Intercalated Cells: More than pH Regulation

Abstract
:1. Introduction: Acid-Base Balance and Role of the Kidney
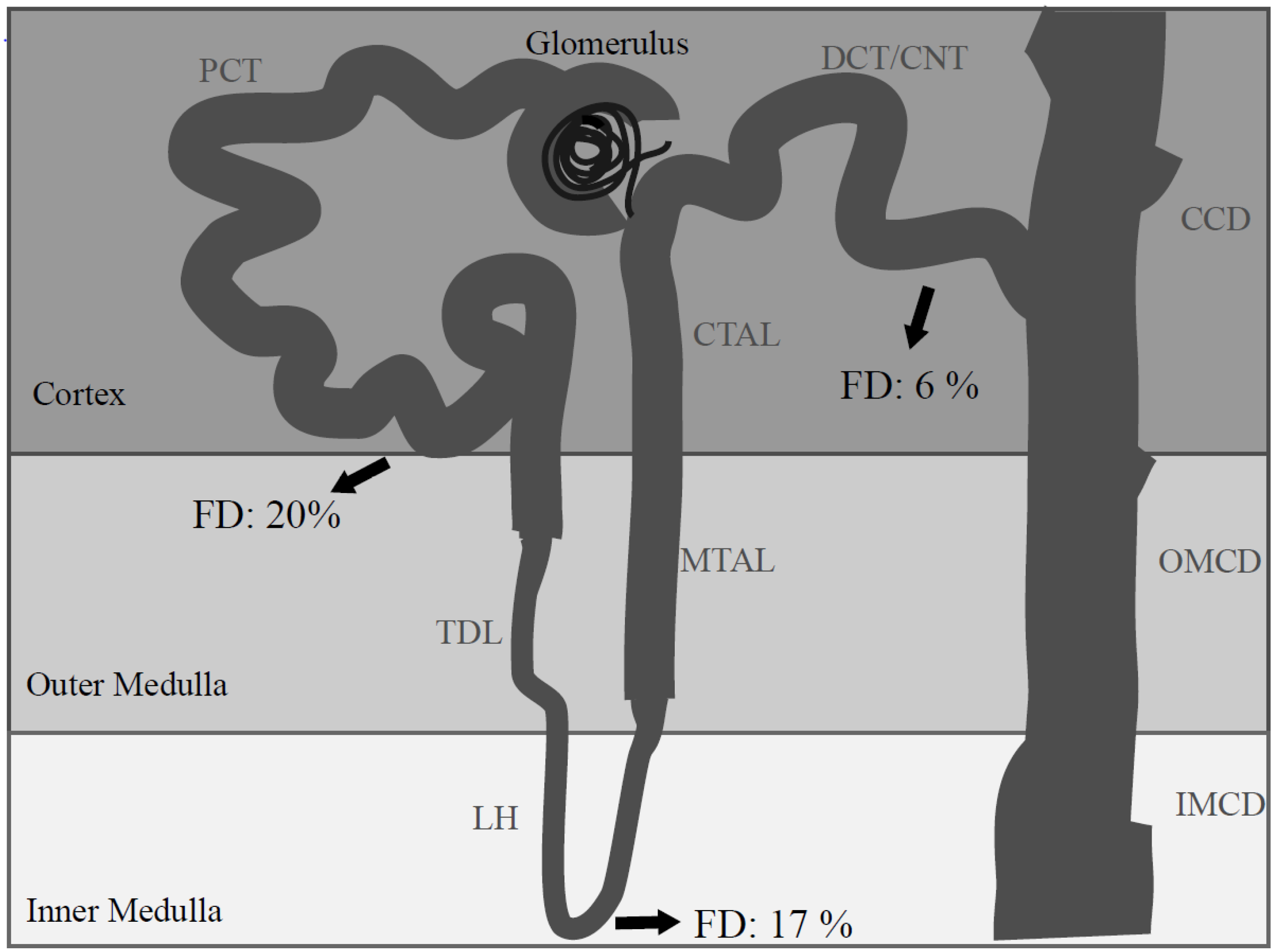
2. Renal Intercalated Cells: Mirror Cells?

{kind=link}
{kind=link}
| Nephron Segmen | Type-A IC | Type-B IC | Non-A, non-B IC |
|---|---|---|---|
| CNT | 35%–40% of IC in mouse | present in mouse | 50% of IC of mouse |
| predominant in rat and rabbit | predominant in the initial CNT | 14% of IC in rat | |
| CCD | 60% of IC in mouse | 20% of IC in mouse | 20% of IC in mouse |
| 45% of IC in rat | 50% of IC in rat | 5% of IC in rat | |
| OMCD | 100% of IC in mouse | 0% in mouse | 0% in mouse |
| predominant in rat and rabbit | |||
| IMCD | 100% of IC in mouse | 0% in mouse | 0% in mouse |
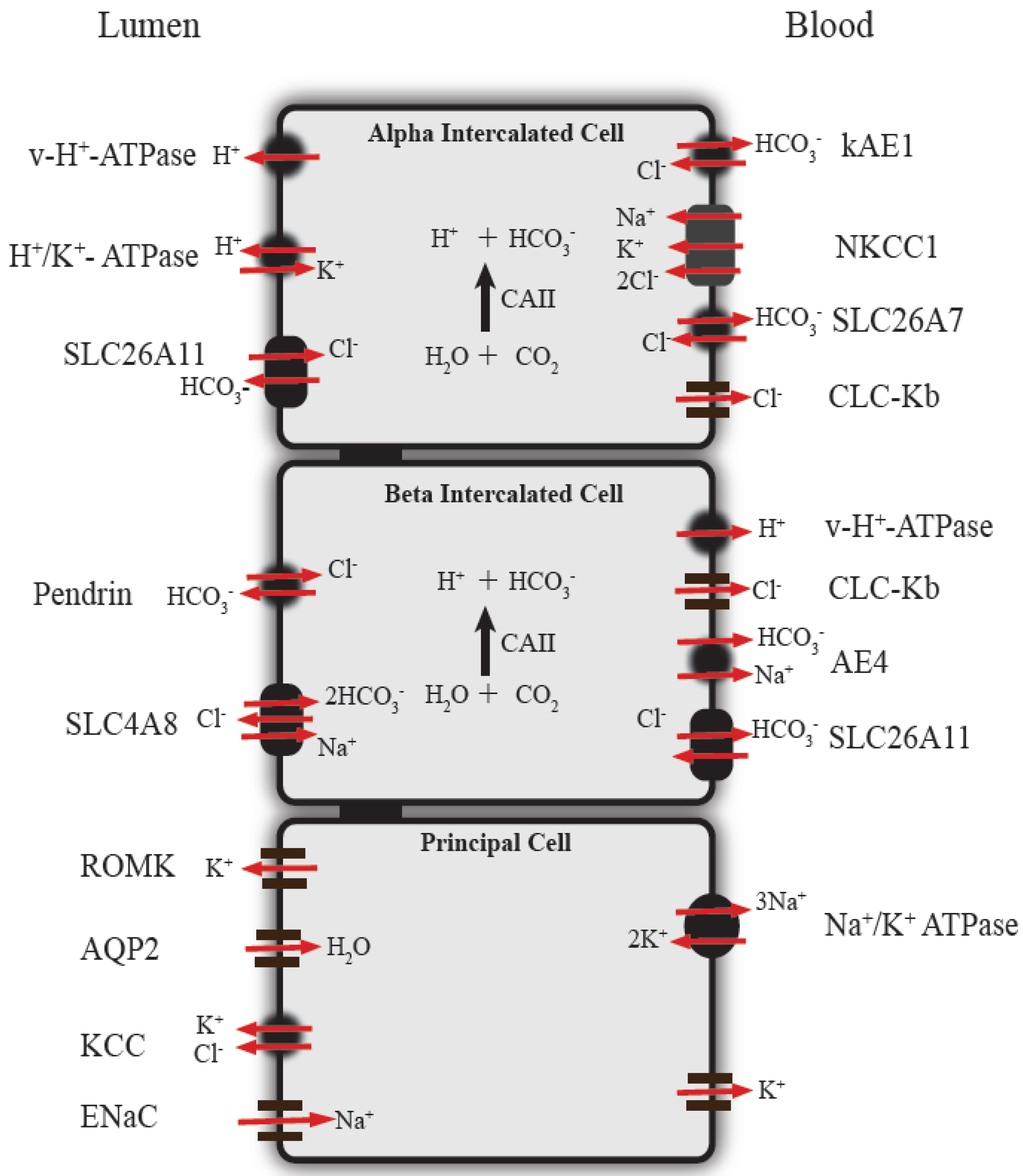
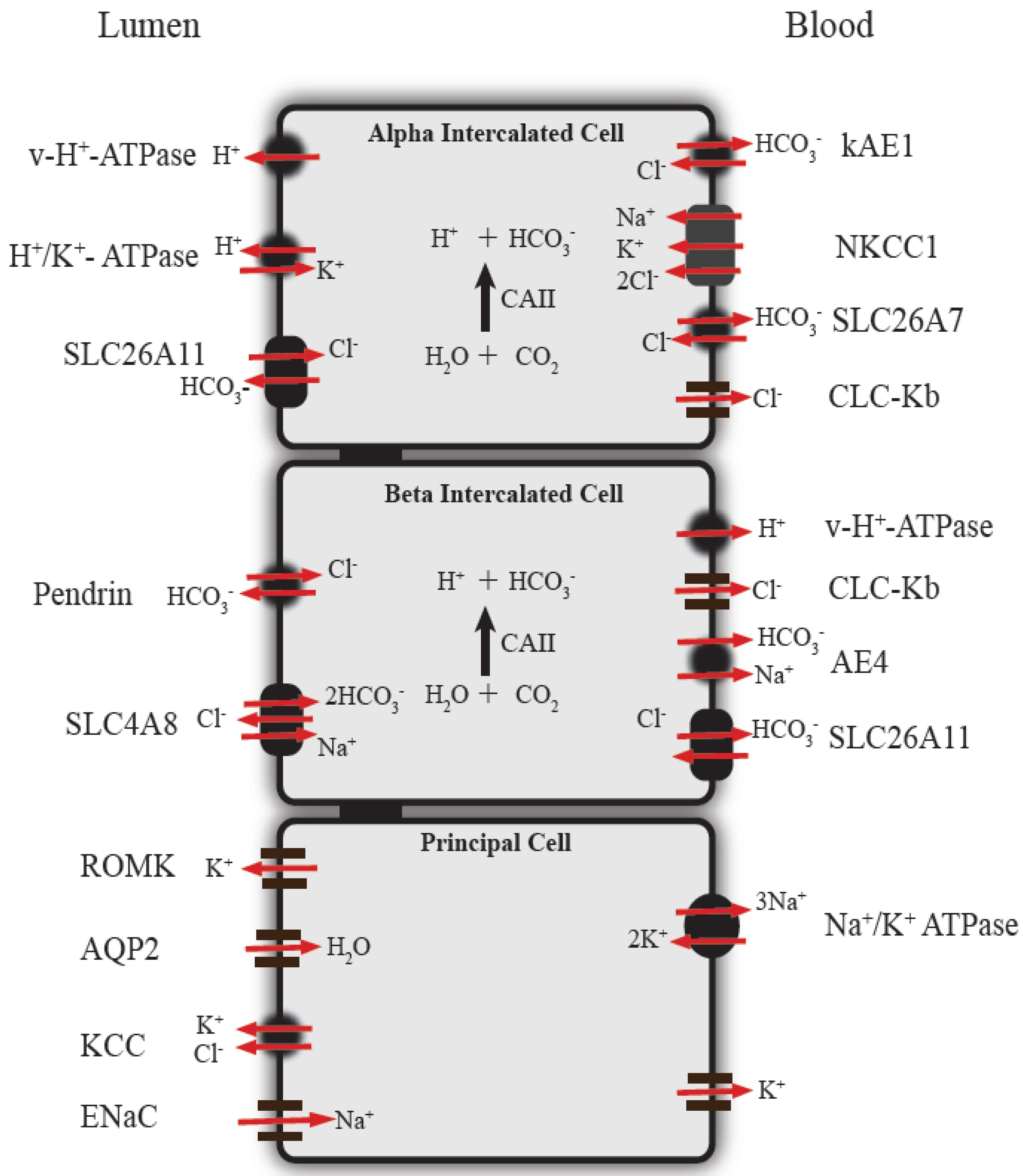
3. Function of Renal Intercalated Cells in Acid-Base Regulation
4. Emerging Roles of Intercalated Cells in the Regulation of Electrolyte Homeostasis
5. Interconversion of Intercalated Cells
6. Regulation of Intercalated Cell Function
7. Diseases Associated with Abnormal Intercalated Cells Function and Animal Models (Table 2)
| Protein | Disease and Symptoms | Mutated gene | Knockout mice symptoms | References |
|---|---|---|---|---|
| Kidney anion exchanger 1 (kAE1) | Distal renal tubular acidosis (dRTA) Metabolic acidosis, alkaline urine, hypokalemia, hyperchloremia, hypercalciuria, kidney stones, mild electrolyte imbalance | SLC4A1 | Severe anemia, complete dRTA, increased serum osmolarity, decreased urine osmolarity, hypercalciuria, dehydration | [90,91,92] |
| vATPase subunit B1 | Recessive dRTA associated with deafness, hypokalemia and dehydration | ATP6V1B1 | Mild metabolic acidosis | [93,94,95] |
| vATPase subunit a4 | Recessive dRTA associated with deafness, hypokalemia and dehydration | ATP6V0A4 | Deafness, dRTA accompanied by unsuspected proximal tubule malfunction, proteinuria and phosphaturia | [95,96,97] |
| SLC26A7 | dRTA | SLC26A7 | Complete dRTA, hypotension, failure to reabsorb chloride | [22,24] |
| Carbonic anhydrase II | Mixture of recessive distal and proximal RTA, can be associated with autosomal recessive osteopetrosis (increased bone density), growth failure, mental retardation and hearing impairment | CA2 | Renal tubular acidosis symptoms, down-regulation of proteins involved in acid-base homeostasis in intercalated cells such as SLC26A7, pendrin and AE1 | [98,99,100,101] |
| Pendrin | Pendred syndrome Goiter and hearing loss | SLC26A4 | Renal inability to excrete the excess bicarbonate into the urine, deafness | [38,56,102,103] |
8. Conclusions and Future Directions
Acknowledgments
Authors Contributions
Conflicts of Interest
References
- Cordat, E.; Casey, J.R. Bicarbonate transport in cell physiology and disease. Biochem. J. 2009, 417, 423–439. [Google Scholar] [CrossRef]
- Gross, E.; Hawkins, K.; Pushkin, A.; Sassani, P.; Dukkipati, R.; Abuladze, N.; Hopfer, U.; Kurtz, I. Phosphorylation of ser(982) in the sodium bicarbonate cotransporter knbc1 shifts the hco(3)(-): Na(+) stoichiometry from 3 : 1 to 2 : 1 in murine proximal tubule cells. J. Physiol. 2001, 537, 659–665. [Google Scholar] [CrossRef]
- Buerkert, J.; Martin, D.; Trigg, D. Segmental analysis of the renal tubule in buffer production and net acid formation. Am. J. Physiol. 1983, 244, F442–F454. [Google Scholar]
- DuBose, T.D., Jr.; Pucacco, L.R.; Lucci, M.S.; Carter, N.W. Micropuncture determination of ph, pco2, and total co2 concentration in accessible structures of the rat renal cortex. J. Clin. Invest. 1979, 64, 476–482. [Google Scholar] [CrossRef]
- Brenner, B.M.; Rector, F.C. Brenner & Rector’s the Kidney, 8th ed.; Saunders Elsevier: Philadelphia, PA, USA, 2008. [Google Scholar]
- Schwartz, G.J.; Barasch, J.; Al-Awqati, Q. Plasticity of functional epithelial polarity. Nature 1985, 318, 368–371. [Google Scholar] [CrossRef]
- Alper, S.L.; Natale, J.; Gluck, S.; Lodish, H.F.; Brown, D. Subtypes of intercalated cells in rat kidney collecting duct defined by antibodies against erythroid band 3 and renal vacuolar h+-atpase. Proc. Natl. Acad. Sci. USA 1989, 86, 5429–5433. [Google Scholar] [CrossRef]
- Holthofer, H.; Schulte, B.A.; Pasternack, G.; Siegel, G.J.; Spicer, S.S. Three distinct cell populations in rat kidney collecting duct. Am. J. Physiol. 1987, 253, C323–C328. [Google Scholar]
