
MOGAT2: A New Therapeutic Target for Metabolic Syndrome

Abstract
:1. Introduction
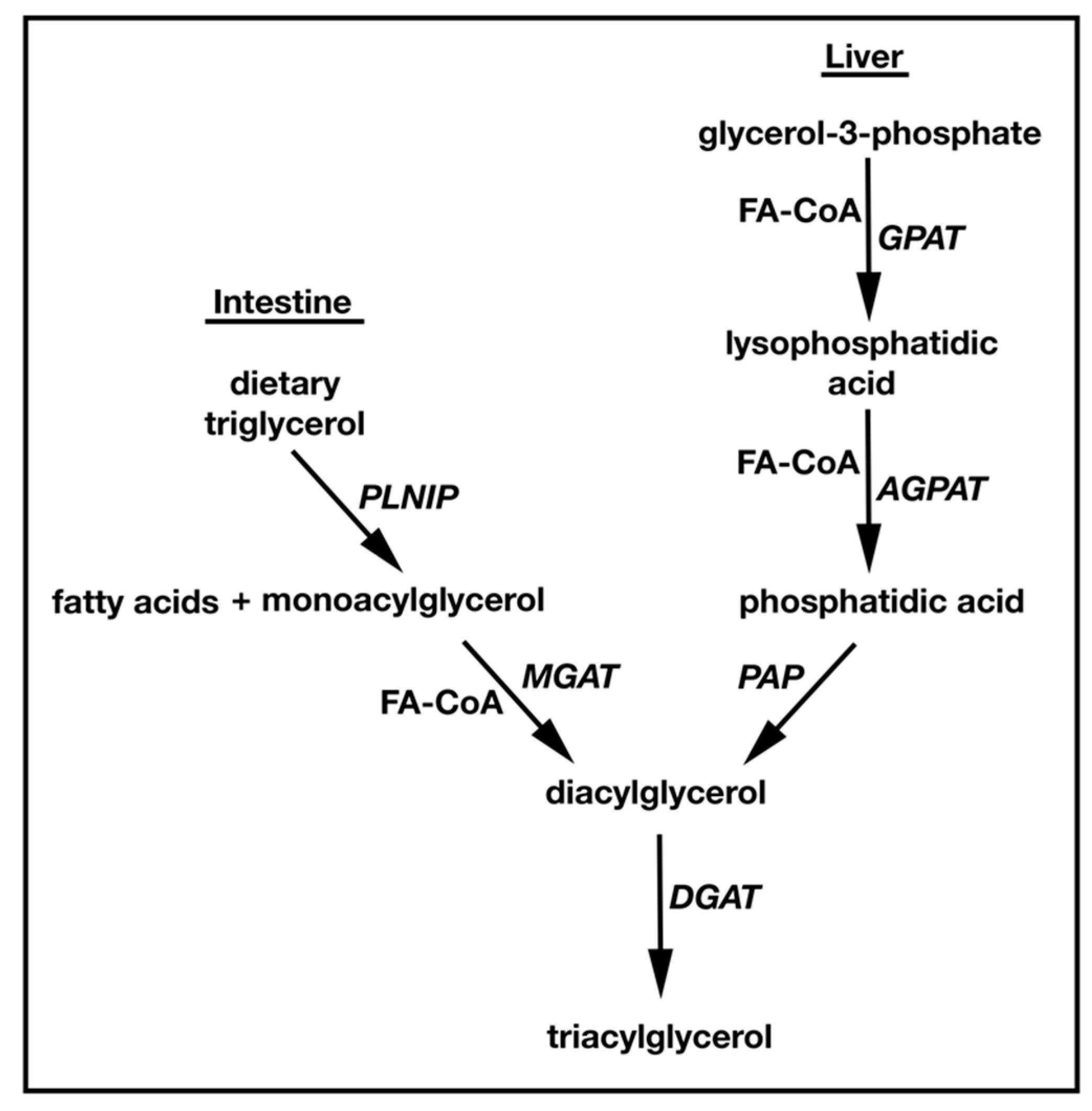
1.1. TAG Biosynthesis
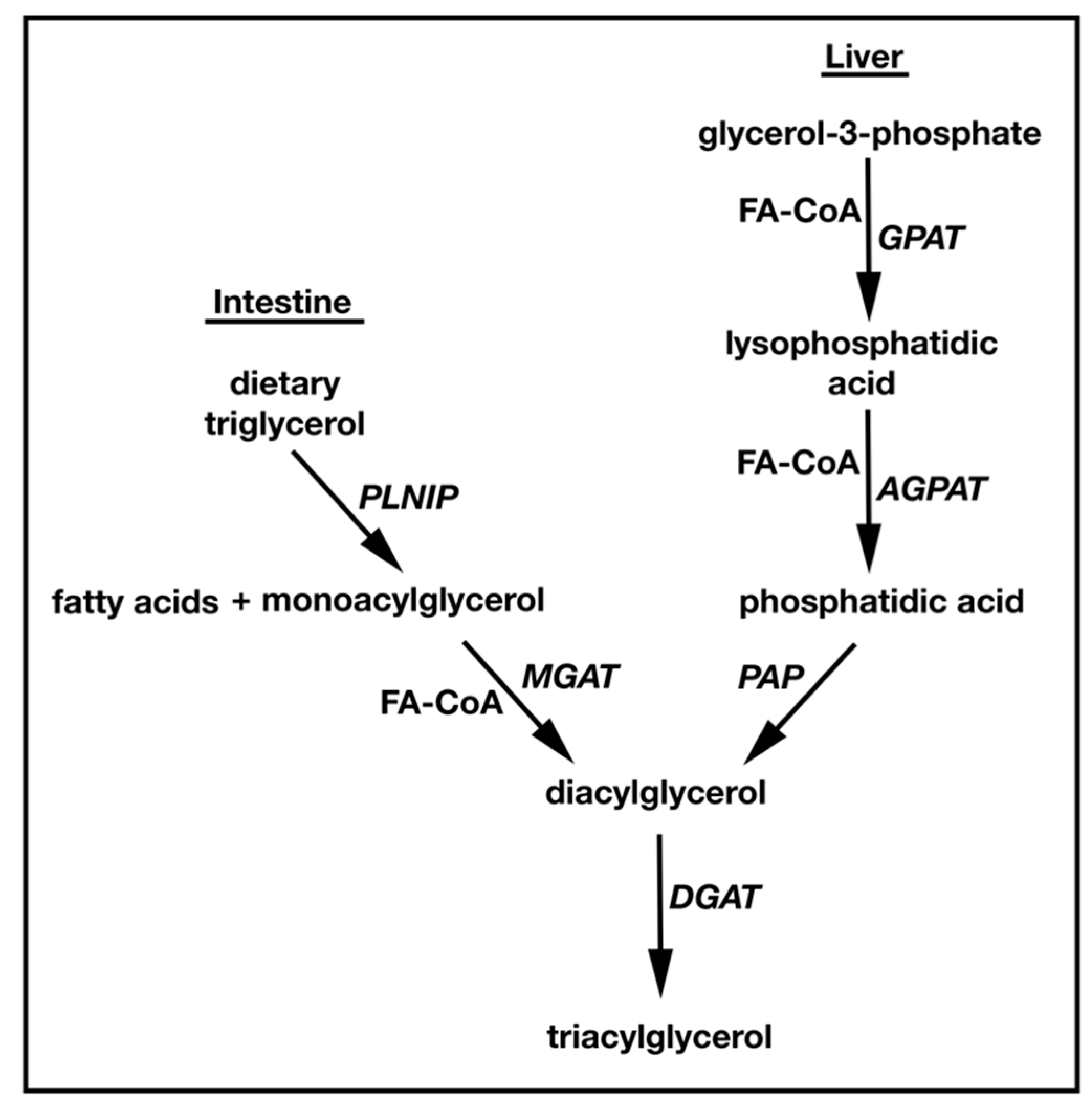
2. Identification and Characterization of MGATs
2.1. MGAT1
2.2. MGAT2

{kind=link}
{kind=link}
| Genetic Manipulation | Mouse Model | Description | TAG Content | Reference | |
|---|---|---|---|---|---|
| Plasma | Liver | ||||
| Mogat1 ASO | DIO | Weight loss was seen in all but DIO model; | n.c. | n.c. | [35,36,37] |
| ob/ob | Fatty acid oxidation was increased; Lipogenesis was suppressed; | ↓ | n.c. | ||
| HFD | Insulin sensitivity was improved; Hepatic inflammation was enhanced. | n.c. | ↓ | ||
| Mogat2 −/− Mogat2iko | HFD | Weight and food intake was decreased; Food preference was changed to carbohydrate from fat; Plasma non-HDL cholesterol was decreased; Hepatic steatosis was prevented; Energy expenditure was increased; GLP-1 secretion was increased; Insulin sensitivity was improved. | ↓ | n.d. | [43,44] |
2.3. MGAT3
2.4. DGATs
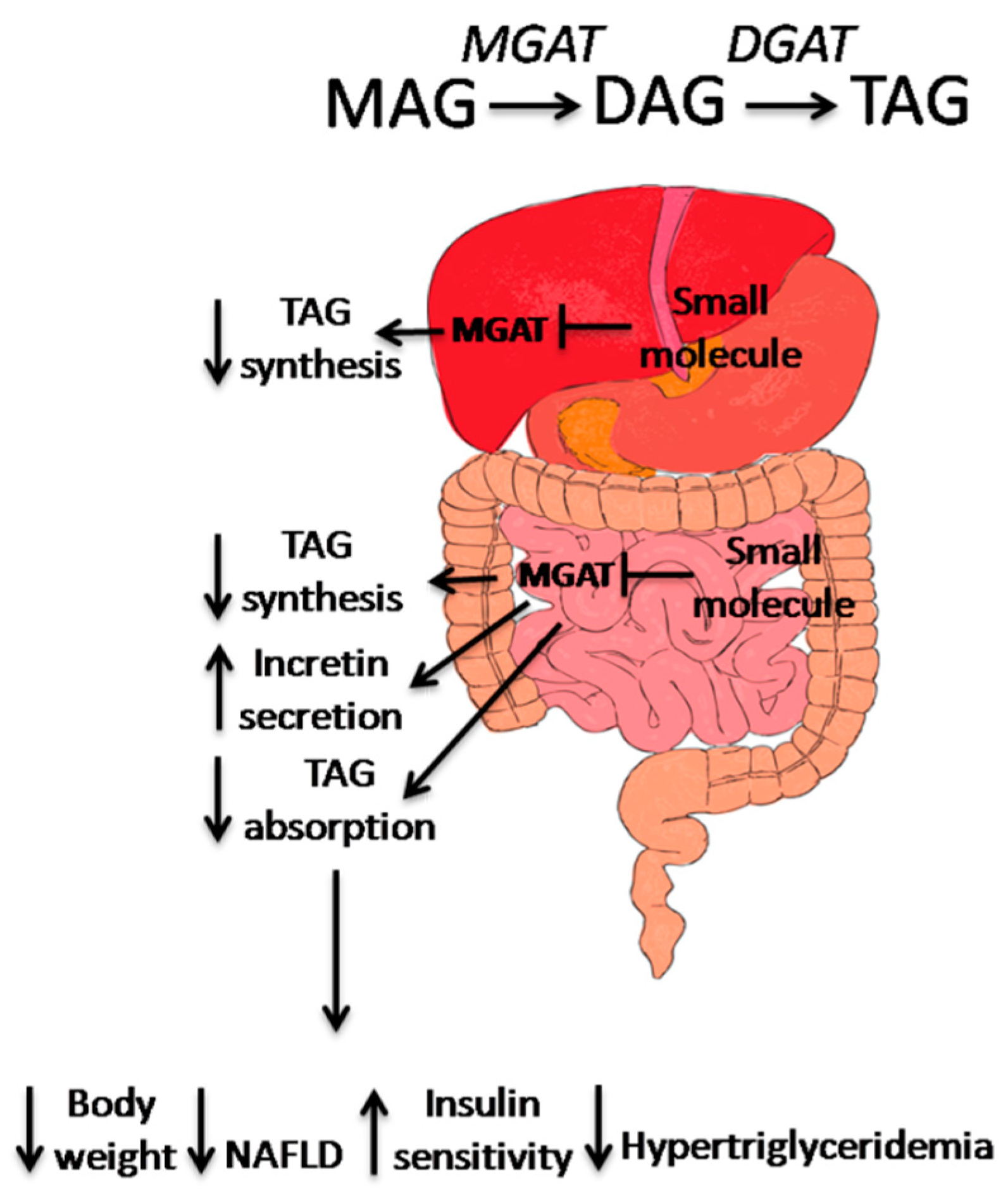
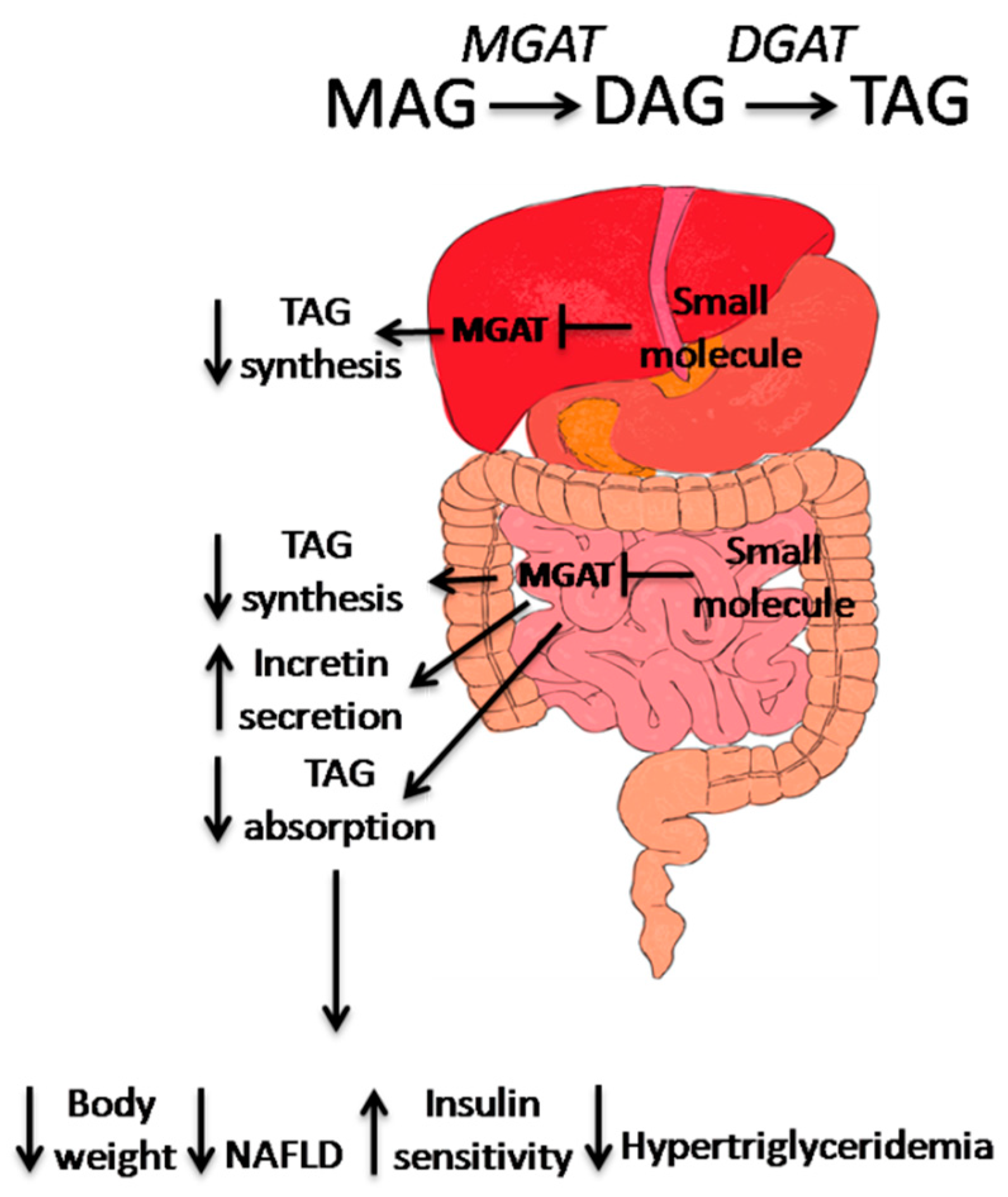
3. Small Molecule MGAT2 Inhibitors
| Company | Description | Reference |
|---|---|---|
| Banyu | Pyrimidine-4(3H)-one derivatives, IC50 = 56 nM, Selectivity to other acyltransferasesare unknown | WO2010/095767 |
| Dainippon Sumitomo | Bicyclic pyrimidine derivative, IC50 = 2 nM, Selectivity to other acyltransferasesare unknown | WO2012091010 A1 |
| Taisho | N-containing heterocyclic derivatives, IC50 = 4.1 nM, Selectivity to other acyltransferasesare unknown | WO2012124744 A1 |
| Eli Lily | Phenyl methanesulfonamide derivatives, IC50 = 12 nM in vitro assay, IC50 = 17.7 nM CaCo2 cell based LC-MS assay, Selectivity to other acyltransferasesare unknown Benzyl sulfonamide derivatives, IC50 = 2.28 nM in vitro assay, IC50 = 3.8 nM CaCo2 cell based LC-MS assay, >70% reduction in TAG excursion in a dog oil bolus model, Selectivity to other acyltransferasesare unknown Morpholynyl derivatives, IC50 = 12 nM in vitro assay, IC50 = 16 nM CaCo2 cell based LC-MS assay, 43% to 64% reduction of TAG excursion in a dog oil bolus model, Selectivity to other acyltransferasesare unknown | WO 2013112323 A1 WO2014074365A1 US20150005305 |
| Bristol-Myers Squibb | Aryl dihydropyridinones and piperidinones derivatives, IC50 = 14 nM in vitro assay, IC50 = 4 nM in STC-1 LC/MS assay, >1000-fold selective over other acyltransferases | WO2013/082345 [65] |
| AstraZeneca | 3-ethyl-3-methyl-2,5-dioxo-
N-phenyl-2,3,4,5-tetra hydro-1H-1,4-benzodiazepine-7-sulphonamide 1, IC50 = 1.6 nMRapidfire LCMS® assay, 68% reduction of TAG excursion in a mice oil oral gavage model, >10,000-fold selective over AWAT, variable selectivity toward MGAT2 and no selectivity toward MGAT1 | [66] |
