discrimination of food composition and quality. | 9 participants with deletion subtype of PWS (2 males, mean 22.9 y) 8 participants with UPD subtype of PWS (3 males, mean 22.4 y) 9 age-matched control participants (4 males, mean 21.7 y) | N1 ERP suggested deletion group early processing by categorising mainly according to quantity, whilst UPD did so by quality and suitability for consumption more similarly to the control group. Later P3 response showed deletion group could discriminate foods by combinational suitability, but this later processing response was greater than that seen in UPD. Later P3 motivational processing of food images not seen in control group. |
| Kim et al. 2006 [49] | Resting-state PET while fasted. | 16 children with PWS (9 males, mean 4.2 y) 7 typically-developing siblings/relatives (4 males, mean 4.0 y) | PWS group showed decreased metabolism in right superior temporal gyrus and left verebellar vermis, and increased metabolism in right OFC, bilateral medial PFC, right inferior and left superior frontal cortex, bilateral ACC, right temporal pole and left uncus. |
| Klabunde et al. 2015 [50] | fMRI: sessions where participants engaged in skin picking compared to sessions where they did no. | 17 participants with PWS (11 males, mean 15.7 y) | 2 main clusters showing greater activation during skin picking: right ACC & right middle frontal gyrus; primary somatosensory cortex, left inferior parietal lobule, supplementary motor area, left middle frontal gyrus and right posterior insula. Self-injury trauma scale scores negatively correlated with right insula and left precentral gyrus activity. |
| Mantoulan et al. 2011 [24] | Resting-state PET | 9 PWS teenagers (6 males; 12.7–18.6y, mean 16.4 y) 9 typically developing young adults (6 males; mean 21.2 y) | Hypoperfusion in PWS, most strongly in ACC and superior temporal regions, but also in right orbitofrontal gyrus and postcentral gyrus. Positive correlations between Child Behaviour Checklist (CBCL) scale scores and rCBF in ACC (activity, social, & attention scales), superior temporal gyrus (attention & social scales), and superior frontal gyrus (activity scale). Negative correlation between rCBF in ACC and CBCL depression scale score. |
| Miller et al. 2007 [51] | fMRI: response to food images following glucose load. | 8 participants with PWS (6 males, mean 25 y) 8 typically-developing siblings (4 males, mean 27 y) | Significantly increased activity ventromedial PFC in PWS group when viewing food images following glucose load. |
| Ogura et al. 2013 [52] | Resting state PET | 12 participants with PWS (6 males, 19–31 y) 13 age- & gender-matched controls (6 males, 19–29 y) | Decrease metabolism in PWs group in the lingual gyri, cerebellum, right thalamus and left insula. Increased metabolism in bilateral angular and inferior frontal gyri and left middle frontal gyrus. Negative correlation between questionnaire score of eating severity and rCBF in left insula. |
| Pujol et al. 2014 [53] | Resting state fMRI | 24 adults with PWS (12 males, mean 26.3 y) 20 adults with Down’s syndrome (10 males, mean 24.5 y) 20 adults with William’s syndrome (11 males, mean 25.2 y) 80 young adults (45 males, mean 26.4 y) 71 children (30 males, mean 9.6 y) 53 older adults (24 males, mean 67.4 y) | Young adults showed sensorimotor system activity positively correlating with motion, suggesting system specific effects. Seen to some extent in children and older adults but also wider effects of motion. Greater motion in genetic disorders, and specific motion-connectivity correlations: PWS group shoed mostly frontal and temporal lobe correlations with motion, but also significant in the dorsal ACC; in Down’s syndrome group was with anterior and dorsal regions; and in William’s syndrome were diffuse correlations across grey matter voxels. |
| Shapira et al. 2005 [54] | fMRI: before and after glucose load. | 3 participants with PWS (1 male, 25–38 y) | Delay in activation of brain areas associated in satiety response in previous study of participants without PWS, including hypothalamus, insula, ventromedial PFC and nucleus accumbens following oral glucose load. |
| Stauder et al. 2002 [55] | EEG: P3 ERP response to visual and auditory oddball tasks. | 10 adults with PWS (5 males, mean 30.8 y) 10 typically-developing controls (3 males, mean 23.2 y) | Markedly decreased P3 response in both visual and auditory tasks in PWS compared to control group, and most strongly for the auditory task. |
