
Early- and Late-Onset Alzheimer’s Disease: Two Sides of the Same Coin?
 , , , and
, , , and
Abstract
1. Introduction
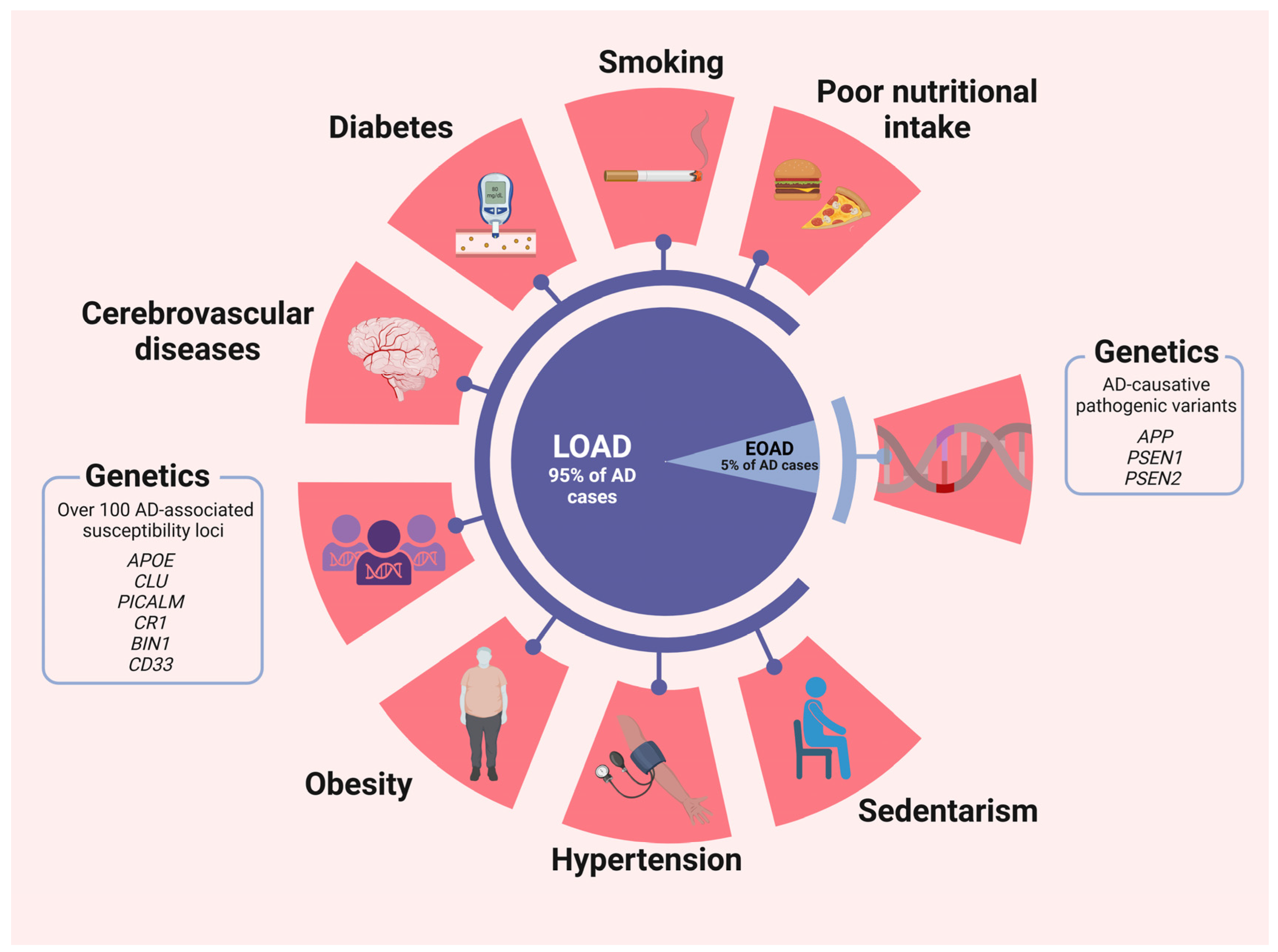
2. Risk Factors: Early-Onset vs. Late-Onset Alzheimer’s Disease
2.1. Genetics Factors
2.2. Non-Genetic Factors
2.3. Other Factors
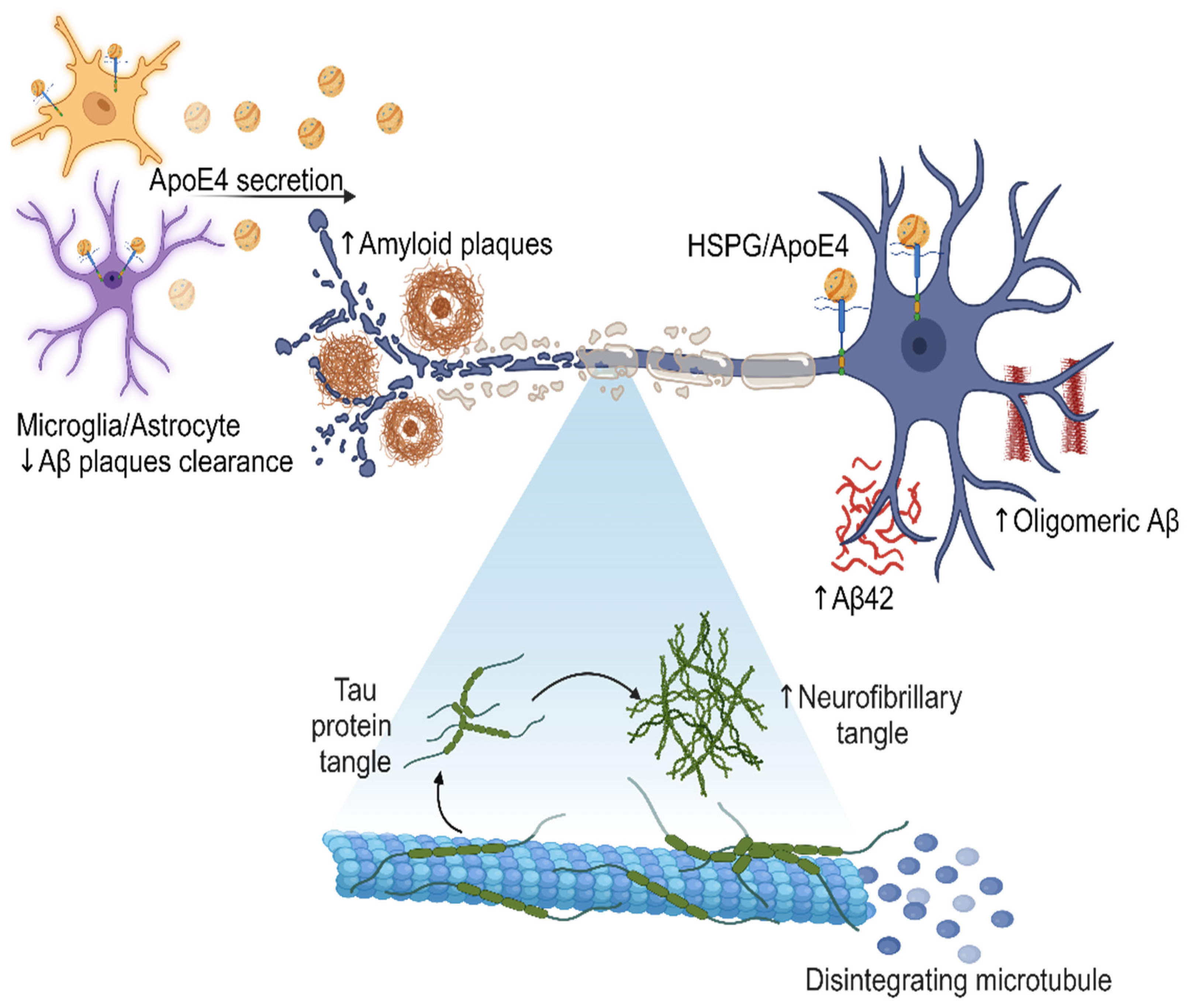
3. Pathological Mechanisms
3.1. Amyloid Plaques
- Non-amyloidogenic pathway: In this pathway, APP is cleaved by α-secretases, such as ADAM10, resulting in the generation of a soluble fraction of alpha-APP (sAPPα) and a carboxy-terminal fragment (CTF-α). Subsequently, γ-secretase also acts on CTF-α, producing the P3 fragment. Importantly, P3 is a soluble peptide that lacks the propensity to aggregate, unlike Aβ [54].