| Takeda | N-phenylindoline-5-sulfonamide derivatives, IC50 = 3.4 nMRapidfire LCMS® assay, Significant reduction of TAG excursion in a mice olive oil oral gavage model, >30,000 fold selective over MGAT3, DGAT1, DGAT2 and ACAT1 | [67] |
| Japan Tobacco Inc. (Minato, TKY) | 7-(4,6-Di-tert-butyl-pyrimidin-2-yl)-3-(4-tri-fluoromethoxy-phenyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine derivative, IC50 = 19 nM in vitro assay, Significant reduction of TAG excursion in a mice olive oil oral gavage model, >300-fold selective over MGAT3, and >1000-fold selective over DGAT2, | [68] |
4. Conclusion
Acknowledgments
Conflicts of Interest
Abbreviations
| NAFLD | nonalcoholic fatty liver disease; |
| TAG | triacylglycerol; |
| MGAT2 | acyl-coA: monoacylglycerolacyltransferase 2; |
| MAG | monoacylglycerol; |
| DAG | diacylglycerol; |
| FA | fatty acid; |
| G3P | sn-glycerol-3-phosphate; |
| DGAT | acyl-coA: diacylglycerolacyltransferase; |
| GPAT | glycerol-3-phosphate acyltransferase; |
| LPA | lysophosphatidic acid; |
| AGPAT | acylglycerol-3-phosphate acyltransferase; |
| Pa | phosphatidic acid; |
| Lipin/PAP | lipins/PA phsphohydrolases; |
| ER | endoplasmic reticulum; |
| DHAP | dihydroxyacetone phosphate; |
| VLDL | very low density lipoprotein; |
| GLP-1 | glucagon-like peptide-1; |
| RYBG | Reux-en-Y gastric bypass; |
| DIO | diet-induced obese; |
| PPAR | peroxisome proliferator-activated receptor; |
| ASO | antisense oligonucleotides; |
| HTF-C diet | high trans FAs, fructose and cholesterol diet; |
| NASH | nonalcoholic steatohepatitis. |
References
- Coleman, R.A.; Mashek, D.G. Mammalian triacylglycerol metabolism: Synthesis, lipolysis, and signaling. Chem. Rev. 2011, 111, 6359–6386. [Google Scholar] [CrossRef]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V., Jr.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef] [PubMed]
- Kopelman, P.G. Obesity as a medical problem. Nature 2000, 404, 635–643. [Google Scholar] [PubMed]
- Mokdad, A.H.; Serdula, M.K.; Dietz, W.H.; Bowman, B.A.; Marks, J.S.; Koplan, J.P. The continuing epidemic of obesity in the United States. JAMA 2000, 284, 1650–1651. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Davidson, M.H.; Hirsh, B.J.; Kathiresan, S.; Gaudet, D. Genetics and causality of triglyceride-rich lipoproteins in atherosclerotic cardiovascular disease. J. Am. Coll Cardiol. 2014, 64, 2525–2540. [Google Scholar] [CrossRef] [PubMed]
- Triglyceride Coronary Disease Genetics Consortium and Emerging Risk Factors Collaboration; Sarwar, N.; Sandhu, M.S.; Ricketts, S.L.; Butterworth, A.S.; di Angelantonio, E.; Boekholdt, S.M.; Ouwehand, W.; Watkins, H.; Samani, N.J.; et al. Triglyceride-mediated pathways and coronary disease: Collaborative analysis of 101 studies. Lancet 2010, 375, 1634–1639. [Google Scholar]