| Stauder et al. 2005 [56] | EEG: N200 and P300 ERPs during response inhibition on Go-Nogo task. | 11 participants with deletion subtype of PWS (7 males, mean 26.7 y) 11 participants with UPD subtype of PWS (4 males, mean 27.7 y) 11 typically-developing control participants (6 males, mean 27.3 y) | Behavioural task performance poorer for both PWS groups compared to control group. N200 amplitude didn’t show normal peak in either PWS group, suggesting impaired early modality specific inhibition in both UPD and deletion, but only UPD group showed impaired P300 modulation, indicative of later general inhibition. |
| Woodcock et al. 2010 [57] | fMRI: set-shifting task. | 8 participants with PWS (5 males, mean 20.7 y) 8 age & gender matched typically-developing control participants (mean 21 y) | Control group showed increased activity compared to the PWS group in frontoparietal regions during switching, including in ventromedial PFC and posterior parietal cortex. PWS tended to show ventromedial PFC deactivation instead. Group interactions: Greater activity in hippocampus, amygdala, thalamus and putamen during switching in control group than PWS group, driven by deactivation of these areas by PWS group during switching alongside activation in the control group. The reverse pattern was found for the anterior poles. |
| Zhang et al. 2013 [58] | Resting state fMRI | 21 participants with PWS (11 males, mean 7.3 y) 18 sibling control participants (8 males, mean 11.1 y) | ALFF greater in PWS than control group in ventrolateral PFC, ACC, inferior parietal lobe and left insula, and decreased in the medial and dorsolateral PFC, hippocampus, pre- and post-central gyri, and left OFC. DMN: reduced functional connectivity pairwise between medial PFC, inferior parietal lobe and precuneus, and between precuneus and inferior parietal lobe. PFC network: reduced functional connectivity between dorsolateral PFC and OFC, and increased functional connectivity between ventrolateral PFC and both OFC and dorsolateral PFC. Core network: increased ACC-insula functional connectivity. Motor sensory network: decrease pre- to post-central gyrus connectivity. |
| Zhang et al. 2012 (see also 2015) [59,60] | Resting state fMRI | 21 participants with PWS (11 males, mean 7.3 y) 18 sibling control participants (8 males, mean 11.1y) | ALFF increased in PWS in ACC, hypothalamus, & left amygdala and decreased in medial PFC and right amygdala. Granger causality analysis of direction of connectivity: Increased causal influence bilaterally from amygdala to the hypothalamus, from the ACC to the medial PFC right amygdala, from the medial PFC to bilateral amygdala, & from ACC to medial PFC in PWS compared to control participants. Altered directionality in effective connectivity in all pairwise analyses except the right amygdala to hypothalamus, although this increased in strength. |
| Study | Methods | Sample | Key findings |
|---|---|---|---|
| Akefeldt et al. 1998 [61] | Lumbar puncture: CSF analysed for metabolites of serotonin (5-HIAA), dopamine (HVA) & noradrenaline (HMPG) | 13 participants with PWS (8 males, 0.4–23 y) 5 control groups: 15 typically-developing participants (11 males, 6 aged 3–8 y, 9 aged 18–25 y); 22 typically-developing children (16 males, 3–14 y); 8 children with autism (5 males, 3–13 y); 7 children with mixed neurological diagnoses (6 males, 0.3–15 y); 4 overweight children with ID but not PWS (0 males, 4–15 y) | Increased levels of 5-HIAA & HVA in PWS compared to all groups, most markedly for serotonin. Significant for 5-HIAA at corrected level compared to all control groups except the overweight and mixed neurological groups at the corrected significance level, but trend found. Significant for HVA at corrected level compared to typically-developing groups and all control participants combined. No association found between increased metabolites in PWS and age, BMI or severity of intellectual disability. |
| Ebert et al. 1997 [62] | Venepuncture: Plasma GABA levels | 14 participants with PWS (6 males, 2–21 y) 9 children with AS (7 males, 2–17 y); 2 control groups: 7 moderately obese participants without ID (5 males, 2–20 y); 5 healthy weight children with ID (4 males, 3–17 y) | Mean GABA levels in plasmas significantly higher (2-3 times) in both PWS and AS groups than either control group, but no significant difference between PWS and AS groups. |
| Fronczek et al. 2005 [63] | Post-mortem: immunocytochemistry and image analysis system estimation of orexin neuron number in the lateral hypothalamus | 7 hypothalami from individuals with PWS (3 males, 5 adults aged 25–64 y, 2 infants aged 6m & 3 y) 11 control hypothalami, matched for age, sex, post-mortem delay, fixation time and premorbid illness duration. | No difference in number of orexin neurons in hypothalamus found. |