- Kim, J.; Kim, Y.H.; Cha, J.H.; Tisher, C.C.; Madsen, K.M. Intercalated cell subtypes in connecting tubule and cortical collecting duct of rat and mouse. J. Am. Soc. Nephrol. 1999, 10, 1–12. [Google Scholar]
- Madsen, K.M.; Tisher, C.C. Structural-functional relationships along the distal nephron. Am. J. Physiol. 1986, 250, F1–F15. [Google Scholar]
- Schuster, V.L. Function and regulation of collecting duct intercalated cells. Annu. Rev. Physiol. 1993, 55, 267–288. [Google Scholar] [CrossRef]
- Verlander, J.W.; Madsen, K.M.; Tisher, C.C. Effect of acute respiratory acidosis on two populations of intercalated cells in rat cortical collecting duct. Am. J. Physiol. 1987, 253, F1142–F1156. [Google Scholar]
- Teng-umnuay, P.; Verlander, J.W.; Yuan, W.; Tisher, C.C.; Madsen, K.M. Identification of distinct subpopulations of intercalated cells in the mouse collecting duct. J. Am. Soc. Nephrol. 1996, 7, 260–274. [Google Scholar]
- Satlin, L.M.; Schwartz, G.J. Cellular remodeling of hco3(-)-secreting cells in rabbit renal collecting duct in response to an acidic environment. J. Cell. Biol. 1989, 109, 1279–1288. [Google Scholar] [CrossRef]
- Brown, D.; Hirsch, S.; Gluck, S. Localization of a proton-pumping atpase in rat kidney. J. Clin. Invest. 1988, 82, 2114–2126. [Google Scholar] [CrossRef]
- Schuster, V.L.; Fejes-Toth, G.; Naray-Fejes-Toth, A.; Gluck, S. Colocalization of h(+)-atpase and band 3 anion exchanger in rabbit collecting duct intercalated cells. Am. J. Physiol. 1991, 260, F506–F517. [Google Scholar]
- Verlander, J.W.; Madsen, K.M.; Cannon, J.K.; Tisher, C.C. Activation of acid-secreting intercalated cells in rabbit collecting duct with ammonium chloride loading. Am. J. Physiol. 1994, 266, F633–F645. [Google Scholar]
- Toei, M.; Saum, R.; Forgac, M. Regulation and isoform function of the v-atpases. Biochemistry 2010, 49, 4715–4723. [Google Scholar] [CrossRef]
- Weiner, I.D.; Hamm, L.L. Regulation of intracellular ph in the rabbit cortical collecting tubule. J. Clin. Invest. 1990, 85, 274–281. [Google Scholar] [CrossRef]
- Alper, S.L. Genetic diseases of acid-base transporters. Annu. Rev. Physiol. 2002, 64, 899–923. [Google Scholar] [CrossRef]
- Amlal, H.; Petrovic, S.; Xu, J.; Wang, Z.; Sun, X.; Barone, S.; Soleimani, M. Deletion of the anion exchanger slc26a4 (pendrin) decreases apical cl(-)/hco3(-) exchanger activity and impairs bicarbonate secretion in kidney collecting duct. Am. J. Physiol. Cell. Physiol. 2010, 299, C33–C41. [Google Scholar] [CrossRef]
- Sun, X.; Petrovic, S. Increased acid load and deletion of ae1 increase slc26a7 expression. Nephron Physiol. 2008, 109, p29–p35. [Google Scholar] [CrossRef]
- Kim, K.H.; Shcheynikov, N.; Wang, Y.; Muallem, S. Slc26a7 is a cl- channel regulated by intracellular ph. J. Biol. Chem. 2005, 280, 6463–6470. [Google Scholar] [CrossRef]
- Xu, J.; Song, P.; Nakamura, S.; Miller, M.; Barone, S.; Alper, S.L.; Riederer, B.; Bonhagen, J.; Arend, L.J.; Amlal, H.; et al. Deletion of the chloride transporter slc26a7 causes distal renal tubular acidosis and impairs gastric acid secretion. J. Biol. Chem. 2009, 284, 29470–29479. [Google Scholar] [CrossRef]
- Leviel, F.; Hubner, C.A.; Houillier, P.; Morla, L.; El Moghrabi, S.; Brideau, G.; Hassan, H.; Parker, M.D.; Kurth, I.; Kougioumtzes, A.; et al. The na+-dependent chloride-bicarbonate exchanger slc4a8 mediates an electroneutral na+ reabsorption process in the renal cortical collecting ducts of mice. J. Clin. Invest. 2010, 120, 1627–1635. [Google Scholar] [CrossRef]
- Procino, G.; Mastrofrancesco, L.; Sallustio, F.; Costantino, V.; Barbieri, C.; Pisani, F.; Schena, F.P.; Svelto, M.; Valenti, G. Aqp5 is expressed in type-b intercalated cells in the collecting duct system of the rat, mouse and human kidney. Cell. Physiol. Biochem. 2011, 28, 683–692. [Google Scholar] [CrossRef]
- Chambrey, R.; Kurth, I.; Peti-Peterdi, J.; Houillier, P.; Purkerson, J.M.; Leviel, F.; Hentschke, M.; Zdebik, A.A.; Schwartz, G.J.; Hubner, C.A.; et al. Renal intercalated cells are rather energized by a proton than a sodium pump. Proc. Natl. Acad. Sci. USA 2013, 110, 7928–7933. [Google Scholar] [CrossRef]