- Weiss, S.B.; Kennedy, E.P.; Kiyasu, J.Y. The enzymatic synthesis of triglycerides. J. Biol. Chem. 1960, 235, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Reue, K.; Brindley, D.N. Thematic Review Series: Glycerolipids. Multiple roles for lipins/phosphatidate phosphatase enzymes in lipid metabolism. J. Lipid. Res. 2008, 49, 2493–2503. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.L.; Stone, S.J.; Koliwad, S.; Harris, C.; Farese, R.V., Jr. Thematic review series: Glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J. Lipid. Res. 2008, 49, 2283–2301. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.W. The structure and functions of human lysophosphatidic acid acyltransferases. Front. Biosci. 2001, 6, D944–D953. [Google Scholar] [CrossRef] [PubMed]
- Wendel, A.A.; Lewin, T.M.; Coleman, R.A. Glycerol-3-phosphate acyltransferases: Rate limiting enzymes of triacylglycerol biosynthesis. Biochim. Biophys. Acta 2009, 1791, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Han, G.S.; Wu, W.I.; Carman, G.M. The Saccharomyces cerevisiae Lipin homolog is a Mg2+/− dependent phosphatidate phosphatase enzyme. J. Biol. Chem. 2006, 281, 9210–9218. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Lockwood, J.; Burn, P.; Shi, Y. Cloning and functional characterization of a mouse intestinal acyl-CoA:monoacylglycerol acyltransferase, MGAT2. J. Biol. Chem. 2003, 278, 13860–13866. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.G.; Chen, Y.Q.; Zhang, Y.; Wang, H.; Qian, Y.W.; Arnold, J.S.; Calley, J.N.; Li, S.D.; Perry, W.L., 3rd; Zhang, H.Y.; et al. The acyl coenzymeA:monoacylglycerol acyltransferase 3 (MGAT3) gene is a pseudogene in mice but encodes a functional enzyme in rats. Lipids 2011, 46, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.M.; Coleman, R.A. Enzymes of glycerolipid synthesis in eukaryotes. Annu. Rev. Biochem. 1980, 49, 459–487. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.A.; Lee, D.P. Enzymes of triacylglycerol synthesis and their regulation. Prog. Lipid. Res. 2004, 43, 134–176. [Google Scholar] [CrossRef]
- Kennedy, E.P. Metabolism of lipides. Annu. Rev. Biochem. 1957, 26, 119–148. [Google Scholar] [CrossRef] [PubMed]
- Lehner, R.; Kuksis, A. Biosynthesis of triacylglycerols. Prog. Lipid Res. 1996, 35, 169–201. [Google Scholar] [CrossRef]
- Montero-Moran, G.; Caviglia, J.M.; McMahon, D.; Rothenberg, A.; Subramanian, V.; Xu, Z.; Lara-Gonzalez, S.; Storch, J.; Carman, G.M.; Brasaemle, D.L. CGI-58/ABHD5 is a coenzyme A-dependent lysophosphatidic acid acyltransferase. J. Lipid Res. 2010, 51, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Czabany, T.; Athenstaedt, K.; Daum, G. Synthesis, storage and degradation of neutral lipids in yeast. Biochim. Biophys. Acta 2007, 1771, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Hajra, A.K. Dihydroxyacetone phosphate acyltransferase. Biochim. Biophys. Acta 1997, 1348, 27–34. [Google Scholar] [CrossRef]