| Goldstone et al. 2002 [64] | Post-mortem: immunocytochemistry & in situ hybridization to study NPY, AGRP, and NPY mRNA expression in the hypothalamus | 6 obese adults with PWS (2 males, 25–64 y) 4 obese adults without PWS (2 males, 66–76 y) 22 control adults (13 males, 28–90 y) | Significant decrease of NPY and tendency to decreased NPY mRNA expression in all obese subjects, including PWS, but consistent with literature on inhibition on NPY in obesity. No significant difference in AGRP and NPY staining, or NPY mRNA expression, between PWS and obese adults without PWS. NPY/AGRP neurons show appropriate functioning. |
| Goldstone et al. 2003 [65] | Post-mortem: immunocytochemistry assessment of GHRH neuron number in infundibular nucleus/median eminence complex of the hypothalamus | 6 adults with PWS (2 males, 25–64 y) 2 children with PWS (1 males, 0.5 & 0.75 y) 6 o6 control participants (13 males, 22 adults aged 28–90y, 4 children aged 0.4–0.75 y) | Higher GHRH neuron number in both control adults and adults with PWS who had prolonged premorbid illness, but no difference between PWS and control or obese adults without PWS. |
| Hayashi et al. 2011 [66] | Post-mortem: immunohistochemical analysis of GABAergic interneurons in superior frontal cortex & OFC, ACh neurons in the nucleus basalis of Meynert & PPN, & orexin-A and vasopressin in the hypothalamus. | 6 month old female with PWS 3 control subjects following fatal pneumonia (2 males, 4 m, 1 y, 6 y) | GABAergic interneurons in cortex and ACh neurons in nucleus basalis similar in PWS and control samples. No clear abnormalities in orexin and vasopressin neurons in hypothalamus. Marked reduction of ACh neurons in PPN. |
| Lucignani et al. 2004 [67] | Resting state PET: 11C-flumazenil binding to evaluate GABAA receptor functioning. | 6 participants with PWS (2 males, 19.3–29.7 y, mean 24.6 y) 9 typically-developing participants (9 males, mean 25.9 y) | Significant binding reduction (7%) in cingulate. Reduced binding of ca. 3-6% in PWS in temporal and frontal cortices, and including insula (7%), but not significant when corrected for multiple comparisons. Trend to reduced binding in amygdala, caudate and thalamus (10-14%). Binding potential very similar in hippocampus, putamen and parietal and occipital cortices. |
| Martin et al. 1998 [68] | Lumbar puncture: CSF analysed for levels of oxytocin and vasopressin | 5 participants with PWS (2 males, 16–21 y) 6 typically-developing participants (0 males, 21–28 y) | Oxytocin levels in CSF significantly higher in PWS, especially in females. Vasopressin levels significant lower in PWS females compared to control females, but not for males or combined sex groups. |
| Pasi et al. 1989 [69] | Post-mortem: immunoradiological assay of beta-endorphin levels in neural tissue | 19 y old female with PWS | No clear beta-endorphin abnormality: rank of levels of beta-endorphin in areas of the brain on which there was prior reference literature (hypothalamus, medulla, periaqueductal grey, pons, & thalamus) was very similar, with the exception of the medulla. |
| Swaab et al. 1995 [70] | Post-mortem: thionine and immunocytochemical staining to assess PVN size and number of oxytocin and vasopressin neurons | 5 adults with PWS (2 males, 22–64 y) 27 control adults (14 males) | PVN significantly smaller (28%) in PWS, with total cell number reduced by 38%. Oxytocin number significantly decreased in PWS (by 42%) as was volume of PVN containing oxytocin cells (by 54%). No significant difference in number vasopressin neurons between PWS and control samples. |
| Talebizadeh et al. 2005 [71] | Post-mortem: RT-PCR evaluation of gene expression of ghrelin, peptide YY and their receptors in the frontal, temporal, and visual cortices, pons, medulla and hypothalamus | 3 individuals with PWS (0 male, 1 infant aged 1 y, 2 adults aged 32 y) 2 individuals with AS (4y old male, 43y old female) 6 control individuals (3 males, 1–72 y) | Expression detected in all brain areas in PWS, AS and control samples, with exception of PYY in pons for 1 PWS & 1 control subject. |
Discussion of Anatomical Findings
4. Functional Studies in the PWS Brain
4.1. Food-Related Function
4.2. Function Relating to Other Aspects of the PWS Phenotype
4.3. Resting State
4.4. Discussion of Functional Findings
5. Neurochemical Investigations and Neuroanatomy at the Cellular Level
Discussion of Neurochemical Findings
6. General Discussion
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Whittington, J.E.; Holland, A.J.; Webb, T.; Butler, J.; Clarke, D.; Boer, H. Population prevalence and estimated birth incidence and mortality rate for people with Prader-Willi syndrome in one UK health region. J. Med. Genet. 2001, 38, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.; Egan, J.; Ridley, G.; Haan, E.; Montgomery, P.; Williams, K.; Elliott, E. Birth prevalence of Prader-Willi syndrome in australia. Arch. Dis. Child. 2003, 88, 263–264. [Google Scholar] [CrossRef] [PubMed]