- Buffin-Meyer, B.; Verbavatz, J.M.; Cheval, L.; Marsy, S.; Younes-Ibrahim, M.; Le Moal, C.; Doucet, A. Regulation of na+, k(+)-atpase in the rat outer medullary collecting duct during potassium depletion. J. Am. Soc. Nephrol. 1998, 9, 538–550. [Google Scholar]
- Kashgarian, M.; Biemesderfer, D.; Caplan, M.; Forbush, B., 3rd. Monoclonal antibody to na,k-atpase: Immunocytochemical localization along nephron segments. Kidney Int. 1985, 28, 899–913. [Google Scholar] [CrossRef]
- Piepenhagen, P.A.; Peters, L.L.; Lux, S.E.; Nelson, W.J. Differential expression of na(+)-k(+)-atpase, ankyrin, fodrin, and e-cadherin along the kidney nephron. Am. J. Physiol. 1995, 269, C1417–C1432. [Google Scholar]
- Sabolic, I.; Herak-Kramberger, C.M.; Breton, S.; Brown, D. Na/k-atpase in intercalated cells along the rat nephron revealed by antigen retrieval. J. Am. Soc. Nephrol. 1999, 10, 913–922. [Google Scholar]
- Chen, Y.; Cann, M.J.; Litvin, T.N.; Iourgenko, V.; Sinclair, M.L.; Levin, L.R.; Buck, J. Soluble adenylyl cyclase as an evolutionarily conserved bicarbonate sensor. Science 2000, 289, 625–628. [Google Scholar] [CrossRef]
- Paunescu, T.G.; Da Silva, N.; Russo, L.M.; McKee, M.; Lu, H.A.; Breton, S.; Brown, D. Association of soluble adenylyl cyclase with the v-atpase in renal epithelial cells. Am. J. Physiol. Renal Physiol. 2008, 294, F130–F138. [Google Scholar]
- Paunescu, T.G.; Ljubojevic, M.; Russo, L.M.; Winter, C.; McLaughlin, M.M.; Wagner, C.A.; Breton, S.; Brown, D. Camp stimulates apical v-atpase accumulation, microvillar elongation, and proton extrusion in kidney collecting duct a-intercalated cells. Am. J. Physiol. Renal Physiol. 2010, 298, F643–F654. [Google Scholar] [CrossRef]
- Bates, C.M.; Merenmies, J.M.; Kelly-Spratt, K.S.; Parada, L.F. Insulin receptor-related receptor expression in non-a intercalated cells in the kidney. Kidney Int. 1997, 52, 674–681. [Google Scholar] [CrossRef]
- Deyev, I.E.; Sohet, F.; Vassilenko, K.P.; Serova, O.V.; Popova, N.V.; Zozulya, S.A.; Burova, E.B.; Houillier, P.; Rzhevsky, D.I.; Berchatova, A.A.; et al. Insulin receptor-related receptor as an extracellular alkali sensor. Cell Metab. 2011, 13, 679–689. [Google Scholar] [CrossRef]
- Brown, D.; Wagner, C.A. Molecular mechanisms of acid-base sensing by the kidney. J. Am. Soc. Nephrol. 2012, 23, 774–780. [Google Scholar] [CrossRef]
- Royaux, I.E.; Wall, S.M.; Karniski, L.P.; Everett, L.A.; Suzuki, K.; Knepper, M.A.; Green, E.D. Pendrin, encoded by the pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc. Natl. Acad. Sci. USA 2001, 98, 4221–4226. [Google Scholar] [CrossRef]
- Schuster, V.L.; Stokes, J.B. Chloride transport by the cortical and outer medullary collecting duct. Am. J. Physiol. 1987, 253, F203–F212. [Google Scholar]
- Wall, S.M.; Kim, Y.H.; Stanley, L.; Glapion, D.M.; Everett, L.A.; Green, E.D.; Verlander, J.W. Nacl restriction upregulates renal slc26a4 through subcellular redistribution: Role in cl- conservation. Hypertension 2004, 44, 982–987. [Google Scholar] [CrossRef]
- Jacques, T.; Picard, N.; Miller, R.L.; Riemondy, K.A.; Houillier, P.; Sohet, F.; Ramakrishnan, S.K.; Busst, C.J.; Jayat, M.; Corniere, N.; et al. Overexpression of pendrin in intercalated cells produces chloride-sensitive hypertension. J. Am. Soc. Nephrol. 2013, 24, 1104–1113. [Google Scholar] [CrossRef]
- Verlander, J.W.; Hassell, K.A.; Royaux, I.E.; Glapion, D.M.; Wang, M.E.; Everett, L.A.; Green, E.D.; Wall, S.M. Deoxycorticosterone upregulates pds (slc26a4) in mouse kidney: Role of pendrin in mineralocorticoid-induced hypertension. Hypertension 2003, 42, 356–362. [Google Scholar] [CrossRef]
- Pech, V.; Zheng, W.; Pham, T.D.; Verlander, J.W.; Wall, S.M. Angiotensin ii activates h+-atpase in type a intercalated cells. J. Am. Soc. Nephrol. 2008, 19, 84–91. [Google Scholar] [CrossRef]
- Terada, Y.; Knepper, M.A. Thiazide-sensitive nacl absorption in rat cortical collecting duct. Am. J. Physiol. 1990, 259, F519–F528. [Google Scholar]
- Rubera, I.; Loffing, J.; Palmer, L.G.; Frindt, G.; Fowler-Jaeger, N.; Sauter, D.; Carroll, T.; McMahon, A.; Hummler, E.; Rossier, B.C. Collecting duct-specific gene inactivation of alphaenac in the mouse kidney does not impair sodium and potassium balance. J. Clin. Invest. 2003, 112, 554–565. [Google Scholar] [CrossRef]