- Phan, C.T.; Tso, P. Intestinal lipid absorption and transport. Front Biosci. 2001, 6, D299–D319. [Google Scholar] [CrossRef] [PubMed]
- Grigor, M.R.; Bell, R.M. Separate monoacylglycerol and diacylglycerol acyltransferases function in intestinal triacylglycerol synthesis. Biochim. Biophys. Acta 1982, 712, 464–472. [Google Scholar] [CrossRef]
- Kayden, H.J.; Senior, J.R.; Mattson, F.H. The monoglyceride pathway of fat absorption in man. J. Clin. Investig. 1967, 46, 1695–1703. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.B., Jr. Assay of acyl-CoA:monoglyceride acyltransferase from rat small intestine using continuous recording spectrophotometry. J. Lipid. Res. 1969, 10, 427–432. [Google Scholar] [PubMed]
- Skipski, V.P.; Morehouse, M.G.; Deuel, H.J., Jr. The absorption in the rat of a 1,3-dioleyl-2-deuteriostearyl glyceride-C14 and a 1-monodeuteriostearyl glyceride-C14. Arch. Biochem. Biophys. 1959, 81, 93–104. [Google Scholar] [CrossRef]
- Hall, A.M.; Kou, K.; Chen, Z.; Pietka, T.A.; Kumar, M.; Korenblat, K.M.; Lee, K.; Ahn, K.; Fabbrini, E.; Klein, S.; et al. Evidence for regulated monoacylglycerol acyltransferase expression and activity in human liver. J. Lipid Res. 2012, 53, 990–999. [Google Scholar] [CrossRef] [PubMed]
- Mul, J.D.; Begg, D.P.; Haller, A.M.; Pressler, J.W.; Sorrell, J.; Woods, S.C.; Farese, R.V., Jr.; Seeley, R.J.; Sandoval, D.A. MGAT2 deficiency and vertical sleeve gastrectomy have independent metabolic effects in the mouse. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E1065–E1072. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Cheng, L.; Shi, Y. Catalytic properties of MGAT3, a putative triacylgycerol synthase. J. Lipid Res. 2007, 48, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.L.; Farese, R.V., Jr. MGAT2, a monoacylglycerol acyltransferase expressed in the small intestine. J. Biol. Chem. 2003, 278, 18532–18537. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.L.; Stone, S.J.; Cases, S.; Zhou, P.; Farese, R.V., Jr. Identification of a gene encoding MGAT1, a monoacylglycerol acyltransferase. Proc. Natl. Acad. Sci. USA 2002, 99, 8512–8517. [Google Scholar] [CrossRef] [PubMed]
- Cases, S.; Stone, S.J.; Zhou, P.; Yen, E.; Tow, B.; Lardizabal, K.D.; Voelker, T.; Farese, R.V., Jr. Cloning of DGAT2, a second mammalian diacylglycerol acyltransferase, and related family members. J. Biol. Chem. 2001, 276, 38870–38876. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Mostafa, N.; Bhat, B.G.; Florant, G.L.; Coleman, R.A. Selective retention of essential fatty acids: The role of hepatic monoacylglycerol acyltransferase. Am. J. Physiol. 1993, 265 2 Pt 2, R414–R419. [Google Scholar] [PubMed]