- Siemensma, E.P.; van Wijngaarden, R.F.T.L.; Festen, D.A.; Troeman, Z.C.; van der Velden, A.A.; Otten, B.J.; Rotteveel, J.; Odink, R.J.; Bindels-de Heus, G.C.; van Leeuwen, M.; et al. Beneficial effects of growth hormone treatment on cognition in children with Prader-Willi syndrome: A randomized controlled trial and longitudinal study. J. Clin. Endocrinol. Metab. 2012, 97, 2307–2314. [Google Scholar] [CrossRef] [PubMed]
- Cahill, L. Why sex matters for neuroscience. Nat. Rev. Neurosci. 2006, 7, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.J.; Whittington, J.E.; Butler, J.; Webb, T.; Boer, H.; Clarke, D. Behavioural phenotypes associated with specific genetic disorders: Evidence from a population-based study of people with Prader-Willi syndrome. Psych. Med. 2003, 33, 141–153. [Google Scholar] [CrossRef]
- Clarke, D.J.; Boer, H.; Whittington, J.; Holland, A.; Butler, J.; Webb, T. Prader-Willi syndrome, compulsive and ritualistic behaviours: The first population-based survey. Brit. J. Psychiat 2002, 180, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Dykens, E.M.; Kasari, C. Maladaptive behavior in children with Prader-Willi syndrome, down syndrome, and nonspecific mental retardation. Am. J. Ment. Retard. 1997, 102, 228–237. [Google Scholar] [CrossRef]
- Haig, D.; Wharton, R. Prader-Willi syndrome and the evolution of human childhood. Am. J. Hum. Biol. 2003, 15, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Cacciari, E.; Zucchini, S.; Carla, G.; Pirazzoli, P.; Cicognani, A.; Mandini, M.; Busacca, M.; Trevisan, C. Endocrine function and morphological findings in patients with disorders of the hypothalamo-pituitary area: A study with magnetic resonance. Arch. Dis. Child. 1990, 65, 1199–1202. [Google Scholar] [CrossRef] [PubMed]
- Crino, A.; di Giorgio, G.; Schiaffini, R.; Fierabracci, A.; Spera, S.; Maggioni, A.; Gattinara, G.C. Central precocious puberty and growth hormone deficiency in a boy with Prader-Willi syndrome. Eur J. Pediatr. 2008, 167, 1455–1458. [Google Scholar] [CrossRef] [PubMed]
- Linnemann, K.; Schroder, C.; Mix, M.; Kruger, G.; Fusch, C. Prader-labhart-willi syndrome with central precocious puberty and empty sella syndrome. Acta. Paediatr. 1999, 88, 1295–1297. [Google Scholar] [CrossRef] [PubMed]
- Iughetti, L.; Bosio, L.; Corrias, A.; Gargantini, L.; Ragusa, L.; Livieri, C.; Predieri, B.; Bruzzi, P.; Caselli, G.; Grugni, G. Pituitary height and neuroradiological alterations in patients with prader-labhart-willi syndrome. Eur. J. Pediatr. 2008, 167, 701–702. [Google Scholar] [CrossRef] [PubMed]
- Tauber, M.; Barbeau, C.; Jouret, B.; Pienkowski, C.; Malzac, P.; Moncla, A.; Rochiccioli, P. Auxological and endocrine evolution of 28 children with Prader-Willi syndrome: Effect of GH therapy in 14 children. Horm. Res. 2000, 53, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Grugni, G.; Guzzaloni, G.; Moro, D.; Mazzilli, G.; Morabito, F. GH secretion and pituitary abnormalities in Prader-Willi syndrome. Int. J. Obes. 2000, 24. [Google Scholar] [CrossRef]
- Fan, Z.; Greenwood, R.; Fisher, A.; Pendyal, S.; Powell, C.M. Characteristics and frequency of seizure disorder in 56 patients with Prader-Willi syndrome. Am. J. Med. Genet. A 2009, 149, 1581–1584. [Google Scholar] [CrossRef] [PubMed]
- Gilboa, T.; Gross-Tsur, V. Epilepsy in Prader-Willi syndrome: Experience of a national referral centre. Dev. Med. Child. Neurol. 2013, 55, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Mori, K.; Yoneda, Y.; Yamaue, T.; Miyazaki, M.; Harada, M.; Miyoshi, H.; Kuroda, Y. Proton magnetic resonance spectroscopy of the brain in patients with Prader-Willi syndrome. Pediatr. Neurol. 1998, 18, 30–35. [Google Scholar] [CrossRef]
- Hayashi, M.; Itoh, M.; Kabasawa, Y.; Hayashi, H.; Satoh, J.; Morimatsu, Y. A neuropathological study of a case of the Prader-Willi syndrome with an interstitial deletion of the proximal long arm of chromosome 15. Brain Dev. 1992, 14, 58–62. [Google Scholar] [CrossRef]
- Honea, R.A.; Holsen, L.M.; Lepping, R.J.; Perea, R.; Butler, M.G.; Brooks, W.M.; Savage, C.R. The neuroanatomy of genetic subtype differences in Prader-Willi syndrome. Am. J. Med. Genet. B 2012, 159B, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Kumada, T.; Ito, M.; Miyajima, T.; Fujii, T.; Okuno, T.; Go, T.; Hattori, H.; Yoshioka, M.; Kobayashi, K.; Kanazawa, O.; et al. Multi-institutional study on the correlation between chromosomal abnormalities and epilepsy. Brain Dev. 2005, 27, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Leonard, C.M.; Williams, C.A.; Nicholls, R.D.; Agee, O.F.; Voeller, K.K.; Honeyman, J.C.; Staab, E.V. Angelman and Prader-Willi syndrome: A magnetic resonance imaging study of differences in cerebral structure. Am. J. Med. Genet. 1993, 46, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Lukoshe, A.; White, T.; Schmidt, M.N.; van der Lugt, A.; Hokken-Koelega, A.C. Divergent structural brain abnormalities between different genetic subtypes of children with Prader-Willi syndrome. J. Neurodev. Dis. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Lukoshe, A.; Hokken-Koelega, A.C.; van der Lugt, A.; White, T. Reduced cortical complexity in children with Prader-Willi syndrome and its association with cognitive impairment and developmental delay. PloS One 2014, 9, e107320. [Google Scholar] [CrossRef] [PubMed]