- Amyloidogenic pathway: APP can also be cleaved at the β-site by a β-secretase (BACE1), resulting in the production of a soluble beta-amyloid precursor protein (sAPPβ) and a carboxy-terminal fragment composed of 99 amino acids (known as CTF-β or C99). Subsequently, γ-secretase acts on CTF-β, releasing the Aβ, which can vary in size (primarily Aβ40, Aβ42, and Aβ43), depending on enzymatic cleavage. The pathway described above leads to the accumulation of Aβ protein and, consequently, the formation of amyloid plaques. Although the 42-amino acid form (Aβ42) is suspected to be a causative agent in AD, the molecular basis of its neurotoxicity remains unknown. However, amyloid plaques can induce synaptic dysfunction and an inflammatory response that ultimately triggers neurodegeneration. In a healthy individual, the non-amyloidogenic pathway predominates, while in a patient with AD, Aβ clearance is lower than its production through the amyloidogenic pathway [55,56].
3.2. Neurofibrillary Tangles
3.3. Differences of Pathological Mechanisms between EOAD and LOAD
4. Diagnosis and Assessment
4.1. Neuroimaging
4.1.1. Positron Emission Tomography (PET)
4.1.2. Magnetic Resonance Imaging (MRI)
4.2. Cognitive Assessment
4.3. Biomarkers
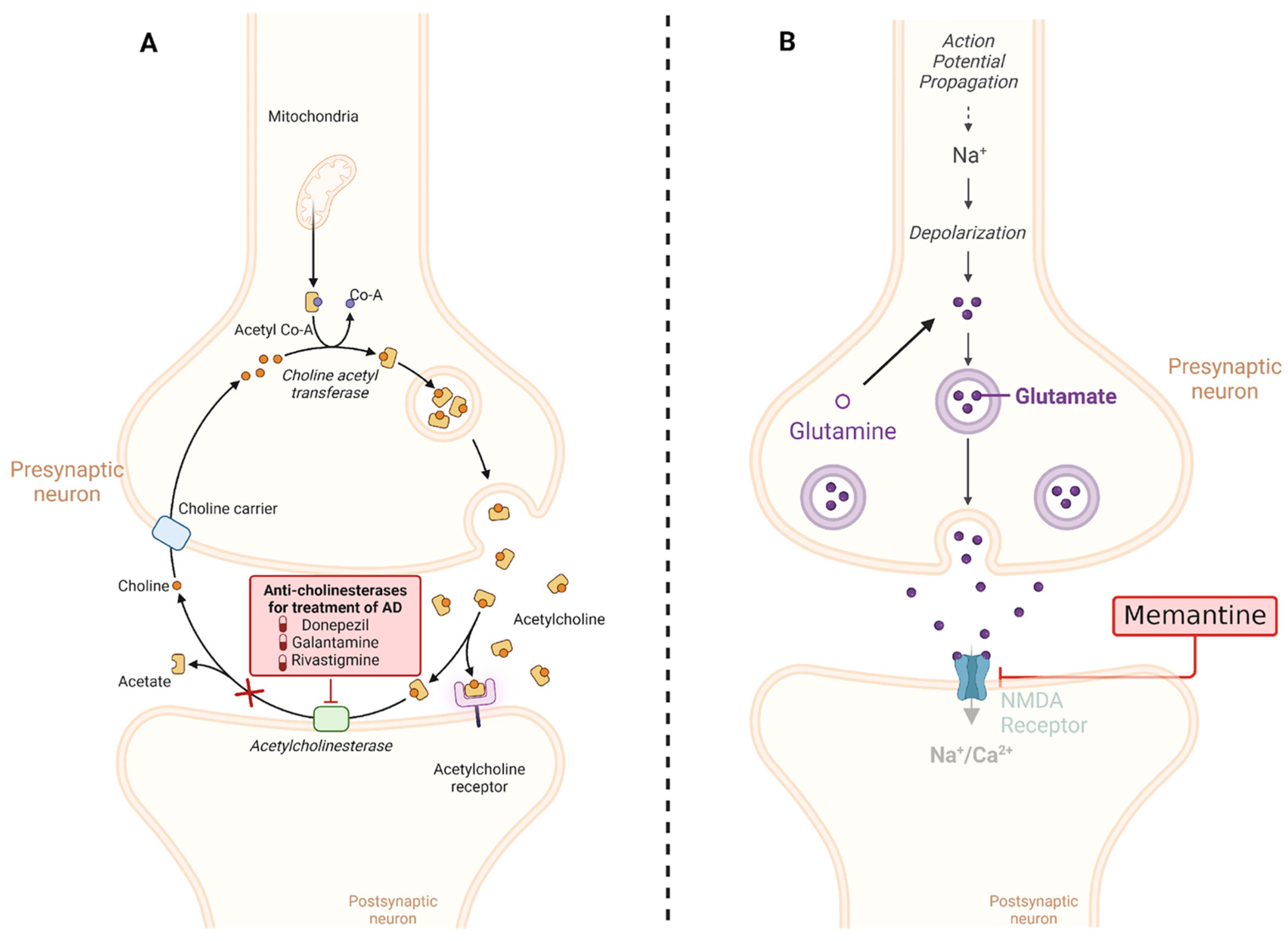
5. Treatments and Therapeutic Approaches
5.1. Conventional Treatments
5.1.1. Cholinesterase Inhibitors
5.1.2. NMDA Receptor Inhibitor
5.2. Emerging Treatments
5.2.1. Monoclonal Antibodies
5.2.2. Hybrid Molecules with Dual Affinity
5.2.3. Gamma-Secretase Modulators
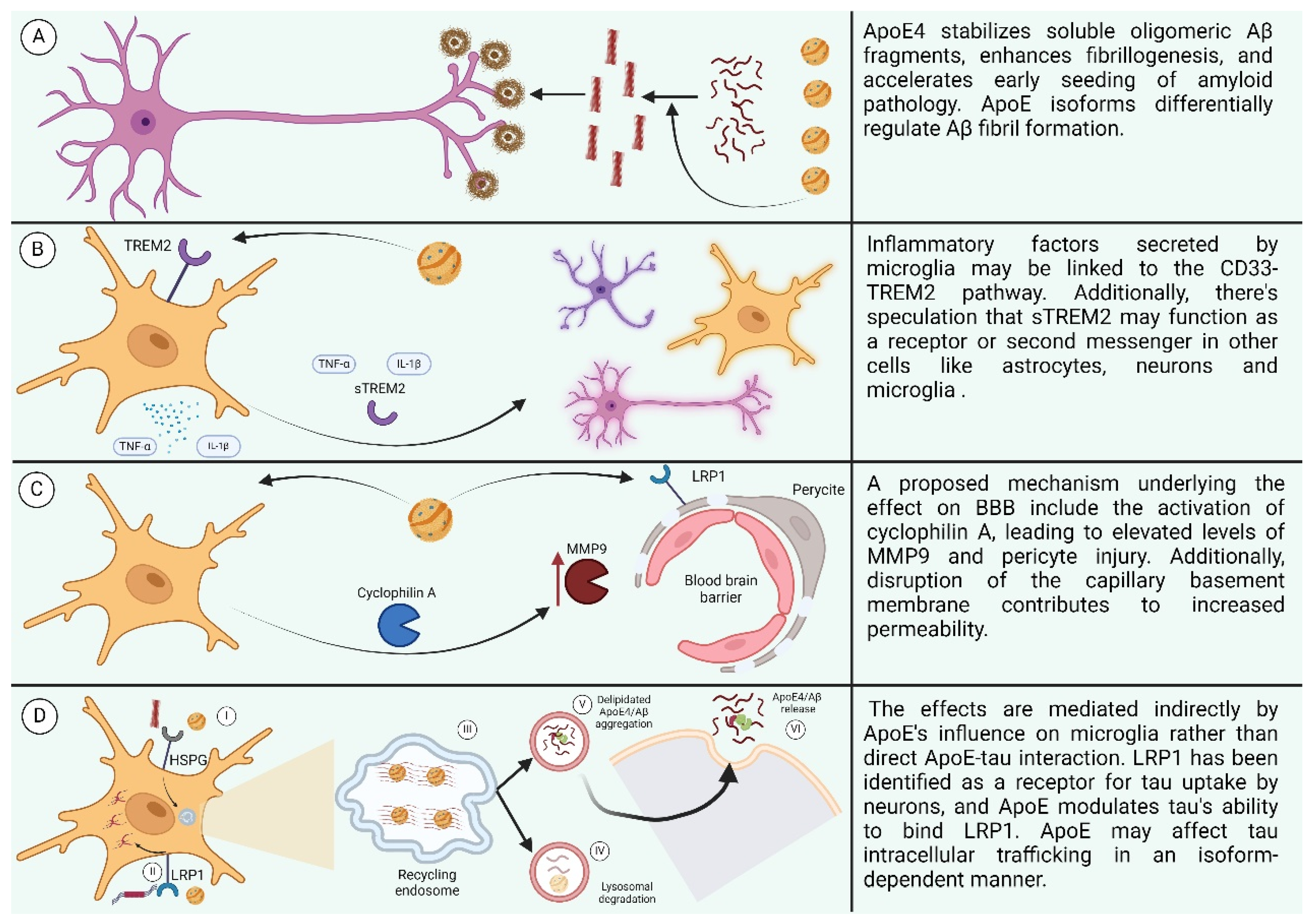
6. The Effect of ApoE
6.1. Effects of ApoE on LOAD
6.1.1. Effect of ApoE on Cognition
6.1.2. Effect of ApoE on Aβ
6.1.3. Effect of ApoE on Neuroinflammation
6.1.4. Effect of APOE on Blood–Brain Barrier
6.1.5. Effect of ApoE on Tau
6.2. Effect of ApoE on EOAD
7. Future Perspectives
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The Global Prevalence of Dementia: A Systematic Review and Metaanalysis. Alzheimers Dement. J. Alzheimers Assoc. 2013, 9, 63–75.e2. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.A.; Arvanitakis, Z.; Bang, W.; Bennett, D.A. Mixed Brain Pathologies Account for Most Dementia Cases in Community-Dwelling Older Persons. Neurology 2007, 69, 2197–2204. [Google Scholar] [CrossRef] [PubMed]
- Gale, S.A.; Acar, D.; Daffner, K.R. Dementia. Am. J. Med. 2018, 131, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Stelzmann, R.A.; Norman Schnitzlein, H.; Reed Murtagh, F. An English Translation of Alzheimer’s 1907 Paper, “Über Eine Eigenartige Erkankung Der Hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Hasana, S.; Hossain, M.F.; Islam, M.S.; Behl, T.; Perveen, A.; Hafeez, A.; Ashraf, G.M. Molecular Genetics of Early- and Late-Onset Alzheimer’s Disease. Curr. Gene Ther. 2021, 21, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, S.; Rosa-Neto, P.; Morais, J.; Webster, C. World Alzheimer Report 2021: Journey through the Diagnosis of Dementia. 2021. Available online: https://cdn.alzheimers.org.nz/wp-content/uploads/2021/09/World-Alzheimer-Report-2021-Web.pdf (accessed on 3 April 2024).