- Turkish, A.R.; Henneberry, A.L.; Cromley, D.; Padamsee, M.; Oelkers, P.; Bazzi, H.; Christiano, A.M.; Billheimer, J.T.; Sturley, S.L. Identification of two novel human acyl-CoA wax alcohol acyltransferases: Members of the diacylglycerol acyltransferase 2 (DGAT2) gene superfamily. J. Biol. Chem. 2005, 280, 14755–14764. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Ko, E.H.; Kim, J.E.; Kim, E.; Lee, H.; Choi, H.; Yu, J.H.; Kim, H.J.; Seong, J.K.; Kim, K.S.; et al. Nuclear receptor PPARgamma-regulated monoacylglycerol O-acyltransferase 1 (MGAT1) expression is responsible for the lipid accumulation in diet-induced hepatic steatosis. Proc. Natl. Acad. Sci. USA 2012, 109, 13656–13661. [Google Scholar] [CrossRef] [PubMed]
- Soufi, N.; Hall, A.M.; Chen, Z.; Yoshino, J.; Collier, S.L.; Mathews, J.C.; Brunt, E.M.; Albert, C.J.; Graham, M.J.; Ford, D.A.; et al. Inhibiting monoacylglycerol acyltransferase 1 ameliorates hepatic metabolic abnormalities but not inflammation and injury in mice. J. Biol. Chem. 2014, 289, 30177–30188. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.M.; Soufi, N.; Chambers, K.T.; Chen, Z.; Schweitzer, G.G.; McCommis, K.S.; Erion, D.M.; Graham, M.J.; Su, X.; Finck, B.N. Abrogating monoacylglycerol acyltransferase activity in liver improves glucose tolerance and hepatic insulin signaling in obese mice. Diabetes 2014, 63, 2284–2296. [Google Scholar] [CrossRef] [PubMed]
- Lockwood, J.F.; Cao, J.; Burn, P.; Shi, Y. Human intestinal monoacylglycerol acyltransferase: Differential features in tissue expression and activity. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E927–E937. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.A.; Haynes, E.B. Hepatic monoacylglycerol acyltransferase. Characterization of an activity associated with the suckling period in rats. J. Biol. Chem. 1984, 259, 8934–8938. [Google Scholar] [PubMed]
- Coleman, R.A.; Haynes, E.B.; Coats, C.D. Ontogeny of microsomal activities of triacylglycerol synthesis in guinea pig liver. J. Lipid Res. 1987, 28, 320–325. [Google Scholar] [PubMed]
- Sansbury, K.; Millington, D.S.; Coleman, R.A. Hepatic monoacylglycerol acyltransferase: Ontogeny and characterization of an activity associated with the chick embryo. J. Lipid Res. 1989, 30, 1251–1258. [Google Scholar] [PubMed]
- Mentlein, R.; Rix-Matzen, H.; Heymann, E. Subcellular localization of non-specific carboxylesterases, acylcarnitine hydrolase, monoacylglycerol lipase and palmitoyl-CoA hydrolase in rat liver. Biochim. Biophys. Acta 1988, 964, 319–328. [Google Scholar] [CrossRef]