- Mantoulan, C.; Payoux, P.; Diene, G.; Glattard, M.; Roge, B.; Molinas, C.; Sevely, A.; Zilbovicius, M.; Celsis, P.; Tauber, M. Pet scan perfusion imaging in the Prader-Willi syndrome: New insights into the psychiatric and social disturbances. J. Cerebr. Blood. F. Met. 2011, 31, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Maski, K.P.; Vendrame, M.; Tan, W.H.; Kothare, S.V. Characterization of epilepsy in Prader Willi syndrome. Epilepsia 2009, 50, 302. [Google Scholar]
- Miller, L.; Angulo, M.; Price, D.; Taneja, S. MR of the pituitary in patients with Prader-Willi syndrome: Size determination and imaging findings. Pediatr. Radiol. 1996, 26, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.; Kranzler, J.; Liu, Y.; Schmalfuss, I.; Theriaque, D.W.; Shuster, J.J.; Hatfield, A.; Mueller, O.T.; Goldstone, A.P.; Sahoo, T.; et al. Neurocognitive findings in Prader-Willi syndrome and early-onset morbid obesity. J. Pediatri. 2006, 149, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; Couch, J.A.; Leonard, C.M.; Schwenk, K.; Towler, S.D.; Shuster, J.; Goldstone, A.P.; He, G.; Driscoll, D.J.; Liu, Y. Sylvian fissure morphology in Prader-Willi syndrome and early-onset morbid obesity. Genet. Med. 2007, 9, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; Couch, J.A.; Schmalfuss, I.; He, G.; Liu, Y.; Driscoll, D.J. Intracranial abnormalities detected by three-dimensional magnetic resonance imaging in Prader-Willi syndrome. Am. J. Med. Genet. A 2007, 143A, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; Goldstone, A.P.; Couch, J.A.; Shuster, J.; He, G.; Driscoll, D.J.; Liu, Y.; Schmalfuss, I.M. Pituitary abnormalities in Prader-Willi syndrome and early onset morbid obesity. Am. J. Med. Genet. A 2008, 146, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; Couch, J.; Schwenk, K.; Long, M.; Towler, S.; Theriaque, D.W.; He, G.; Liu, Y.; Driscoll, D.J.; Leonard, C.M. Early childhood obesity is associated with compromised cerebellar development. Dev. Neuropsychol. 2009, 34, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Ogura, K.; Fujii, T.; Abe, N.; Hosokai, Y.; Shinohara, M.; Takahashi, S.; Mori, E. Small gray matter volume in orbitofrontal cortex in Prader-Willi syndrome: A voxel-based mri study. Hum. Brain Mapp. 2011, 32, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, D.A.; Anaya, T.M.; Clayton-Smith, J.; Hall, B.D.; van Allen, M.I.; Zori, R.T.; Zackai, E.H.; Frank, G.; Clericuzio, C.L. Unexpected death and critical illness in Prader-Willi syndrome: Report of ten individuals. Am. J. Med. Genet. A 2004, 124, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, E.; Murakami, N.; Sakuta, R.; Nagai, T. Evaluating the frequency and characteristics of seizures in 142 japanese patients with Prader-Willi syndrome. Am. J. Med. Genet. A 2013, 161, 2052–2055. [Google Scholar] [CrossRef] [PubMed]
- Van Nieuwpoort, I.C.; Sinnema, M.; Castelijns, J.A.; Twisk, J.W.; Curfs, L.M.; Drent, M.L. The GH/IGF-I axis and pituitary function and size in adults with Prader-Willi syndrome. Horm. Res. Paediatr. 2011, 75, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Vendrame, M.; Maski, K.P.; Chatterjee, M.; Heshmati, A.; Krishnamoorthy, K.; Tan, W.H.; Kothare, S.V. Epilepsy in Prader-Willi syndrome: Clinical characteristics and correlation to genotype. Epilepsy Behav. 2010, 19, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Verrotti, A.; Cusmai, R.; Laino, D.; Carotenuto, M.; Esposito, M.; Falsaperla, R.; Margari, L.; Rizzo, R.; Savasta, S.; Grosso, S.; et al. Long-term outcome of epilepsy in patients with Prader-Willi syndrome. J. Neurol. 2015, 262, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Matsuzawa, H.; Uchiyama, M.; Kwee, I.L.; Nakada, T. Brain developmental abnormalities in Prader-Willi syndrome detected by diffusion tensor imaging. Pediatrics 2006, 118, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, A.; Krishnamoorthy, K.S.; Grant, P.E. Abnormal cortical development shown by 3D MRI in Prader-Willi syndrome. Neurology 2002, 59, 644–645. [Google Scholar] [CrossRef] [PubMed]
- Akefeldt, A.; Akefeldt, B.; Gillberg, C. Voice, speech and language characteristics of children with Prader-Willi syndrome. J. Intell. Disabil. Res. 1997, 41, 302–311. [Google Scholar] [CrossRef]