- Langa, K.M. Is the Risk of Alzheimer’s Disease and Dementia Declining? Alzheimers Res. Ther. 2015, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Skaria, A.P. The Economic and Societal Burden of Alzheimer Disease: Managed Care Considerations. Am. J. Manag. Care 2022, 28 (Suppl. S10), S188–S196. [Google Scholar] [CrossRef]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The Genetic Landscape of Alzheimer Disease: Clinical Implications and Perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef]
- Larner, A.J. Presenilin-1 Mutations in Alzheimer’s Disease: An Update on Genotype-Phenotype Relationships. J. Alzheimers Dis. 2013, 37, 653–659. [Google Scholar] [CrossRef]
- Mendez, M.F. Early-Onset Alzheimer’s Disease: Nonamnestic Subtypes and Type 2 AD. Arch. Med. Res. 2012, 43, 677–685. [Google Scholar] [CrossRef]
- Ayodele, T.; Rogaeva, E.; Kurup, J.T.; Beecham, G.; Reitz, C. Early-Onset Alzheimer’s Disease: What Is Missing in Research? Curr. Neurol. Neurosci. Rep. 2021, 21, 4. [Google Scholar] [CrossRef] [PubMed]
- Cacace, R.; Sleegers, K.; Van Broeckhoven, C. Molecular Genetics of Early-onset Alzheimer’s Disease Revisited. Alzheimers Dement. 2016, 12, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Lanoiselée, H.-M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.-C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 Mutations in Early-Onset Alzheimer Disease: A Genetic Screening Study of Familial and Sporadic Cases. PLOS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.S.; Hou, C.E. Early-Onset Alzheimer Disease: When Is Genetic Testing Appropriate? Alzheimer Dis. Assoc. Disord. 2004, 18, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Nudelman, K.N.H.; Jackson, T.; Rumbaugh, M.; Eloyan, A.; Abreu, M.; Dage, J.L.; Snoddy, C.; Faber, K.M.; Foroud, T.; Hammers, D.B.; et al. Pathogenic Variants in the Longitudinal Early-onset Alzheimer’s Disease Study Cohort. Alzheimers Dement. 2023, 19 (Suppl. S9), S64–S73. [Google Scholar] [CrossRef] [PubMed]
- Bellenguez, C.; Charbonnier, C.; Grenier-Boley, B.; Quenez, O.; Le Guennec, K.; Nicolas, G.; Chauhan, G.; Wallon, D.; Rousseau, S.; Richard, A.C.; et al. Contribution to Alzheimer’s Disease Risk of Rare Variants in TREM2, SORL1, and ABCA7 in 1779 Cases and 1273 Controls. Neurobiol. Aging 2017, 59, 220.e1–220.e9. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, S.; Patel, T.; Barber, I.S.; Guetta-Baranes, T.; Brookes, K.J.; Chappell, S.; Turton, J.; Guerreiro, R.; Bras, J.; Hernandez, D.; et al. Polygenic Risk Score in Postmortem Diagnosed Sporadic Early-Onset Alzheimer’s Disease. Neurobiol. Aging 2018, 62, 244.e1–244.e8. [Google Scholar] [CrossRef] [PubMed]
- Raman, S.; Brookhouser, N.; Brafman, D.A. Using Human Induced Pluripotent Stem Cells (hiPSCs) to Investigate the Mechanisms by Which Apolipoprotein E (APOE) Contributes to Alzheimer’s Disease (AD) Risk. Neurobiol. Dis. 2020, 138, 104788. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.J.; Fulton-Howard, B.; Goate, A. Interpretation of Risk Loci from Genome-Wide Association Studies of Alzheimer’s Disease. Lancet Neurol. 2020, 19, 326–335. [Google Scholar] [CrossRef]
- Van Acker, Z.P.; Bretou, M.; Annaert, W. Endo-Lysosomal Dysregulations and Late-Onset Alzheimer’s Disease: Impact of Genetic Risk Factors. Mol. Neurodegener. 2019, 14, 20. [Google Scholar] [CrossRef]
- Park, D.C.; Reuter-Lorenz, P. The Adaptive Brain: Aging and Neurocognitive Scaffolding. Annu. Rev. Psychol. 2009, 60, 173–196. [Google Scholar] [CrossRef]
- Rao, S.M.; Bonner-Jackson, A.; Nielson, K.A.; Seidenberg, M.; Smith, J.C.; Woodard, J.L.; Durgerian, S. Genetic Risk for Alzheimer’s Disease Alters the Five-Year Trajectory of Semantic Memory Activation in Cognitively Intact Elders. NeuroImage 2015, 111, 136–146. [Google Scholar] [CrossRef]
- Guo, Z.; Cupples, L.A.; Kurz, A.; Auerbach, S.H.; Volicer, L.; Chui, H.; Green, R.C.; Sadovnick, A.D.; Duara, R.; DeCarli, C.; et al. Head Injury and the Risk of AD in the MIRAGE Study. Neurology 2000, 54, 1316–1323. [Google Scholar] [CrossRef]
- Yaffe, K.; Vittinghoff, E.; Hoang, T.; Matthews, K.; Golden, S.H.; Zeki Al Hazzouri, A. Cardiovascular Risk Factors Across the Life Course and Cognitive Decline: A Pooled Cohort Study. Neurology 2021, 96, e2212–e2219. [Google Scholar] [CrossRef]
- Diniz Pereira, J.; Gomes Fraga, V.; Morais Santos, A.L.; Carvalho, M.D.G.; Caramelli, P.; Braga Gomes, K. Alzheimer’s Disease and Type 2 Diabetes Mellitus: A Systematic Review of Proteomic Studies. J. Neurochem. 2021, 156, 753–776. [Google Scholar] [CrossRef]
- Durazzo, T.C.; Mattsson, N.; Weiner, M.W. Alzheimer’s Disease Neuroimaging Initiative. Smoking and Increased Alzheimer’s Disease Risk: A Review of Potential Mechanisms. Alzheimers Dement. 2014, 10 (Suppl. S3), S122–S145. [Google Scholar] [CrossRef]
- Xu, C.; Apostolova, L.G.; Oblak, A.L.; Gao, S. Association of Hypercholesterolemia with Alzheimer’s Disease Pathology and Cerebral Amyloid Angiopathy. J. Alzheimers Dis. 2020, 73, 1305–1311. [Google Scholar] [CrossRef]
- Di Liegro, C.M.; Schiera, G.; Proia, P.; Di Liegro, I. Physical Activity and Brain Health. Genes 2019, 10, 720. [Google Scholar] [CrossRef]
- Olufunmilayo, E.O.; Holsinger, R.M.D. Roles of Non-Coding RNA in Alzheimer’s Disease Pathophysiology. Int. J. Mol. Sci. 2023, 24, 12498. [Google Scholar] [CrossRef]
- Kumar, S.; Reddy, A.P.; Yin, X.; Reddy, P.H. Novel MicroRNA-455-3p and Its Protective Effects against Abnormal APP Processing and Amyloid Beta Toxicity in Alzheimer’s Disease. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2019, 1865, 2428–2440. [Google Scholar] [CrossRef]
- Ke, S.; Yang, Z.; Yang, F.; Wang, X.; Tan, J.; Liao, B. Long Noncoding RNA NEAT1 Aggravates Aβ-Induced Neuronal Damage by Targeting miR-107 in Alzheimer’s Disease. Yonsei Med. J. 2019, 60, 640. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Luan, W.; Shen, X.; Wang, Z.; Cao, Y. LncRNA BDNF-AS as ceRNA Regulates the miR-9-5p/BACE1 Pathway Affecting Neurotoxicity in Alzheimer’s Disease. Arch. Gerontol. Geriatr. 2022, 99, 104614. [Google Scholar] [CrossRef]
- Smith, R.G.; Hannon, E.; De Jager, P.L.; Chibnik, L.; Lott, S.J.; Condliffe, D.; Smith, A.R.; Haroutunian, V.; Troakes, C.; Al-Sarraj, S.; et al. Elevated DNA Methylation across a 48-kb Region Spanning the HOXA Gene Cluster Is Associated with Alzheimer’s Disease Neuropathology. Alzheimers Dement. 2018, 14, 1580–1588. [Google Scholar] [CrossRef] [PubMed]
- De Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimer’s Disease: Early Alterations in Brain DNA Methylation at ANK1, BIN1, RHBDF2 and Other Loci. Nat. Neurosci. 2014, 17, 1156–1163. [Google Scholar] [CrossRef]