- Yen, C.L.; Cheong, M.L.; Grueter, C.; Zhou, P.; Moriwaki, J.; Wong, J.S.; Hubbard, B.; Marmor, S.; Farese, R.V., Jr. Deficiency of the intestinal enzyme acyl CoA:monoacylglycerol acyltransferase-2 protects mice from metabolic disorders induced by high-fat feeding. Nat. Med. 2009, 15, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.W.; Gao, Y.; Yen, M.I.; Yen, C.L. Intestine-specific deletion of acyl-CoA:monoacylglycerol acyltransferase (MGAT) 2 protects mice from diet-induced obesity and glucose intolerance. J. Biol. Chem. 2014, 289, 17338–17349. [Google Scholar] [CrossRef] [PubMed]
- Heppner, K.M.; Perez-Tilve, D. GLP-1 based therapeutics: Simultaneously combating T2DM and obesity. Front Neurosci. 2015, 9, 92. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Nelson, D.W.; Banh, T.; Yen, M.I.; Yen, C.L. Intestine-specific expression of MOGAT2 partially restores metabolic efficiency in Mogat2-deficient mice. J. Lipid Res. 2013, 54, 1644–1652. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.W.; Gao, Y.; Spencer, N.M.; Banh, T.; Yen, C.L. Deficiency of MGAT2 increases energy expenditure without high-fat feeding and protects genetically obese mice from excessive weight gain. J. Lipid Res. 2011, 52, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Banh, T.; Nelson, D.W.; Gao, Y.; Huang, T.N.; Yen, M.I.; Yen, C.L. Adult-onset deficiency of acyl CoA:monoacylglycerol acyltransferase 2 protects mice from diet-induced obesity and glucose intolerance. J. Lipid Res. 2015, 56, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.B.; Petersen, K.F.; Shulman, G.I. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol. Rev. 2007, 87, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, J.E. Lipotoxicity: When tissues overeat. Curr. Opin. Lipidol. 2003, 14, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Nelson, T.C.; Chen, J.; Walker, S.G.; Wardwell-Swanson, J.; Meegalla, R.; Taub, R.; Billheimer, J.T.; Ramaker, M.; Feder, J.N. Identification of acyl coenzyme A:monoacylglycerol acyltransferase 3, an intestinal specific enzyme implicated in dietary fat absorption. J. Biol. Chem. 2003, 278, 13611–13614. [Google Scholar] [CrossRef] [PubMed]
- Cases, S.; Smith, S.J.; Zheng, Y.W.; Myers, H.M.; Lear, S.R.; Sande, E.; Novak, S.; Collins, C.; Welch, C.B.; Lusis, A.J.; et al. Identification of a gene encoding an acyl CoA:diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc. Natl. Acad. Sci. USA 1998, 95, 13018–13023. [Google Scholar] [CrossRef] [PubMed]
- Stone, S.J.; Myers, H.M.; Watkins, S.M.; Brown, B.E.; Feingold, K.R.; Elias, P.M.; Farese, R.V., Jr. Lipopenia and skin barrier abnormalities in DGAT2-deficient mice. J. Biol. Chem. 2004, 279, 11767–11776. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Siloto, R.M.; Lehner, R.; Stone, S.J.; Weselake, R.J. Acyl-CoA:diacylglycerol acyltransferase: Molecular biology, biochemistry and biotechnology. Prog. Lipid Res. 2012, 51, 350–377. [Google Scholar] [CrossRef] [PubMed]