- Dimitropoulos, A.; Schultz, R.T. Food-related neural circuitry in Prader-Willi syndrome: Response to high- versus low-calorie foods. J. Autism. Dev. Disord. 2008, 38, 1642–1653. [Google Scholar] [CrossRef] [PubMed]
- Halit, H.; Grice, S.J.; Bolton, R.; Johnson, M.H. Face and gaze processing in Prader-Willi syndrome. J. Neuropsychol. 2008, 2, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Hinton, E.C.; Holland, A.J.; Gellatly, M.S.; Soni, S.; Patterson, M.; Ghatei, M.A.; Owen, A.M. Neural representations of hunger and satiety in Prader-Willi syndrome. Int. J. Obes. 2006, 30, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Hinton, E.C.; Holland, A.J.; Gellatly, M.S.; Soni, S.; Owen, A.M. An investigation into food preferences and the neural basis of food-related incentive motivation in Prader-Willi syndrome. J. Intell Disabil Res. 2006, 50, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Holsen, L.M.; Zarcone, J.R.; Brooks, W.M.; Butler, M.G.; Thompson, T.I.; Ahluwalia, J.S.; Nollen, N.L.; Savage, C.R. Neural mechanisms underlying hyperphagia in Prader-Willi syndrome. Obes. 2006, 14, 1028–1037. [Google Scholar] [CrossRef] [PubMed]
- Holsen, L.M.; Zarcone, J.R.; Chambers, R.; Butler, M.G.; Bittel, D.C.; Brooks, W.M.; Thompson, T.I.; Savage, C.R. Genetic subtype differences in neural circuitry of food motivation in Prader-Willi syndrome. Int. J. Obes. 2009, 33, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Holsen, L.M.; Savage, C.R.; Martin, L.E.; Bruce, A.S.; Lepping, R.J.; Ko, E.; Brooks, W.M.; Butler, M.G.; Zarcone, J.R.; Goldstein, J.M. Importance of reward and prefrontal circuitry in hunger and satiety: Prader-Willi syndrome vs simple obesity. Int. J. Obes. 2012, 36, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Key, A.P.; Dykens, E.M. “Hungry eyes”: Visual processing of food images in adults with Prader-Willi syndrome. J. Intell. Disabil. Res. 2008, 52, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.E.; Jin, D.K.; Cho, S.S.; Kim, J.H.; Hong, S.D.; Paik, K.H.; Oh, Y.J.; Kim, A.H.; Kwon, E.K.; Choe, Y.H. Regional cerebral glucose metabolic abnormality in Prader-Willi syndrome: A 18F-FDG pet study under sedation. J. Nucl. Med. 2006, 47, 1088–1092. [Google Scholar] [PubMed]
- Klabunde, M.; Saggar, M.; Hustyi, K.M.; Hammond, J.L.; Reiss, A.L.; Hall, S.S. Neural correlates of self-injurious behavior in Prader-Willi syndrome. Hum. Brain Mapp. 2015, 36, 4135–4143. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; James, G.A.; Goldstone, A.P.; Couch, J.A.; He, G.; Driscoll, D.J.; Liu, Y. Enhanced activation of reward mediating prefrontal regions in response to food stimuli in Prader-Willi syndrome. J. Neurol. Neurosur. Ps. 2007, 78, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Ogura, K.; Fujii, T.; Abe, N.; Hosokai, Y.; Shinohara, M.; Fukuda, H.; Mori, E. Regional cerebral blood flow and abnormal eating behavior in Prader-Willi syndrome. Brain Dev. 2013, 35, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Pujol, J.; Macia, D.; Blanco-Hinojo, L.; Martinez-Vilavella, G.; Sunyer, J.; de la Torre, R.; Caixas, A.; Martin-Santos, R.; Deus, J.; Harrison, B.J. Does motion-related brain functional connectivity reflect both artifacts and genuine neural activity? NeuroImage 2014, 101, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Shapira, N.A.; Lessig, M.C.; He, A.G.; James, G.A.; Driscoll, D.J.; Liu, Y. Satiety dysfunction in Prader-Willi syndrome demonstrated by FMRI. J. Neurol. Neurosur. Ps. 2005, 76, 260–262. [Google Scholar] [CrossRef] [PubMed]
- Stauder, J.E.; Brinkman, M.J.; Curfs, L.M. Multi-modal P3 deflation of event-related brain activity in Prader-Willi syndrome. Neurosci. lett. 2002, 327, 99–102. [Google Scholar] [CrossRef]
- Stauder, J.E.; Boer, H.; Gerits, R.H.; Tummers, A.; Whittington, J.; Curfs, L.M. Differences in behavioural phenotype between parental deletion and maternal uniparental disomy in Prader-Willi syndrome: An ERP study. Clin. Neurophysiol. 2005, 116, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, K.A.; Humphreys, G.W.; Oliver, C.; Hansen, P.C. Neural correlates of task switching in paternal 15Q11-Q13 deletion Prader-Willi syndrome. Brain Res. 2010, 1363, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, H.; Qiu, S.; Tian, J.; Wen, X.; Miller, J.L.; von Deneen, K.M.; Zhou, Z.; Gold, M.S.; Liu, Y. Altered functional brain networks in Prader-Willi syndrome. NMR Biomed. 2013, 26, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tian, J.; von Deneen, K.M.; Gold, M.S.; Liu, Y. Altered “driving” effect from amygdala to hypothalamus in Prader-Willi syndrome during resting state. In Proceedings of Organisation for Human Brain Mapping 2012 Conference, Beijing, China, 10–14 June 2012.