- Gueutin, V.; Vallet, M.; Jayat, M.; Peti-Peterdi, J.; Corniere, N.; Leviel, F.; Sohet, F.; Wagner, C.A.; Eladari, D.; Chambrey, R. Renal beta-intercalated cells maintain body fluid and electrolyte balance. J. Clin. Invest. 2013, 123, 4219–4231. [Google Scholar] [CrossRef] [Green Version]
- Sebastian, A.; McSherry, E.; Morris, R.C., Jr. Impaired renal conservation of sodium and chloride during sustained correction of systemic acidosis in patients with type 1, classic renal tubular acidosis. J. Clin. Invest. 1976, 58, 454–469. [Google Scholar] [CrossRef]
- Edwards, J.C.; van Adelsberg, J.; Rater, M.; Herzlinger, D.; Lebowitz, J.; al-Awqati, Q. Conditional immortalization of bicarbonate-secreting intercalated cells from rabbit. Am. J. Physiol. 1992, 263, C521–C529. [Google Scholar]
- van Adelsberg, J.; Edwards, J.C.; Takito, J.; Kiss, B.; al-Awqati, Q. An induced extracellular matrix protein reverses the polarity of band 3 in intercalated epithelial cells. Cell 1994, 76, 1053–1061. [Google Scholar] [CrossRef]
- Gao, X.; Eladari, D.; Leviel, F.; Tew, B.Y.; Miro-Julia, C.; Cheema, F.H.; Miller, L.; Nelson, R.; Paunescu, T.G.; McKee, M.; et al. Deletion of hensin/dmbt1 blocks conversion of beta- to alpha-intercalated cells and induces distal renal tubular acidosis. Proc. Natl. Acad. Sci. USA 2010, 107, 21872–21877. [Google Scholar] [CrossRef]
- Hikita, C.; Takito, J.; Vijayakumar, S.; Al-Awqati, Q. Only multimeric hensin located in the extracellular matrix can induce apical endocytosis and reverse the polarity of intercalated cells. J. Biol. Chem. 1999, 274, 17671–17676. [Google Scholar] [CrossRef]
- Hikita, C.; Vijayakumar, S.; Takito, J.; Erdjument-Bromage, H.; Tempst, P.; Al-Awqati, Q. Induction of terminal differentiation in epithelial cells requires polymerization of hensin by galectin 3. J. Cell Biol. 2000, 151, 1235–1246. [Google Scholar] [CrossRef]
- Watanabe, S.; Tsuruoka, S.; Vijayakumar, S.; Fischer, G.; Zhang, Y.; Fujimura, A.; Al-Awqati, Q.; Schwartz, G.J. Cyclosporin a produces distal renal tubular acidosis by blocking peptidyl prolyl cis-trans isomerase activity of cyclophilin. Am. J. Physiol. Renal Physiol. 2005, 288, F40–F47. [Google Scholar]
- Liu, Y.; Pathak, N.; Kramer-Zucker, A.; Drummond, I.A. Notch signaling controls the differentiation of transporting epithelia and multiciliated cells in the zebrafish pronephros. Development 2007, 134, 1111–1122. [Google Scholar] [CrossRef]
- Jeong, H.W.; Jeon, U.S.; Koo, B.K.; Kim, W.Y.; Im, S.K.; Shin, J.; Cho, Y.; Kim, J.; Kong, Y.Y. Inactivation of notch signaling in the renal collecting duct causes nephrogenic diabetes insipidus in mice. J. Clin. Invest. 2009, 119, 3290–3300. [Google Scholar]
- Hulander, M.; Kiernan, A.E.; Blomqvist, S.R.; Carlsson, P.; Samuelsson, E.J.; Johansson, B.R.; Steel, K.P.; Enerback, S. Lack of pendrin expression leads to deafness and expansion of the endolymphatic compartment in inner ears of foxi1 null mutant mice. Development 2003, 130, 2013–2025. [Google Scholar] [CrossRef]
- Blomqvist, S.R.; Vidarsson, H.; Fitzgerald, S.; Johansson, B.R.; Ollerstam, A.; Brown, R.; Persson, A.E.; Bergstrom, G.G.; Enerback, S. Distal renal tubular acidosis in mice that lack the forkhead transcription factor foxi1. J. Clin. Invest. 2004, 113, 1560–1570. [Google Scholar]
- Kurth, I.; Hentschke, M.; Hentschke, S.; Borgmeyer, U.; Gal, A.; Hubner, C.A. The forkhead transcription factor foxi1 directly activates the ae4 promoter. Biochem. J. 2006, 393, 277–283. [Google Scholar] [CrossRef]
- Vidarsson, H.; Westergren, R.; Heglind, M.; Blomqvist, S.R.; Breton, S.; Enerback, S. The forkhead transcription factor foxi1 is a master regulator of vacuolar h-atpase proton pump subunits in the inner ear, kidney and epididymis. PLoS One 2009, 4, e4471. [Google Scholar]
- Fejes-Toth, G.; Naray-Fejes-Toth, A. Differentiation of renal beta-intercalated cells to alpha-intercalated and principal cells in culture. Proc. Natl. Acad. Sci. USA 1992, 89, 5487–5491. [Google Scholar] [CrossRef]
- Wallin, L.; Alling, C.; Aurell, M. Impairment of renal function in patients on long-term lithium treatment. Clin. Nephrol. 1982, 18, 23–28. [Google Scholar]