- Marzi, S.J.; Leung, S.K.; Ribarska, T.; Hannon, E.; Smith, A.R.; Pishva, E.; Poschmann, J.; Moore, K.; Troakes, C.; Al-Sarraj, S.; et al. A Histone Acetylome-Wide Association Study of Alzheimer’s Disease Identifies Disease-Associated H3K27ac Differences in the Entorhinal Cortex. Nat. Neurosci. 2018, 21, 1618–1627. [Google Scholar] [CrossRef]
- Chen, L.; Guo, X.; Li, Z.; He, Y. Relationship between Long Non-Coding RNAs and Alzheimer’s Disease: A Systematic Review. Pathol.-Res. Pract. 2019, 215, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Takousis, P.; Sadlon, A.; Schulz, J.; Wohlers, I.; Dobricic, V.; Middleton, L.; Lill, C.M.; Perneczky, R.; Bertram, L. Differential Expression of microRNAs in Alzheimer’s Disease Brain, Blood, and Cerebrospinal Fluid. Alzheimers Dement. 2019, 15, 1468–1477. [Google Scholar] [CrossRef]
- Ivannikov, M.V.; Sugimori, M.; Llinás, R.R. Synaptic Vesicle Exocytosis in Hippocampal Synaptosomes Correlates Directly with Total Mitochondrial Volume. J. Mol. Neurosci. 2013, 49, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; McInnes, J.; Wierda, K.; Holt, M.; Herrmann, A.G.; Jackson, R.J.; Wang, Y.-C.; Swerts, J.; Beyens, J.; Miskiewicz, K.; et al. Tau Association with Synaptic Vesicles Causes Presynaptic Dysfunction. Nat. Commun. 2017, 8, 15295. [Google Scholar] [CrossRef]
- Peric, A.; Annaert, W. Early Etiology of Alzheimer’s Disease: Tipping the Balance toward Autophagy or Endosomal Dysfunction? Acta Neuropathol. 2015, 129, 363–381. [Google Scholar] [CrossRef]
- Vaz-Silva, J.; Gomes, P.; Jin, Q.; Zhu, M.; Zhuravleva, V.; Quintremil, S.; Meira, T.; Silva, J.; Dioli, C.; Soares-Cunha, C.; et al. Endolysosomal Degradation of Tau and Its Role in Glucocorticoid-driven Hippocampal Malfunction. EMBO J. 2018, 37, e99084. [Google Scholar] [CrossRef] [PubMed]
- Giovedì, S.; Ravanelli, M.M.; Parisi, B.; Bettegazzi, B.; Guarnieri, F.C. Dysfunctional Autophagy and Endolysosomal System in Neurodegenerative Diseases: Relevance and Therapeutic Options. Front. Cell. Neurosci. 2020, 14, 602116. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shi, C.; He, M.; Xiong, S.; Xia, X. Endoplasmic Reticulum Stress: Molecular Mechanism and Therapeutic Targets. Signal Transduct. Target. Ther. 2023, 8, 352. [Google Scholar] [CrossRef] [PubMed]
- Ajoolabady, A.; Lindholm, D.; Ren, J.; Pratico, D. ER Stress and UPR in Alzheimer’s Disease: Mechanisms, Pathogenesis, Treatments. Cell Death Dis. 2022, 13, 706. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Sanjo, N.; Uchihara, T.; Watabe, K.; George-Hyslop, P.S.; Fraser, P.E.; Mizusawa, H. Presenilin-1 Holoprotein Is an Interacting Partner of Sarco Endoplasmic Reticulum Calcium-ATPase and Confers Resistance to Endoplasmic Reticulum Stress. J. Alzheimers Dis. 2010, 20, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E.; Hyman, B.T.; Flory, J.; Damasio, A.R.; Van Hoesen, G.W. The Topographical and Neuroanatomical Distribution of Neurofibrillary Tangles and Neuritic Plaques in the Cerebral Cortex of Patients with Alzheimer’s Disease. Cereb. Cortex 1991, 1, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging–Alzheimer’s Association Guidelines for the Neuropathologic Assessment of Alzheimer’s Disease: A Practical Approach. Acta Neuropathol. 2012, 123, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Raskin, J.; Cummings, J.; Hardy, J.; Schuh, K.; Dean, R. Neurobiology of Alzheimer’s Disease: Integrated Molecular, Physiological, Anatomical, Biomarker, and Cognitive Dimensions. Curr. Alzheimer Res. 2015, 12, 712–722. [Google Scholar] [CrossRef]
- Snow, A.D.; Sekiguchi, R.T.; Nochlin, D.; Kalaria, R.N.; Kimata, K. Heparan Sulfate Proteoglycan in Diffuse Plaques of Hippocampus but Not of Cerebellum in Alzheimer’s Disease Brain. Am. J. Pathol. 1994, 144, 337–347. [Google Scholar]
- Fukuchi, K. Alzheimer s Disease and Heparan Sulfate Proteoglycan. Front. Biosci. 1998, 3, d327–d337. [Google Scholar] [CrossRef]
- Zhu, Y.; Gandy, L.; Zhang, F.; Liu, J.; Wang, C.; Blair, L.J.; Linhardt, R.J.; Wang, L. Heparan Sulfate Proteoglycans in Tauopathy. Biomolecules 2022, 12, 1792. [Google Scholar] [CrossRef]
- Coronel, R.; Bernabeu-Zornoza, A.; Palmer, C.; Muñiz-Moreno, M.; Zambrano, A.; Cano, E.; Liste, I. Role of Amyloid Precursor Protein (APP) and Its Derivatives in the Biology and Cell Fate Specification of Neural Stem Cells. Mol. Neurobiol. 2018, 55, 7107–7117. [Google Scholar] [CrossRef] [PubMed]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted Amyloid β–Protein Similar to That in the Senile Plaques of Alzheimer’s Disease Is Increased in Vivo by the Presenilin 1 and 2 and APP Mutations Linked to Familial Alzheimer’s Disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Gao, Y.; Winblad, B.; Tjernberg, L.O.; Schedin-Weiss, S. A Super-Resolved View of the Alzheimer’s Disease-Related Amyloidogenic Pathway in Hippocampal Neurons. J. Alzheimers Dis. 2021, 83, 833–852. [Google Scholar] [CrossRef]
- Hatters, D.M.; Peters-Libeu, C.A.; Weisgraber, K.H. Apolipoprotein E Structure: Insights into Function. Trends Biochem. Sci. 2006, 31, 445–454. [Google Scholar] [CrossRef]
- Gallardo, G.; Holtzman, D.M. Amyloid-β and Tau at the Crossroads of Alzheimer’s Disease. In Tau Biology; Takashima, A., Wolozin, B., Buee, L., Eds.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; Volume 1184, pp. 187–203. [Google Scholar] [CrossRef]
- Eftekharzadeh, B.; Daigle, J.G.; Kapinos, L.E.; Coyne, A.; Schiantarelli, J.; Carlomagno, Y.; Cook, C.; Miller, S.J.; Dujardin, S.; Amaral, A.S.; et al. Tau Protein Disrupts Nucleocytoplasmic Transport in Alzheimer’s Disease. Neuron 2018, 99, 925–940.e7. [Google Scholar] [CrossRef]
- Yamada, K.; Holth, J.K.; Liao, F.; Stewart, F.R.; Mahan, T.E.; Jiang, H.; Cirrito, J.R.; Patel, T.K.; Hochgräfe, K.; Mandelkow, E.-M.; et al. Neuronal Activity Regulates Extracellular Tau in Vivo. J. Exp. Med. 2014, 211, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Tan, L.; Yu, J.-T.; Tan, L. Tau in Alzheimer’s Disease: Mechanisms and Therapeutic Strategies. Curr. Alzheimer Res. 2018, 15, 283–300. [Google Scholar] [CrossRef]
- Wu, J.W.; Hussaini, S.A.; Bastille, I.M.; Rodriguez, G.A.; Mrejeru, A.; Rilett, K.; Sanders, D.W.; Cook, C.; Fu, H.; Boonen, R.A.; et al. Neuronal Activity Enhances Tau Propagation and Tau Pathology In Vivo. Nat. Neurosci. 2016, 19, 1085–1092. [Google Scholar] [CrossRef]
- Wei, Y.; Liu, M.; Wang, D. The Propagation Mechanisms of Extracellular Tau in Alzheimer’s Disease. J. Neurol. 2022, 269, 1164–1181. [Google Scholar] [CrossRef]