- Ables, G.P.; Yang, K.J.; Vogel, S.; Hernandez-Ono, A.; Yu, S.; Yuen, J.J.; Birtles, S.; Buckett, L.K.; Turnbull, A.V.; Goldberg, I.J.; et al. Intestinal DGAT1 deficiency reduces postprandial triglyceride and retinyl ester excursions by inhibiting chylomicron secretion and delaying gastric emptying. J. Lipid Res. 2012, 53, 2364–2379. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Zhou, Y.; Peng, H.; Huang, X.; Stahler, S.; Suri, V.; Qadri, A.; Gareski, T.; Jones, J.; Hahm, S.; et al. Targeting Acyl-CoA:diacylglycerol acyltransferase 1 (DGAT1) with small molecule inhibitors for the treatment of metabolic diseases. J. Biol. Chem. 2011, 286, 41838–41851. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Iqbal, J.; Devenny, J.; Chu, C.H.; Chen, L.; Dong, J.; Seethala, R.; Keim, W.J.; Azzara, A.V.; Lawrence, R.M.; et al. Acylation of acylglycerols by acyl coenzyme A:diacylglycerol acyltransferase 1 (DGAT1). Functional importance of DGAT1 in the intestinal fat absorption. J. Biol. Chem. 2008, 283, 29802–29811. [Google Scholar] [CrossRef] [PubMed]
- Dow, R.L.; Li, J.C.; Pence, M.P.; Gibbs, E.M.; LaPerle, J.L.; Litchfield, J.; Piotrowski, D.W.; Munchhof, M.J.; Manion, T.B.; Zavadoski, W.J.; et al. Discovery of PF-04620110, a Potent, Selective, and Orally Bioavailable Inhibitor of DGAT-1. ACS Med. Chem. Lett. 2011, 2, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Schober, G.; Arnold, M.; Birtles, S.; Buckett, L.K.; Pacheco-Lopez, G.; Turnbull, A.V.; Langhans, W.; Mansouri, A. Diacylglycerol acyltransferase-1 inhibition enhances intestinal fatty acid oxidation and reduces energy intake in rats. J. Lipid Res. 2013, 54, 1369–1384. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Yamaguchi, H.; Miki, H.; Kitamura, S.; Nakada, Y.; Aicher, T.D.; Pratt, S.A.; Kato, K. A novel coenzyme A:diacylglycerol acyltransferase 1 inhibitor stimulates lipid metabolism in muscle and lowers weight in animal models of obesity. Eur. J. Pharmacol. 2011, 650, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Souers, A.J.; Voorbach, M.; Falls, H.D.; Droz, B.; Brodjian, S.; Lau, Y.Y.; Iyengar, R.R.; Gao, J.; Judd, A.S.; et al. Validation of diacyl glycerolacyltransferase I as a novel target for the treatment of obesity and dyslipidemia using a potent and selective small molecule inhibitor. J. Med. Chem. 2008, 51, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.T.; Winter, H.S.; Lim, E.; Kirby, A.; Blumenstiel, B.; DeFelice, M.; Gabriel, S.; Jalas, C.; Branski, D.; Grueter, C.A.; et al. DGAT1 mutation is linked to a congenital diarrheal disorder. J. Clin. Investig. 2012, 122, 4680–4684. [Google Scholar] [CrossRef] [PubMed]