- Kortenoeven, M.L.; Li, Y.; Shaw, S.; Gaeggeler, H.P.; Rossier, B.C.; Wetzels, J.F.; Deen, P.M. Amiloride blocks lithium entry through the sodium channel thereby attenuating the resultant nephrogenic diabetes insipidus. Kidney Int. 2009, 76, 44–53. [Google Scholar] [CrossRef]
- de Groot, T.; Alsady, M.; Jaklofsky, M.; Otte-Holler, I.; Baumgarten, R.; Giles, R.H.; Deen, P.M. Lithium causes g2 arrest of renal principal cells. J. Am. Soc. Nephrol. 2014, 25, 501–510. [Google Scholar] [CrossRef]
- Trepiccione, F.; Capasso, G.; Nielsen, S.; Christensen, B.M. Evaluation of cellular plasticity in the collecting duct during recovery from lithium-induced nephrogenic diabetes insipidus. Am. J. Physiol. Renal Physiol. 2013, 305, F919–F929. [Google Scholar] [CrossRef]
- Wall, S.M.; Pech, V. The interaction of pendrin and the epithelial sodium channel in blood pressure regulation. Curr. Opin. Nephrol. Hypertens. 2008, 17, 18–24. [Google Scholar] [CrossRef]
- Thumova, M.; Pech, V.; Froehlich, O.; Agazatian, D.; Wang, X.; Verlander, J.W.; Kim, Y.H.; Wall, S.M. Pendrin protein abundance in the kidney is regulated by nitric oxide and camp. Am. J. Physiol. Renal Physiol. 2012, 303, F812–F820. [Google Scholar] [CrossRef]
- DuBose, T.D., Jr.; Caflisch, C.R. Effect of selective aldosterone deficiency on acidification in nephron segments of the rat inner medulla. J. Clin. Invest. 1988, 82, 1624–1632. [Google Scholar] [CrossRef]
- Garg, L.C.; Narang, N. Effects of aldosterone on nem-sensitive atpase in rabbit nephron segments. Kidney Int. 1988, 34, 13–17. [Google Scholar] [CrossRef]
- Hays, S.R. Mineralocorticoid modulation of apical and basolateral membrane h+/oh-/hco3- transport processes in the rabbit inner stripe of outer medullary collecting duct. J. Clin. Invest. 1992, 90, 180–187. [Google Scholar] [CrossRef]
- Khadouri, C.; Marsy, S.; Barlet-Bas, C.; Doucet, A. Short-term effect of aldosterone on nem-sensitive atpase in rat collecting tubule. Am. J. Physiol. 1989, 257, F177–F181. [Google Scholar]
- McKinney, T.D.; Davidson, K.K. Bicarbonate transport in collecting tubules from outer stripe of outer medulla of rabbit kidneys. Am. J. Physiol. 1987, 253, F816–F822. [Google Scholar]
- Mujais, S.K. Effects of aldosterone on rat collecting tubule n-ethylmaleimide-sensitive adenosine triphosphatase. J. Lab. Clin. Med. 1987, 109, 34–39. [Google Scholar]
- Wagner, C.A.; Mohebbi, N.; Uhlig, U.; Giebisch, G.H.; Breton, S.; Brown, D.; Geibel, J.P. Angiotensin ii stimulates h(+)-atpase activity in intercalated cells from isolated mouse connecting tubules and cortical collecting ducts. Cell. Physiol. Biochem. 2011, 28, 513–520. [Google Scholar] [CrossRef] [Green Version]
- Rothenberger, F.; Velic, A.; Stehberger, P.A.; Kovacikova, J.; Wagner, C.A. Angiotensin ii stimulates vacuolar h+-atpase activity in renal acid-secretory intercalated cells from the outer medullary collecting duct. J. Am. Soc. Nephrol. 2007, 18, 2085–2093. [Google Scholar] [CrossRef]
- Winter, C.; Kampik, N.B.; Vedovelli, L.; Rothenberger, F.; Paunescu, T.G.; Stehberger, P.A.; Brown, D.; John, H.; Wagner, C.A. Aldosterone stimulates vacuolar h(+)-atpase activity in renal acid-secretory intercalated cells mainly via a protein kinase c-dependent pathway. Am. J. Physiol. Cell Physiol. 2011, 301, C1251–C1261. [Google Scholar] [CrossRef]
- Lu, X.; Garrelds, I.M.; Wagner, C.A.; Danser, A.H.; Meima, M.E. (Pro)renin receptor is required for prorenin-dependent and -independent regulation of vacuolar h(+)-atpase activity in mdck.C11 collecting duct cells. Am. J. Physiol. Renal Physiol. 2013, 305, F417–F425. [Google Scholar] [CrossRef]
- Mohebbi, N.; Perna, A.; van der Wijst, J.; Becker, H.M.; Capasso, G.; Wagner, C.A. Regulation of two renal chloride transporters, ae1 and pendrin, by electrolytes and aldosterone. PLoS One 2013, 8, e55286. [Google Scholar]
- Lifton, R.P.; Gharavi, A.G.; Geller, D.S. Molecular mechanisms of human hypertension. Cell 2001, 104, 545–556. [Google Scholar] [CrossRef]
- Shibata, S.; Rinehart, J.; Zhang, J.; Moeckel, G.; Castaneda-Bueno, M.; Stiegler, A.L.; Boggon, T.J.; Gamba, G.; Lifton, R.P. Mineralocorticoid receptor phosphorylation regulates ligand binding and renal response to volume depletion and hyperkalemia. Cell Metab. 2013, 18, 660–671. [Google Scholar] [CrossRef]