- Annadurai, N.; De Sanctis, J.B.; Hajdúch, M.; Das, V. Tau Secretion and Propagation: Perspectives for Potential Preventive Interventions in Alzheimer’s Disease and Other Tauopathies. Exp. Neurol. 2021, 343, 113756. [Google Scholar] [CrossRef]
- Vogels, T.; Leuzy, A.; Cicognola, C.; Ashton, N.J.; Smolek, T.; Novak, M.; Blennow, K.; Zetterberg, H.; Hromadka, T.; Zilka, N.; et al. Propagation of Tau Pathology: Integrating Insights From Postmortem and In Vivo Studies. Biol. Psychiatry 2020, 87, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.; Avila, J.; Hernández, F. Propagation of Tau via Extracellular Vesicles. Front. Neurosci. 2019, 13, 698. [Google Scholar] [CrossRef]
- Leake, I. Oligomeric Tau Might Spread Trans-Synaptically in Alzheimer Disease. Nat. Rev. Neurosci. 2023, 24, 393. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wu, H.; Tang, X. Tau Internalization: A Complex Step in Tau Propagation. Ageing Res. Rev. 2021, 67, 101272. [Google Scholar] [CrossRef]
- Fleeman, R.M.; Proctor, E.A. Astrocytic Propagation of Tau in the Context of Alzheimer’s Disease. Front. Cell. Neurosci. 2021, 15, 645233. [Google Scholar] [CrossRef]
- Sirkis, D.W.; Bonham, L.W.; Johnson, T.P.; La Joie, R.; Yokoyama, J.S. Dissecting the Clinical Heterogeneity of Early-Onset Alzheimer’s Disease. Mol. Psychiatry 2022, 27, 2674–2688. [Google Scholar] [CrossRef]
- Middleton, L.E.; Grinberg, L.T.; Miller, B.; Kawas, C.; Yaffe, K. Neuropathologic Features Associated with Alzheimer Disease Diagnosis: Age Matters. Neurology 2011, 77, 1737–1744. [Google Scholar] [CrossRef] [PubMed]
- Palasí, A.; Gutiérrez-Iglesias, B.; Alegret, M.; Pujadas, F.; Olabarrieta, M.; Liébana, D.; Quintana, M.; Álvarez-Sabín, J.; Boada, M. Differentiated Clinical Presentation of Early and Late-Onset Alzheimer’s Disease: Is 65 Years of Age Providing a Reliable Threshold? J. Neurol. 2015, 262, 1238–1246. [Google Scholar] [CrossRef]
- Squire, L.R.; Genzel, L.; Wixted, J.T.; Morris, R.G. Memory Consolidation. Cold Spring Harb. Perspect. Biol. 2015, 7, a021766. [Google Scholar] [CrossRef]
- Murray, M.E.; Graff-Radford, N.R.; Ross, O.A.; Petersen, R.C.; Duara, R.; Dickson, D.W. Neuropathologically Defined Subtypes of Alzheimer’s Disease with Distinct Clinical Characteristics: A Retrospective Study. Lancet Neurol. 2011, 10, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.; Lin, C.-L.; Chiang, M.-C. Exploring the Frontiers of Neuroimaging: A Review of Recent Advances in Understanding Brain Functioning and Disorders. Life 2023, 13, 1472. [Google Scholar] [CrossRef] [PubMed]
- Tanner, J.A.; Iaccarino, L.; Edwards, L.; Asken, B.M.; Gorno-Tempini, M.L.; Kramer, J.H.; Pham, J.; Perry, D.C.; Possin, K.; Malpetti, M.; et al. Amyloid, Tau and Metabolic PET Correlates of Cognition in Early and Late-Onset Alzheimer’s Disease. Brain 2022, 145, 4489–4505. [Google Scholar] [CrossRef]
- Atri, A. The Alzheimer’s Disease Clinical Spectrum. Med. Clin. North Am. 2019, 103, 263–293. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jeong, M.; Stiles, W.R.; Choi, H.S. Neuroimaging Modalities in Alzheimer’s Disease: Diagnosis and Clinical Features. Int. J. Mol. Sci. 2022, 23, 6079. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.M.; Murray, A.D. Alzheimer’s Dementia: The Emerging Role of Positron Emission Tomography. Neurosci. 2022, 28, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Rowley, P.A.; Samsonov, A.A.; Betthauser, T.J.; Pirasteh, A.; Johnson, S.C.; Eisenmenger, L.B. Amyloid and Tau PET Imaging of Alzheimer Disease and Other Neurodegenerative Conditions. Semin. Ultrasound CT MRI 2020, 41, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Brier, M.R.; Gordon, B.; Friedrichsen, K.; McCarthy, J.; Stern, A.; Christensen, J.; Owen, C.; Aldea, P.; Su, Y.; Hassenstab, J.; et al. Tau and Aβ Imaging, CSF Measures, and Cognition in Alzheimer’s Disease. Sci. Transl. Med. 2016, 8, 338ra66. [Google Scholar] [CrossRef]
- Mosconi, L.; McHugh, P.F. FDG- and Amyloid-PET in Alzheimer’s Disease: Is the Whole Greater than the Sum of the Parts? Q. J. Nucl. Med. Mol. Imaging 2011, 55, 250–264. [Google Scholar]
- Meysami, S.; Raji, C.A.; Merrill, D.A.; Porter, V.R.; Mendez, M.F. Quantitative MRI Differences Between Early versus Late Onset Alzheimer’s Disease. Am. J. Alzheimers Dis. Dement. 2021, 36, 153331752110553. [Google Scholar] [CrossRef] [PubMed]
- Filley, C.M.; Kelly, J.; Heaton, R.K. Neuropsychologic Features of Early- and Late-Onset Alzheimer’s Disease. Arch. Neurol. 1986, 43, 574–576. [Google Scholar] [CrossRef] [PubMed]
- for the Alzheimer’s Disease Neuroimaging Initiative; Chandra, A.; Dervenoulas, G.; Politis, M. Magnetic Resonance Imaging in Alzheimer’s Disease and Mild Cognitive Impairment. J. Neurol. 2019, 266, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.K. Clinical Diagnosis of Alzheimer’s Disease. In Biomarkers in Alzheimer’s Disease; Elsevier: Amsterdam, The Netherlands, 2016; pp. 27–48. [Google Scholar] [CrossRef]
- Folstein, M.F.; Folstein, S.E.; McHugh, P.R. Mini-Mental State. J. Psychiatr. Res. 1975, 12, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Molloy, D.W.; Alemayehu, E.; Roberts, R. Reliability of a Standardized Mini-Mental State Examination Compared with the Traditional Mini-Mental State Examination. Am. J. Psychiatry 1991, 148, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.C.; Ernesto, C.; Schafer, K.; Coats, M.; Leon, S.; Sano, M.; Thal, L.J.; Woodbury, P. Clinical Dementia Rating Training and Reliability in Multicenter Studies: The Alzheimer’s Disease Cooperative Study Experience. Neurology 1997, 48, 1508–1510. [Google Scholar] [CrossRef] [PubMed]
- Duara, R.; Loewenstein, D.A.; Greig-Custo, M.T.; Raj, A.; Barker, W.; Potter, E.; Schofield, E.; Small, B.; Schinka, J.; Wu, Y.; et al. Diagnosis and Staging of Mild Cognitive Impairment, Using a Modification of the Clinical Dementia Rating Scale: The mCDR. Int. J. Geriatr. Psychiatry 2010, 25, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Rockwood, K.; Strang, D.; MacKnight, C.; Downer, R.; Morris, J.C. Interrater Reliability of the Clinical Dementia Rating in a Multicenter Trial. J. Am. Geriatr. Soc. 2000, 48, 558–559. [Google Scholar] [CrossRef] [PubMed]
- Nystuen, K.L.; McNamee, S.M.; Akula, M.; Holton, K.M.; DeAngelis, M.M.; Haider, N.B. Alzheimer’s Disease: Models and Molecular Mechanisms Informing Disease and Treatments. Bioengineering 2024, 11, 45. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Zetterberg, H. The Past and the Future of Alzheimer’s Disease Fluid Biomarkers. J. Alzheimers Dis. 2018, 62, 1125–1140. [Google Scholar] [CrossRef]