- Zhang, Y.; Wang, J.; Zhang, G.; Zhu, Q.; Cai, W.; Tian, J.; Zhang, Y.E.; Miller, J.L.; Wen, X.; Ding, M.; et al. The neurobiological drive for overeating implicated in Prader-Willi syndrome. Brain Res. 2015, 1620, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Akefeldt, A.; Ekman, R.; Gillberg, C.; Mansson, J.E. Cerebrospinal fluid monoamines in Prader-Willi syndrome. Biol. Psych. 1998, 44, 1321–1328. [Google Scholar] [CrossRef]
- Ebert, M.H.; Schmidt, D.E.; Thompson, T.; Butler, M.G. Elevated plasma gamma-aminobutyric acid (GABA) levels in individuals with either Prader-Willi syndrome or angelman syndrome. J. Neuropsychiatry Clin. Neurosci. 1997, 9, 75–80. [Google Scholar] [PubMed]
- Fronczek, R.; Lammers, G.J.; Balesar, R.; Unmehopa, U.A.; Swaab, D.F. The number of hypothalamic hypocretin (orexin) neurons is not affected in Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 2005, 90, 5466–5470. [Google Scholar] [CrossRef] [PubMed]
- Goldstone, A.P.; Unmehopa, U.A.; Bloom, S.R.; Swaab, D.F. Hypothalamic npy and agouti-related protein are increased in human illness but not in Prader-Willi syndrome and other obese subjects. J. Clin. Endocrinol. Metab. 2002, 87, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Goldstone, A.P.; Unmehopa, U.A.; Swaab, D.F. Hypothalamic growth hormone-releasing hormone (GHRH) cell number is increased in human illness, but is not reduced in Prader-Willi syndrome or obesity. Clin. Endocrinol. 2003, 58, 743–755. [Google Scholar] [CrossRef]
- Hayashi, M.; Miyata, R.; Tanuma, N. Decrease in acetylcholinergic neurons in the pedunculopontine tegmental nucleus in a patient with Prader-Willi syndrome. Neuropathology 2011, 31, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Lucignani, G.; Panzacchi, A.; Bosio, L.; Moresco, R.M.; Ravasi, L.; Coppa, I.; Chiumello, G.; Frey, K.; Koeppe, R.; Fazio, F. Gaba a receptor abnormalities in Prader-Willi syndrome assessed with positron emission tomography and [11C]flumazenil. NeuroImage 2004, 22, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; State, M.; Anderson, G.M.; Kaye, W.M.; Hanchett, J.M.; McConaha, C.W.; North, W.G.; Leckman, J.F. Cerebrospinal fluid levels of oxytocin in Prader-Willi syndrome: A preliminary report. Biol. Psych. 1998, 44, 1349–1352. [Google Scholar] [CrossRef]
- Pasi, A.; Mehraein, P.; Gramsch, C.; Jehle, A.; Briner, J.; Hani, M.; Kulling, P.; Hauri, R.; Messiha, F.S. Cerebral beta-endorphin levels in a woman with prader-labhart-willi syndrome. Physiol. Behavior. 1989, 46, 17–18. [Google Scholar] [CrossRef]
- Swaab, D.F.; Purba, J.S.; Hofman, M.A. Alterations in the hypothalamic paraventricular nucleus and its oxytocin neurons (putative satiety cells) in Prader-Willi syndrome: A study of five cases. J. Clin. Endocrinol. Metab. 1995, 80, 573–579. [Google Scholar] [PubMed]
- Talebizadeh, Z.; Kibiryeva, N.; Bittel, D.C.; Butler, M.G. Ghrelin, peptide yy and their receptors: Gene expression in brain from subjects with and without Prader-Willi syndrome. Int. J. Mol. Med. 2005, 15, 707–711. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Afif, A.; Bouvier, R.; Buenerd, A.; Trouillas, J.; Mertens, P. Development of the human fetal insular cortex: Study of the gyration from 13 to 28 gestational weeks. Brain Struct. Funct. 2007, 212, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, Y.; Yu, C.; Lin, L.; Li, C.; Jiang, T. Reduced cortical folding in mental retardation. Am. J. Neuroradiol. 2010, 31, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Stunkard, A.J.; Messick, S. The three-factor eating questionnaire to measure dietary restraint, disinhibition and hunger. J. Psychosom. Res. 1985, 29, 71–83. [Google Scholar] [CrossRef]
- Woodcock, K.A.; Oliver, C.; Humphreys, G.W. The relationship between specific cognitive impairment and behaviour in Prader-Willi syndrome. J. Intell. Disabil. Res. 2011, 55, 152–171. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, K.A.; Oliver, C.; Humphreys, G.W. A specific pathway can be identified between genetic characteristics and behaviour profiles in Prader-Willi syndrome via cognitive, environmental and physiological mechanisms. J. Intell. Disabil. Res. 2009, 53, 493–500. [Google Scholar] [CrossRef]
- Woodcock, K.A.; Oliver, C.; Humphreys, G.W. Task-switching deficits and repetitive behaviour in genetic neurodevelopmental disorders: Data from children with Prader-Willi syndrome chromosome 15 Q11-Q13 deletion and boys with fragile X syndrome. Cognitive Neuropsych. 2009, 26, 172–194. [Google Scholar] [CrossRef] [PubMed]
- Bull, L.E.; Woodcock, K.A.; Holland, A.; Oliver, C. Temper outbursts in Prader-Willi syndrome: Early intervention and environmental management. J. Intell. Disabil. Res. 2011, 55, 950. [Google Scholar] [CrossRef] [Green Version]
- Whittington, J.; Holland, A.; Webb, T.; Butler, J.; Clarke, D.; Boer, H. Cognitive abilities and genotype in a population-based sample of people with Prader-Willi syndrome. J. Intell. Disabil. Res. 2004, 48, 172–187. [Google Scholar] [CrossRef]
- Dickinson, D. Digit symbol coding and general cognitive ability in schizophrenia: Worth another look? Brit. J. Psychiat. 2008, 193, 354–356. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Niendam, T.A.; Bearden, C.E.; Rosso, I.M.; Sanchez, L.E.; Hadley, T.; Nuechterlein, K.H.; Cannon, T.D. A prospective study of childhood neurocognitive functioning in schizophrenic patients and their siblings. Am. J. Psychiat. 2003, 160, 2060–2062. [Google Scholar] [CrossRef] [PubMed]