- Eladari, D.; Chambrey, R.; Peti-Peterdi, J. A new look at electrolyte transport in the distal tubule. Annu. Rev. Physiol. 2012, 74, 325–349. [Google Scholar] [CrossRef]
- Rieg, T.; Bundey, R.A.; Chen, Y.; Deschenes, G.; Junger, W.; Insel, P.A.; Vallon, V. Mice lacking p2y2 receptors have salt-resistant hypertension and facilitated renal na+ and water reabsorption. FASEB J. 2007, 21, 3717–3726. [Google Scholar] [CrossRef]
- McCulloch, F.; Chambrey, R.; Eladari, D.; Peti-Peterdi, J. Localization of connexin 30 in the luminal membrane of cells in the distal nephron. Am. J. Physiol. Renal Physiol. 2005, 289, F1304–F1312. [Google Scholar] [CrossRef]
- Lu, M.; MacGregor, G.G.; Wang, W.; Giebisch, G. Extracellular atp inhibits the small-conductance k channel on the apical membrane of the cortical collecting duct from mouse kidney. J. Gen. Physiol. 2000, 116, 299–310. [Google Scholar] [CrossRef]
- Holtzclaw, J.D.; Cornelius, R.J.; Hatcher, L.I.; Sansom, S.C. Coupled atp and potassium efflux from intercalated cells. Am. J. Physiol. Renal Physiol. 2011, 300, F1319–F1326. [Google Scholar] [CrossRef]
- Peti-Peterdi, J. High glucose and renin release: The role of succinate and gpr91. Kidney Int. 2010, 78, 1214–1217. [Google Scholar] [CrossRef]
- Tokonami, N.; Morla, L.; Centeno, G.; Mordasini, D.; Ramakrishnan, S.K.; Nikolaeva, S.; Wagner, C.A.; Bonny, O.; Houillier, P.; Doucet, A.; et al. Alpha-ketoglutarate regulates acid-base balance through an intrarenal paracrine mechanism. J. Clin. Invest. 2013, 123, 3166–3171. [Google Scholar] [CrossRef] [Green Version]
- Bhoola, K.D.; Figueroa, C.D.; Worthy, K. Bioregulation of kinins: Kallikreins, kininogens, and kininases. Pharmacol. Rev. 1992, 44, 1–80. [Google Scholar]
- Figueroa, C.D.; Gonzalez, C.B.; Grigoriev, S.; Abd Alla, S.A.; Haasemann, M.; Jarnagin, K.; Muller-Esterl, W. Probing for the bradykinin b2 receptor in rat kidney by anti-peptide and anti-ligand antibodies. J. Histochem. Cytochem. 1995, 43, 137–148. [Google Scholar] [CrossRef]
- Boedtkjer, E.; Aalkjaer, C. Disturbed acid-base transport: An emerging cause of hypertension. Front. Physiol. 2013, 4, 388. [Google Scholar]
- Alper, S.L. Familial renal tubular acidosis. J. Nephrol. 2010, 23 (Suppl. S16), S57–S76. [Google Scholar]
- Stehberger, P.A.; Shmukler, B.E.; Stuart-Tilley, A.K.; Peters, L.L.; Alper, S.L.; Wagner, C.A. Distal renal tubular acidosis in mice lacking the ae1 (band3) cl-/hco3- exchanger (slc4a1). J. Am. Soc. Nephrol. 2007, 18, 1408–1418. [Google Scholar] [CrossRef]
- Fry, A.C.; Karet, F.E. Inherited renal acidoses. Physiology (Bethesda) 2007, 22, 202–211. [Google Scholar] [CrossRef]
- Dou, H.; Finberg, K.; Cardell, E.L.; Lifton, R.; Choo, D. Mice lacking the b1 subunit of h+ -atpase have normal hearing. Hear. Res. 2003, 180, 76–84. [Google Scholar] [CrossRef]
- Finberg, K.E.; Wagner, C.A.; Bailey, M.A.; Paunescu, T.G.; Breton, S.; Brown, D.; Giebisch, G.; Geibel, J.P.; Lifton, R.P. The b1-subunit of the h(+) atpase is required for maximal urinary acidification. Proc. Natl. Acad. Sci. USA 2005, 102, 13616–13621. [Google Scholar] [CrossRef]
- Karet, F.E. Inherited distal renal tubular acidosis. J. Am. Soc. Nephrol. 2002, 13, 2178–2184. [Google Scholar] [CrossRef]
- Hennings, J.C.; Picard, N.; Huebner, A.K.; Stauber, T.; Maier, H.; Brown, D.; Jentsch, T.J.; Vargas-Poussou, R.; Eladari, D.; Hubner, C.A. A mouse model for distal renal tubular acidosis reveals a previously unrecognized role of the v-atpase a4 subunit in the proximal tubule. EMBO Mol. Med. 2012, 4, 1057–1071. [Google Scholar] [CrossRef]
- Norgett, E.E.; Golder, Z.J.; Lorente-Canovas, B.; Ingham, N.; Steel, K.P.; Karet Frankl, F.E. Atp6v0a4 knockout mouse is a model of distal renal tubular acidosis with hearing loss, with additional extrarenal phenotype. Proc. Natl. Acad. Sci. USA 2012, 109, 13775–13780. [Google Scholar]
- Vainsel, M.; Fondu, P.; Cadranel, S.; Rocmans, C.; Gepts, W. Osteopetrosis associated with proximal and distal tubular acidosis. Acta Paediatr. Scand. 1972, 61, 429–434. [Google Scholar] [CrossRef]
- Whyte, M.P.; Murphy, W.A.; Fallon, M.D.; Sly, W.S.; Teitelbaum, S.L.; McAlister, W.H.; Avioli, L.V. Osteopetrosis, renal tubular acidosis and basal ganglia calcification in three sisters. Am. J. Med. 1980, 69, 64–74. [Google Scholar] [CrossRef]