- Shaw, L.M.; Vanderstichele, H.; Knapik-Czajka, M.; Clark, C.M.; Aisen, P.S.; Petersen, R.C.; Blennow, K.; Soares, H.; Simon, A.; Lewczuk, P.; et al. Cerebrospinal Fluid Biomarker Signature in Alzheimer’s Disease Neuroimaging Initiative Subjects. Ann. Neurol. 2009, 65, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Becker, B.; Nazir, F.H.; Brinkmalm, G.; Camporesi, E.; Kvartsberg, H.; Portelius, E.; Boström, M.; Kalm, M.; Höglund, K.; Olsson, M.; et al. Alzheimer-Associated Cerebrospinal Fluid Fragments of Neurogranin Are Generated by Calpain-1 and Prolyl Endopeptidase. Mol. Neurodegener. 2018, 13, 47. [Google Scholar] [CrossRef] [PubMed]
- Willemse, E.A.J.; De Vos, A.; Herries, E.M.; Andreasson, U.; Engelborghs, S.; Van Der Flier, W.M.; Scheltens, P.; Crimmins, D.; Ladenson, J.H.; Vanmechelen, E.; et al. Neurogranin as Cerebrospinal Fluid Biomarker for Alzheimer Disease: An Assay Comparison Study. Clin. Chem. 2018, 64, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Engelborghs, S.; Niemantsverdriet, E.; Struyfs, H.; Blennow, K.; Brouns, R.; Comabella, M.; Dujmovic, I.; Van Der Flier, W.; Frölich, L.; Galimberti, D.; et al. Consensus Guidelines for Lumbar Puncture in Patients with Neurological Diseases. Alzheimers Dement. Diagn. Assess. Dis. Monit. 2017, 8, 111–126. [Google Scholar] [CrossRef]
- Simrén, J.; Elmgren, A.; Blennow, K.; Zetterberg, H. Fluid Biomarkers in Alzheimer’s Disease. In Advances in Clinical Chemistry; Elsevier: Amsterdam, The Netherlands, 2023; Volume 112, pp. 249–281. [Google Scholar] [CrossRef]
- Moscoso, A.; Grothe, M.J.; Ashton, N.J.; Karikari, T.K.; Lantero Rodríguez, J.; Snellman, A.; Suárez-Calvet, M.; Blennow, K.; Zetterberg, H.; Schöll, M.; et al. Longitudinal Associations of Blood Phosphorylated Tau181 and Neurofilament Light Chain with Neurodegeneration in Alzheimer Disease. JAMA Neurol. 2021, 78, 396. [Google Scholar] [CrossRef] [PubMed]
- Tabet, N. Acetylcholinesterase Inhibitors for Alzheimer’s Disease: Anti-Inflammatories in Acetylcholine Clothing! Age Ageing 2006, 35, 336–338. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The Cholinergic System in the Pathophysiology and Treatment of Alzheimer’s Disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Bartus, R.T.; Dean, R.L.; Beer, B.; Lippa, A.S. The Cholinergic Hypothesis of Geriatric Memory Dysfunction. Science 1982, 217, 408–414. [Google Scholar] [CrossRef]
- Sharma, K. Cholinesterase Inhibitors as Alzheimer’s Therapeutics (Review). Mol. Med. Rep. 2019, 2, 1479–1487. [Google Scholar] [CrossRef]
- Birks, J.S. Cholinesterase Inhibitors for Alzheimer’s Disease. Cochrane Database Syst. Rev. 2006, 2016, CD005593. [Google Scholar] [CrossRef]
- Ogura, H.; Kosasa, T.; Kuriya, Y.; Yamanishi, Y. Comparison of Inhibitory Activities of Donepezil and Othercholinesterase Inhibitors on Acetylcholinesterase Andbutylcholinesterase in Vitro. Methods Find. Exp. Clin. Pharmacol. 2000, 22, 609. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.A.; Sabbagh, M.N. Donepezil: Potential Neuroprotective and Disease-Modifying Effects. Expert Opin. Drug Metab. Toxicol. 2008, 4, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Bar-On, P.; Millard, C.B.; Harel, M.; Dvir, H.; Enz, A.; Sussman, J.L.; Silman, I. Kinetic and Structural Studies on the Interaction of Cholinesterases with the Anti-Alzheimer Drug Rivastigmine. Biochemistry 2002, 41, 3555–3564. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Singh, B. A Review on Cholinesterase Inhibitors for Alzheimer’s Disease. Arch. Pharm. Res. 2013, 36, 375–399. [Google Scholar] [CrossRef] [PubMed]
- Kishi, T.; Matsunaga, S.; Oya, K.; Nomura, I.; Ikuta, T.; Iwata, N. Memantine for Alzheimer’s Disease: An Updated Systematic Review and Meta-Analysis. J. Alzheimers Dis. 2017, 60, 401–425. [Google Scholar] [CrossRef] [PubMed]
- Balázs, N.; Bereczki, D.; Kovács, T. Cholinesterase Inhibitors and Memantine for the Treatment of Alzheimer and Non-Alzheimer Dementias. Ideggyógy. Szle. 2021, 74, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, V.K.; Day, G.S. Anti-Amyloid Therapies for Alzheimer Disease: Finally, Good News for Patients. Mol. Neurodegener. 2023, 18, 42. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, M.; Dibello, V.; Sardone, R.; Castellana, F.; Zupo, R.; Lampignano, L.; Bortone, I.; Stallone, R.; Altamura, M.; Bellomo, A.; et al. Lessons Learned from the Failure of Solanezumab as a Prospective Treatment Strategy for Alzheimer’s Disease. Expert Opin. Drug Discov. 2024, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The Antibody Aducanumab Reduces Aβ Plaques in Alzheimer’s Disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Budd Haeberlein, S.; Aisen, P.S.; Barkhof, F.; Chalkias, S.; Chen, T.; Cohen, S.; Dent, G.; Hansson, O.; Harrison, K.; Von Hehn, C.; et al. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. J. Prev. Alzheimers Dis. 2022, 9, 197–210. [Google Scholar] [CrossRef]
- Howard, R.; Liu, K.Y. Questions EMERGE as Biogen Claims Aducanumab Turnaround. Nat. Rev. Neurol. 2020, 16, 63–64. [Google Scholar] [CrossRef] [PubMed]
- Van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Shcherbinin, S.; Evans, C.D.; Lu, M.; Andersen, S.W.; Pontecorvo, M.J.; Willis, B.A.; Gueorguieva, I.; Hauck, P.M.; Brooks, D.A.; Mintun, M.A.; et al. Association of Amyloid Reduction After Donanemab Treatment with Tau Pathology and Clinical Outcomes: The TRAILBLAZER-ALZ Randomized Clinical Trial. JAMA Neurol. 2022, 79, 1015. [Google Scholar] [CrossRef] [PubMed]
- Biber, K.; Bhattacharya, A.; Campbell, B.M.; Piro, J.R.; Rohe, M.; Staal, R.G.W.; Talanian, R.V.; Möller, T. Microglial Drug Targets in AD: Opportunities and Challenges in Drug Discovery and Development. Front. Pharmacol. 2019, 10, 840. [Google Scholar] [CrossRef] [PubMed]
- Jhee, S.; Shiovitz, T.; Crawford, A.W.; Cutler, N.R. β-Amyloid Therapies in Alzheimer’s Disease. Expert Opin. Investig. Drugs 2001, 10, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Rampa, A.; Belluti, F.; Gobbi, S.; Bisi, A. Hybrid-Based Multi-Target Ligands for the Treatment of Alzheimer’s Disease. Curr. Top. Med. Chem. 2011, 11, 2716–2730. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Kumar, A.; Panda, G. Anti-Cholinesterase Hybrids as Multi-Target-Directed Ligands against Alzheimer’s Disease (1998–2018). Bioorg. Med. Chem. 2019, 27, 895–930. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-Target-Directed Ligands to Combat Neurodegenerative Diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef] [PubMed]
- Jana, A.; Bhattacharjee, A.; Das, S.S.; Srivastava, A.; Choudhury, A.; Bhattacharjee, R.; De, S.; Perveen, A.; Iqbal, D.; Gupta, P.K.; et al. Molecular Insights into Therapeutic Potentials of Hybrid Compounds Targeting Alzheimer’s Disease. Mol. Neurobiol. 2022, 59, 3512–3528. [Google Scholar] [CrossRef]