- Denison, H.; Nilsson, C.; Lofgren, L.; Himmelmann, A.; Martensson, G.; Knutsson, M.; Al-Shurbaji, A.; Tornqvist, H.; Eriksson, J.W. Diacylglycerol acyltransferase 1 inhibition with AZD7687 alters lipid handling and hormone secretion in the gut with intolerable side effects: A randomized clinical trial. Diabetes Obes. Metab. 2014, 16, 334–343. [Google Scholar] [CrossRef] [PubMed]
- DeVita, R.J.; Pinto, S. Current status of the research and development of diacylglycerol O-acyltransferase 1 (DGAT1) inhibitors. J. Med. Chem. 2013, 56, 9820–9825. [Google Scholar] [CrossRef] [PubMed]
- Onorato, J.M.; Chu, C.H.; Ma, Z.; Kopcho, L.M.; Chao, H.J.; Lawrence, R.M.; Cheng, D. Cell-based assay of MGAT2-driven diacylglycerol synthesis for profiling inhibitors: Use of a stable isotope-labeled substrate and high-resolution LC/MS. J. Lipid Res. 2015, 56, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Barlind, J.G.; Buckett, L.K.; Crosby, S.G.; Davidsson, O.; Emtenas, H.; Ertan, A.; Jurva, U.; Lemurell, M.; Gutierrez, P.M.; Nilsson, K.; et al. Identification and design of a novel series of MGAT2 inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 2721–2726. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Takahagi, H.; Yoshikawa, T.; Morimoto, S.; Takai, T.; Hidaka, K.; Kamaura, M.; Kubo, O.; Adachi, R.; Ishii, T. Discovery of a Novel Series of N-Phenylindoline-5-sulfonamide Derivatives as Potent, Selective, and Orally Bioavailable Acyl CoA:Monoacylglycerol Acyltransferase-2 Inhibitors. J. Med. Chem. 2015, 58, 3892–3909. [Google Scholar] [CrossRef] [PubMed]
- Okuma, C.; Takahagi, H.; Yoshikawa, T.; Morimoto, S.; Takai, T.; Hidaka, K.; Kamaura, M.; Kubo, O.; Adachi, R.; Ishii, T.; et al. JTP-103237, a novel monoacylglycerol acyltransferase inhibitor, modulates fat absorption and prevents diet-induced obesity. Eur. J. Pharmacol. 2015, 758, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Yanovski, S.Z.; Yanovski, J.A. Long-term drug treatment for obesity: A systematic and clinical review. JAMA 2014, 311, 74–86. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, M.; Nickels, J.T., Jr. MOGAT2: A New Therapeutic Target for Metabolic Syndrome. Diseases 2015, 3, 176-192. https://doi.org/10.3390/diseases3030176
Yang M, Nickels JT Jr. MOGAT2: A New Therapeutic Target for Metabolic Syndrome. Diseases. 2015; 3(3):176-192. https://doi.org/10.3390/diseases3030176
Chicago/Turabian StyleYang, Muhua, and Joseph T. Nickels, Jr. 2015. "MOGAT2: A New Therapeutic Target for Metabolic Syndrome" Diseases 3, no. 3: 176-192. https://doi.org/10.3390/diseases3030176
APA StyleYang, M., & Nickels, J. T., Jr. (2015). MOGAT2: A New Therapeutic Target for Metabolic Syndrome. Diseases, 3(3), 176-192. https://doi.org/10.3390/diseases3030176