- Zackai, E.H.; Sly, W.S.; McAlister, W.G. Microcephaly, mild mental retardation, short stature, and skeletal anomalies in siblings. Am. J. Dis. Child. 1972, 124, 111–115. [Google Scholar]
- Lewis, S.E.; Erickson, R.P.; Barnett, L.B.; Venta, P.J.; Tashian, R.E. N-ethyl-n-nitrosourea-induced null mutation at the mouse car-2 locus: An animal model for human carbonic anhydrase ii deficiency syndrome. Proc. Natl. Acad. Sci. USA 1988, 85, 1962–1966. [Google Scholar] [CrossRef]
- Everett, L.A.; Glaser, B.; Beck, J.C.; Idol, J.R.; Buchs, A.; Heyman, M.; Adawi, F.; Hazani, E.; Nassir, E.; Baxevanis, A.D.; et al. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (pds). Nat. Genet. 1997, 17, 411–422. [Google Scholar] [CrossRef]
- Nakaya, K.; Harbidge, D.G.; Wangemann, P.; Schultz, B.D.; Green, E.D.; Wall, S.M.; Marcus, D.C. Lack of pendrin hco3- transport elevates vestibular endolymphatic [ca2+] by inhibition of acid-sensitive trpv5 and trpv6 channels. Am. J. Physiol. Renal Physiol. 2007, 292, F1314–F1321. [Google Scholar] [CrossRef]
- Escobar, L.; Mejia, N.; Gil, H.; Santos, F. Distal renal tubular acidosis: A hereditary disease with an inadequate urinary h(+) excretion. Nefrologia 2013, 33, 289–296. [Google Scholar]
- Alexander, R.T.; Dimke, H.; Cordat, E. Proximal tubular nhes: Sodium, protons and calcium? Am. J. Physiol. Renal Physiol. 2013, 305, F229–F236. [Google Scholar] [CrossRef]
- Borthwick, K.J.; Kandemir, N.; Topaloglu, R.; Kornak, U.; Bakkaloglu, A.; Yordam, N.; Ozen, S.; Mocan, H.; Shah, G.N.; Sly, W.S.; et al. A phenocopy of caii deficiency: A novel genetic explanation for inherited infantile osteopetrosis with distal renal tubular acidosis. J. Med. Genet. 2003, 40, 115–121. [Google Scholar] [CrossRef]
- Cordat, E.; Kittanakom, S.; Yenchitsomanus, P.T.; Li, J.; Du, K.; Lukacs, G.L.; Reithmeier, R.A. Dominant and recessive distal renal tubular acidosis mutations of kidney anion exchanger 1 induce distinct trafficking defects in mdck cells. Traffic 2006, 7, 117–128. [Google Scholar] [CrossRef]
- Devonald, M.A.; Smith, A.N.; Poon, J.P.; Ihrke, G.; Karet, F.E. Non-polarized targeting of ae1 causes autosomal dominant distal renal tubular acidosis. Nat. Genet. 2003, 33, 125–127. [Google Scholar] [CrossRef]
- Toye, A.M.; Banting, G.; Tanner, M.J. Regions of human kidney anion exchanger 1 (kae1) required for basolateral targeting of kae1 in polarised kidney cells: Mis-targeting explains dominant renal tubular acidosis (drta). J. Cell Sci. 2004, 117, 1399–1410. [Google Scholar] [CrossRef]
- Quilty, J.A.; Li, J.; Reithmeier, R.A. Impaired trafficking of distal renal tubular acidosis mutants of the human kidney anion exchanger kae1. Am. J. Physiol. Renal Physiol. 2002, 282, F810–F820. [Google Scholar]
- Cordat, E.; Reithmeier, R.A. Expression and interaction of two compound heterozygous distal renal tubular acidosis mutants of kidney anion exchanger 1 in epithelial cells. Am. J. Physiol. Renal Physiol. 2006, 291, F1354–F1361. [Google Scholar] [CrossRef]
- Quilty, J.A.; Cordat, E.; Reithmeier, R.A. Impaired trafficking of human kidney anion exchanger (kae1) caused by hetero-oligomer formation with a truncated mutant associated with distal renal tubular acidosis. Biochem. J. 2002, 368, 895–903. [Google Scholar] [CrossRef]
- Caruana, R.J.; Barish, C.F.; Buckalew, V.M., Jr. Complete distal renal tubular acidosis in systemic lupus: Clinical and laboratory findings. Am. J. Kidney Dis. 1985, 6, 59–63. [Google Scholar]
- Rodriguez Soriano, J. Renal tubular acidosis: The clinical entity. J. Am. Soc. Nephrol. 2002, 13, 2160–2170. [Google Scholar] [CrossRef]
- Karet, F.E.; Finberg, K.E.; Nayir, A.; Bakkaloglu, A.; Ozen, S.; Hulton, S.A.; Sanjad, S.A.; Al-Sabban, E.A.; Medina, J.F.; Lifton, R.P. Localization of a gene for autosomal recessive distal renal tubular acidosis with normal hearing (rdrta2) to 7q33–34. Am. J. Hum. Genet. 1999, 65, 1656–1665. [Google Scholar] [CrossRef]
- Stover, E.H.; Borthwick, K.J.; Bavalia, C.; Eady, N.; Fritz, D.M.; Rungroj, N.; Giersch, A.B.; Morton, C.C.; Axon, P.R.; Akil, I.; et al. Novel atp6v1b1 and atp6v0a4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J. Med. Genet. 2002, 39, 796–803. [Google Scholar] [CrossRef]