- Liu, L.; Lauro, B.M.; He, A.; Lee, H.; Bhattarai, S.; Wolfe, M.S.; Bennett, D.A.; Karch, C.M.; Young-Pearse, T.; Dominantly Inherited Alzheimer Network (DIAN). Identification of the Aβ37/42 Peptide Ratio in CSF as an Improved Aβ Biomarker for Alzheimer’s Disease. Alzheimers Dement. 2023, 19, 79–96. [Google Scholar] [CrossRef]
- Petit, D.; Fernández, S.G.; Zoltowska, K.M.; Enzlein, T.; Ryan, N.S.; O’Connor, A.; Szaruga, M.; Hill, E.; Vandenberghe, R.; Fox, N.C.; et al. Aβ Profiles Generated by Alzheimer’s Disease Causing PSEN1 Variants Determine the Pathogenicity of the Mutation and Predict Age at Disease Onset. Mol. Psychiatry 2022, 27, 2821–2832. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.E.; Carrieri, C.; Dela Cruz, F.; Fullerton, T.; Hajos-Korcsok, E.; He, P.; Kantaridis, C.; Leurent, C.; Liu, R.; Mancuso, J.; et al. Pharmacokinetic and Pharmacodynamic Effects of a γ-Secretase Modulator, PF -06648671, on CSF Amyloid-β Peptides in Randomized Phase I Studies. Clin. Pharmacol. Ther. 2020, 107, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wei, W.; Zhao, M.; Ma, L.; Jiang, X.; Pei, H.; Cao, Y.; Li, H. Interaction between Aβ and Tau in the Pathogenesis of Alzheimer’s Disease. Int. J. Biol. Sci. 2021, 17, 2181–2192. [Google Scholar] [CrossRef] [PubMed]
- Sturchio, A.; Dwivedi, A.K.; Malm, T.; Wood, M.J.A.; Cilia, R.; Sharma, J.S.; Hill, E.J.; Schneider, L.S.; Graff-Radford, N.R.; Mori, H.; et al. High Soluble Amyloid-Β42 Predicts Normal Cognition in Amyloid-Positive Individuals with Alzheimer’s Disease-Causing Mutations. J. Alzheimers Dis. 2022, 90, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Tudorache, I.F.; Trusca, V.G.; Gafencu, A.V. Apolipoprotein E—A Multifunctional Protein with Implications in Various Pathologies as a Result of Its Structural Features. Comput. Struct. Biotechnol. J. 2017, 15, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Hudry, E.; Klickstein, J.; Cannavo, C.; Jackson, R.; Muzikansky, A.; Gandhi, S.; Urick, D.; Sargent, T.; Wrobleski, L.; Roe, A.D.; et al. Opposing Roles of Apolipoprotein E in Aging and Neurodegeneration. Life Sci. Alliance 2019, 2, e201900325. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2023 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2023, 19, 1598–1695. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Risch, N.J.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Rimmler, J.B.; Locke, P.A.; Conneally, P.M.; Schmader, K.E. Protective Effect of Apolipoprotein E Type 2 Allele for Late Onset Alzheimer Disease. Nat. Genet. 1994, 7, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.R.; Liu, P.; Agrawal, A.; Yip, O.; Blumenfeld, J.; Traglia, M.; Kim, M.J.; Koutsodendris, N.; Rao, A.; Grone, B.; et al. The APOE-R136S Mutation Protects against APOE4-Driven Tau Pathology, Neurodegeneration and Neuroinflammation. Nat. Neurosci. 2023, 26, 2104–2121. [Google Scholar] [CrossRef]
- O’Bryant, S.E.; Barber, R.C.; Philips, N.; Johnson, L.A.; Hall, J.R.; Subasinghe, K.; Petersen, M.; Toga, A.W.; Yaffe, K.; Rissman, R.A.; et al. The Link between APOE4 Presence and Neuropsychological Test Performance among Mexican Americans and Non-Hispanic Whites of the Multiethnic Health & Aging Brain Study—Health Disparities Cohort. Dement. Geriatr. Cogn. Disord. 2022, 51, 26–31. [Google Scholar] [CrossRef]
- Kerchner, G.A.; Berdnik, D.; Shen, J.C.; Bernstein, J.D.; Fenesy, M.C.; Deutsch, G.K.; Wyss-Coray, T.; Rutt, B.K. APOE Ε4 Worsens Hippocampal CA1 Apical Neuropil Atrophy and Episodic Memory. Neurology 2014, 82, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, M.N.; Malek-Ahmadi, M.; Dugger, B.N.; Lee, K.; Sue, L.I.; Serrano, G.; Walker, D.G.; Davis, K.; Jacobson, S.A.; Beach, T.G. The Influence of Apolipoprotein E Genotype on Regional Pathology in Alzheimer’s Disease. BMC Neurol. 2013, 13, 44. [Google Scholar] [CrossRef]
- Shinohara, M.; Sato, N. The Roles of Apolipoprotein E, Lipids, and Glucose in the Pathogenesis of Alzheimer’s Disease. In Diabetes Mellitus; Nakabeppu, Y., Ninomiya, T., Eds.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; Volume 1128, pp. 85–101. [Google Scholar] [CrossRef]
- Morgen, K.; Frölich, L.; Tost, H.; Plichta, M.M.; Kölsch, H.; Rakebrandt, F.; Rienhoff, O.; Jessen, F.; Peters, O.; Jahn, H.; et al. APOE-Dependent Phenotypes in Subjects with Mild Cognitive Impairment Converting to Alzheimer’s Disease. J. Alzheimers Dis. 2013, 37, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Sluimer, J.D.; Vrenken, H.; Blankenstein, M.A.; Fox, N.C.; Scheltens, P.; Barkhof, F.; Van Der Flier, W.M. Whole-Brain Atrophy Rate in Alzheimer Disease: Identifying Fast Progressors. Neurology 2008, 70 Pt 2, 1836–1841. [Google Scholar] [CrossRef]
- Van Der Flier, W.M.; Pijnenburg, Y.A.; Fox, N.C.; Scheltens, P. Early-Onset versus Late-Onset Alzheimer’s Disease: The Case of the Missing APOE Ɛ4 Allele. Lancet Neurol. 2011, 10, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Van Der Vlies, A.E.; Koedam, E.L.G.E.; Pijnenburg, Y.A.L.; Twisk, J.W.R.; Scheltens, P.; Van Der Flier, W.M. Most Rapid Cognitive Decline in APOE Ε4 Negative Alzheimer’s Disease with Early Onset. Psychol. Med. 2009, 39, 1907–1911. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Qian, J.; Monsell, S.E.; Betensky, R.A.; Hyman, B.T. APOE Ε2 Is Associated with Milder Clinical and Pathological A Lzheimer Disease. Ann. Neurol. 2015, 77, 917–929. [Google Scholar] [CrossRef]
- William Rebeck, G.; Reiter, J.S.; Strickland, D.K.; Hyman, B.T. Apolipoprotein E in Sporadic Alzheimer’s Disease: Allelic Variation and Receptor Interactions. Neuron 1993, 11, 575–580. [Google Scholar] [CrossRef]
- Jones, P.B.; Adams, K.W.; Rozkalne, A.; Spires-Jones, T.L.; Hshieh, T.T.; Hashimoto, T.; Von Armin, C.A.F.; Mielke, M.; Bacskai, B.J.; Hyman, B.T. Apolipoprotein E: Isoform Specific Differences in Tertiary Structure and Interaction with Amyloid-β in Human Alzheimer Brain. PLoS ONE 2011, 6, e14586. [Google Scholar] [CrossRef]
- Verghese, P.B.; Castellano, J.M.; Garai, K.; Wang, Y.; Jiang, H.; Shah, A.; Bu, G.; Frieden, C.; Holtzman, D.M. ApoE Influences Amyloid-β (Aβ) Clearance despite Minimal apoE/Aβ Association in Physiological Conditions. Proc. Natl. Acad. Sci. USA 2013, 110, E1807–E1816. [Google Scholar] [CrossRef]
- Strickland, M.R.; Holtzman, D.M. Dr. Jekyll and Mr. Hyde: ApoE Explains Opposing Effects of Neuronal LRP1. J. Clin. Investig. 2019, 129, 969–971. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, G.A.; Tai, L.M.; LaDu, M.J.; Rebeck, G.W. Human APOE4 Increases Microglia Reactivity at Aβ Plaques in a Mouse Model of Aβ Deposition. J. Neuroinflamm. 2014, 11, 111. [Google Scholar] [CrossRef] [PubMed]