- Milner, K.M.; Craig, E.E.; Thompson, R.J.; Veltman, M.W.; Thomas, N.S.; Roberts, S.; Bellamy, M.; Curran, S.R.; Sporikou, C.M.; Bolton, P.F. Prader-Willi syndrome: Intellectual abilities and behavioural features by genetic subtype. J. Child. Psychol. Psyc. 2005, 46, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Chaves, V.E.; Tilelli, C.Q.; Brito, N.A.; Brito, M.N. Role of oxytocin in energy metabolism. Peptides 2013, 45, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.S. Oxytocin pathways and the evolution of human behavior. Annu. Rev. Psychol. 2014, 65, 17–39. [Google Scholar] [CrossRef] [PubMed]
- Wassef, A.; Baker, J.; Kochan, L.D. Gaba and schizophrenia: A review of basic science and clinical studies. J. Clin. Psychopharm. 2003, 23, 601–640. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.; Gil, R.; Seibyl, J.; Sewell, R.A.; D’Souza, D.C. Probing gaba receptor function in schizophrenia with iomazenil. Neuropsychopharmacology 2011, 36, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Price, J.L.; Drevets, W.C. Neural circuits underlying the pathophysiology of mood disorders. Trends Cogn. Sci. 2012, 16, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Price, J.L.; Drevets, W.C. Neurocircuitry of mood disorders. Neuropsychopharmacology 2010, 35, 192–216. [Google Scholar] [CrossRef] [PubMed]
- Ressler, K.J.; Mayberg, H.S. Targeting abnormal neural circuits in mood and anxiety disorders: From the laboratory to the clinic. Nat. Neurosci. 2007, 10, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Diener, C.; Kuehner, C.; Brusniak, W.; Ubl, B.; Wessa, M.; Flor, H. A meta-analysis of neurofunctional imaging studies of emotion and cognition in major depression. NeuroImage 2012, 61, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Rive, M.M.; van Rooijen, G.; Veltman, D.J.; Phillips, M.L.; Schene, A.H.; Ruhe, H.G. Neural correlates of dysfunctional emotion regulation in major depressive disorder. A systematic review of neuroimaging studies. Neurosci. Biobehav. R 2013, 37, 2529–2553. [Google Scholar] [CrossRef] [PubMed]
- Campbell-Sills, L.; Simmons, A.N.; Lovero, K.L.; Rochlin, A.A.; Paulus, M.P.; Stein, M.B. Functioning of neural systems supporting emotion regulation in anxiety-prone individuals. NeuroImage 2011, 54, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Jung, W.H.; Kang, D.H.; Kim, E.; Shin, K.S.; Jang, J.H.; Kwon, J.S. Abnormal corticostriatal-limbic functional connectivity in obsessive-compulsive disorder during reward processing and resting-state. NeuroImage 2013, 3, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Berthier, M.L.; Kulisevsky, J.; Gironell, A.; Heras, J.A. Obsessive-compulsive disorder associated with brain lesions: Clinical phenomenology, cognitive function, and anatomic correlates. Neurology 1996, 47, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Davidson, R.J.; Putnam, K.M.; Larson, C.L. Dysfunction in the neural circuitry of emotion regulation--a possible prelude to violence. Science 2000, 289, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Bechara, A.; Damasio, H.; Damasio, A.R. Emotion, decision making and the orbitofrontal cortex. Cereb. Cortex 2000, 10, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Porges, S.W.; Furman, S.A. The early development of the autonomic nervous system provides a neural platform for social behavior: A polyvagal perspective. Infant. Child. Dev. 2011, 20, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, G.; Dahnke, R.; Yeragani, V.K.; Bar, K.J. The relation of ventromedial prefrontal cortex activity and heart rate fluctuations at rest. Eur. J. Neurosci. 2009, 30, 2205–2210. [Google Scholar] [CrossRef] [PubMed]
- Manning, K.E.; McAllister, C.J.; Ring, H.A.; Finer, N.; Kelly, C.L.; Sylvester, K.P.; Fletcher, P.C.; Morrell, N.W.; Garnett, M.R.; Manford, M.R.; et al. Novel insights into maladaptive behaviours in Prader-Willi syndrome: Serendipitous findings from an open trial of vagus nerve stimulation. J. Intell. Disabil. Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Chae, J.H.; Nahas, Z.; Lomarev, M.; Denslow, S.; Lorberbaum, J.P.; Bohning, D.E.; George, M.S. A review of functional neuroimaging studies of vagus nerve stimulation (VNS). J. Psychiat. Res. 2003, 37, 443–455. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manning, K.E.; Holland, A.J. Puzzle Pieces: Neural Structure and Function in Prader-Willi Syndrome. Diseases 2015, 3, 382-415. https://doi.org/10.3390/diseases3040382
Manning KE, Holland AJ. Puzzle Pieces: Neural Structure and Function in Prader-Willi Syndrome. Diseases. 2015; 3(4):382-415. https://doi.org/10.3390/diseases3040382
Chicago/Turabian StyleManning, Katherine E., and Anthony J. Holland. 2015. "Puzzle Pieces: Neural Structure and Function in Prader-Willi Syndrome" Diseases 3, no. 4: 382-415. https://doi.org/10.3390/diseases3040382
APA StyleManning, K. E., & Holland, A. J. (2015). Puzzle Pieces: Neural Structure and Function in Prader-Willi Syndrome. Diseases, 3(4), 382-415. https://doi.org/10.3390/diseases3040382