- Paunescu, T.G.; Russo, L.M.; Da Silva, N.; Kovacikova, J.; Mohebbi, N.; Van Hoek, A.N.; McKee, M.; Wagner, C.A.; Breton, S.; Brown, D. Compensatory membrane expression of the v-atpase b2 subunit isoform in renal medullary intercalated cells of b1-deficient mice. Am. J. Physiol. Renal Physiol. 2007, 293, F1915–F1926. [Google Scholar] [CrossRef] [Green Version]
- Lorente-Canovas, B.; Ingham, N.; Norgett, E.E.; Golder, Z.J.; Karet Frankl, F.E.; Steel, K.P. Mice deficient in h+-atpase a4 subunit have severe hearing impairment associated with enlarged endolymphatic compartments within the inner ear. Dis. Model. Mech. 2013, 6, 434–442. [Google Scholar] [CrossRef]
- Paunescu, T.G.; Rodriguez, S.; Benz, E.; McKee, M.; Tyszkowski, R.; Albers, M.W.; Brown, D. Loss of the v-atpase b1 subunit isoform expressed in non-neuronal cells of the mouse olfactory epithelium impairs olfactory function. PLoS One 2012, 7, e45395. [Google Scholar]
- Peters, L.L.; Shivdasani, R.A.; Liu, S.C.; Hanspal, M.; John, K.M.; Gonzalez, J.M.; Brugnara, C.; Gwynn, B.; Mohandas, N.; Alper, S.L.; et al. Anion exchanger 1 (band 3) is required to prevent erythrocyte membrane surface loss but not to form the membrane skeleton. Cell 1996, 86, 917–927. [Google Scholar] [CrossRef]
- Southgate, C.D.; Chishti, A.H.; Mitchell, B.; Yi, S.J.; Palek, J. Targeted disruption of the murine erythroid band 3 gene results in spherocytosis and severe haemolytic anaemia despite a normal membrane skeleton. Nat. Genet. 1996, 14, 227–230. [Google Scholar] [CrossRef]
- Sun, X.; Soleimani, M.; Petrovic, S. Decreased expression of slc26a4 (pendrin) and slc26a7 in the kidneys of carbonic anhydrase ii-deficient mice. Cell. Physiol. Biochem. 2008, 21, 95–108. [Google Scholar] [CrossRef]
- Pela, I.; Bigozzi, M.; Bianchi, B. Profound hypokalemia and hypochloremic metabolic alkalosis during thiazide therapy in a child with pendred syndrome. Clin. Nephrol. 2008, 69, 450–453. [Google Scholar] [CrossRef]
- Kandasamy, N.; Fugazzola, L.; Evans, M.; Chatterjee, K.; Karet, F. Life-threatening metabolic alkalosis in pendred syndrome. Eur. J. Endocrinol. 2011, 165, 167–170. [Google Scholar] [CrossRef]
- Kim, Y.H.; Pech, V.; Spencer, K.B.; Beierwaltes, W.H.; Everett, L.A.; Green, E.D.; Shin, W.; Verlander, J.W.; Sutliff, R.L.; Wall, S.M. Reduced enac protein abundance contributes to the lower blood pressure observed in pendrin-null mice. Am. J. Physiol. Renal Physiol. 2007, 293, F1314–F1324. [Google Scholar] [CrossRef]
- Xu, J.; Barone, S.; Brooks, M.B.; Soleimani, M. Double knockout of carbonic anhydrase ii (caii) and na-cl(-) cotransporter (ncc) causes salt wasting and volume depletion. Cell. Physiol. Biochem. 2013, 32, 173–183. [Google Scholar] [CrossRef]
- Soleimani, M.; Barone, S.; Xu, J.; Shull, G.E.; Siddiqui, F.; Zahedi, K.; Amlal, H. Double knockout of pendrin and na-cl cotransporter (ncc) causes severe salt wasting, volume depletion, and renal failure. Proc. Natl. Acad. Sci. USA 2012, 109, 13368–13373. [Google Scholar]
- Kokubo, Y.; Tomoike, H.; Tanaka, C.; Banno, M.; Okuda, T.; Inamoto, N.; Kamide, K.; Kawano, Y.; Miyata, T. Association of sixty-one non-synonymous polymorphisms in forty-one hypertension candidate genes with blood pressure variation and hypertension. Hypertens. Res. 2006, 29, 611–619. [Google Scholar] [CrossRef]
- Le Moellic, C.; Boulkroun, S.; Gonzalez-Nunez, D.; Dublineau, I.; Cluzeaud, F.; Fay, M.; Blot-Chabaud, M.; Farman, N. Aldosterone and tight junctions: Modulation of claudin-4 phosphorylation in renal collecting duct cells. Am. J. Physiol. Cell Physiol. 2005, 289, C1513–C1521. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Almomani, E.Y.; Kaur, S.; Alexander, R.T.; Cordat, E. Intercalated Cells: More than pH Regulation. Diseases 2014, 2, 71-92. https://doi.org/10.3390/diseases2020071
Almomani EY, Kaur S, Alexander RT, Cordat E. Intercalated Cells: More than pH Regulation. Diseases. 2014; 2(2):71-92. https://doi.org/10.3390/diseases2020071
Chicago/Turabian StyleAlmomani, Ensaf Y., Sumanpreet Kaur, R. Todd Alexander, and Emmanuelle Cordat. 2014. "Intercalated Cells: More than pH Regulation" Diseases 2, no. 2: 71-92. https://doi.org/10.3390/diseases2020071
APA StyleAlmomani, E. Y., Kaur, S., Alexander, R. T., & Cordat, E. (2014). Intercalated Cells: More than pH Regulation. Diseases, 2(2), 71-92. https://doi.org/10.3390/diseases2020071