- Atagi, Y.; Liu, C.-C.; Painter, M.M.; Chen, X.-F.; Verbeeck, C.; Zheng, H.; Li, X.; Rademakers, R.; Kang, S.S.; Xu, H.; et al. Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). J. Biol. Chem. 2015, 290, 26043–26050. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-J.; Wang, M.; Li, R.-Y.; Wei, T.; Yang, H.-C.; Yin, Y.-S.; Mi, Y.-X.; Qin, Q.; Tang, Y. TREM2 and Microglia Contribute to the Synaptic Plasticity: From Physiology to Pathology. Mol. Neurobiol. 2023, 60, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Keene, C.D.; Cudaback, E.; Li, X.; Montine, K.S.; Montine, T.J. Apolipoprotein E Isoforms and Regulation of the Innate Immune Response in Brain of Patients with Alzheimer’s Disease. Curr. Opin. Neurobiol. 2011, 21, 920–928. [Google Scholar] [CrossRef] [PubMed]
- De Leeuw, S.M.; Kirschner, A.W.T.; Lindner, K.; Rust, R.; Budny, V.; Wolski, W.E.; Gavin, A.-C.; Nitsch, R.M.; Tackenberg, C. APOE2, E3, and E4 Differentially Modulate Cellular Homeostasis, Cholesterol Metabolism, and Inflammatory Response in Isogenic iPSC-Derived Astrocytes. Stem Cell Rep. 2022, 17, 110–126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wu, L.-M.; Wu, J. Cross-Talk between Apolipoprotein E and Cytokines. Mediators Inflamm. 2011, 2011, 949072. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Shinohara, M.; Yamazaki, A.; Ren, Y.; Asmann, Y.W.; Kanekiyo, T.; Bu, G. ApoE (Apolipoprotein E) in Brain Pericytes Regulates Endothelial Function in an Isoform-Dependent Manner by Modulating Basement Membrane Components. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 128–144. [Google Scholar] [CrossRef]
- Jackson, R.J.; Meltzer, J.C.; Nguyen, H.; Commins, C.; Bennett, R.E.; Hudry, E.; Hyman, B.T. APOE4 Derived from Astrocytes Leads to Blood–Brain Barrier Impairment. Brain 2022, 145, 3582–3593. [Google Scholar] [CrossRef]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E Controls Cerebrovascular Integrity via Cyclophilin A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef]
- Liu, C.; Yamazaki, Y.; Heckman, M.G.; Martens, Y.A.; Jia, L.; Yamazaki, A.; Diehl, N.N.; Zhao, J.; Zhao, N.; DeTure, M.; et al. Tau and Apolipoprotein E Modulate Cerebrovascular Tight Junction Integrity Independent of Cerebral Amyloid Angiopathy in Alzheimer’s Disease. Alzheimers Dement. 2020, 16, 1372–1383. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 Leads to Blood–Brain Barrier Dysfunction Predicting Cognitive Decline. Nature 2020, 581, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Kadry, H.; Noorani, B.; Cucullo, L. A Blood–Brain Barrier Overview on Structure, Function, Impairment, and Biomarkers of Integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Zhao, J.; Fu, Y.; Inoue, Y.; Ren, Y.; Chen, Y.; Doss, S.V.; Shue, F.; Jeevaratnam, S.; Bastea, L.; et al. Peripheral apoE4 Enhances Alzheimer’s Pathology and Impairs Cognition by Compromising Cerebrovascular Function. Nat. Neurosci. 2022, 25, 1020–1033. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes Regulate the Blood–Brain Barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.E.; Esparza, T.J.; Lewis, H.A.; Kim, E.; Mac Donald, C.L.; Sullivan, P.M.; Brody, D.L. Human Apolipoprotein E4 Worsens Acute Axonal Pathology but Not Amyloid-β Immunoreactivity After Traumatic Brain Injury in 3×TG-AD Mice. J. Neuropathol. Exp. Neurol. 2013, 72, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Harris, F.M.; Brecht, W.J.; Xu, Q.; Tesseur, I.; Kekonius, L.; Wyss-Coray, T.; Fish, J.D.; Masliah, E.; Hopkins, P.C.; Scearce-Levie, K.; et al. Carboxyl-Terminal-Truncated Apolipoprotein E4 Causes Alzheimer’s Disease-like Neurodegeneration and Behavioral Deficits in Transgenic Mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10966–10971. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Disease Neuroimaging Initiative; Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; et al. ApoE4 Markedly Exacerbates Tau-Mediated Neurodegeneration in a Mouse Model of Tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef]
- Zhao, N.; Liu, C.-C.; Van Ingelgom, A.J.; Linares, C.; Kurti, A.; Knight, J.A.; Heckman, M.G.; Diehl, N.N.; Shinohara, M.; Martens, Y.A.; et al. APOE Ε2 Is Associated with Increased Tau Pathology in Primary Tauopathy. Nat. Commun. 2018, 9, 4388. [Google Scholar] [CrossRef]
- Rauch, J.N.; Luna, G.; Guzman, E.; Audouard, M.; Challis, C.; Sibih, Y.E.; Leshuk, C.; Hernandez, I.; Wegmann, S.; Hyman, B.T.; et al. LRP1 Is a Master Regulator of Tau Uptake and Spread. Nature 2020, 580, 381–385. [Google Scholar] [CrossRef]
- Xian, X.; Pohlkamp, T.; Durakoglugil, M.S.; Wong, C.H.; Beck, J.K.; Lane-Donovan, C.; Plattner, F.; Herz, J. Reversal of ApoE4-Induced Recycling Block as a Novel Prevention Approach for Alzheimer’s Disease. eLife 2018, 7, e40048. [Google Scholar] [CrossRef]
- De Luca, V.; Orfei, M.D.; Gaudenzi, S.; Caltagirone, C.; Spalletta, G. Inverse Effect of the APOE Epsilon4 Allele in Late- and Early-Onset Alzheimer’s Disease. Eur. Arch. Psychiatry Clin. Neurosci. 2016, 266, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Valdez-Gaxiola, C.A.; Maciel-Cruz, E.J.; Hernández-Peña, R.; Dumois-Petersen, S.; Rosales-Leycegui, F.; Gallegos-Arreola, M.P.; Moreno-Ortiz, J.M.; Figuera, L.E. Potential Modifying Effect of the APOEε4 Allele on Age of Onset and Clinical Manifestations in Patients with Early-Onset Alzheimer’s Disease with and without a Pathogenic Variant in PSEN1 in a Sample of the Mexican Population. Int. J. Mol. Sci. 2023, 24, 15687. [Google Scholar] [CrossRef] [PubMed]
- Langella, S.; Barksdale, N.G.; Vasquez, D.; Aguillon, D.; Chen, Y.; Su, Y.; Acosta-Baena, N.; Acosta-Uribe, J.; Baena, A.Y.; Garcia-Ospina, G.; et al. Effect of Apolipoprotein Genotype and Educational Attainment on Cognitive Function in Autosomal Dominant Alzheimer’s Disease. Nat. Commun. 2023, 14, 5120. [Google Scholar] [CrossRef]
- Ryan, N.S.; Nicholas, J.M.; Weston, P.S.J.; Liang, Y.; Lashley, T.; Guerreiro, R.; Adamson, G.; Kenny, J.; Beck, J.; Chavez-Gutierrez, L.; et al. Clinical Phenotype and Genetic Associations in Autosomal Dominant Familial Alzheimer’s Disease: A Case Series. Lancet Neurol. 2016, 15, 1326–1335. [Google Scholar] [CrossRef]
- Van Der Vlies, A.E.; Pijnenburg, Y.A.L.; Koene, T.; Klein, M.; Kok, A.; Scheltens, P.; Van Der Flier, W.M. Cognitive Impairment in Alzheimer’s Disease Is Modified by APOE Genotype. Dement. Geriatr. Cogn. Disord. 2007, 24, 98–103. [Google Scholar] [CrossRef]
- Smits, L.L.; Pijnenburg, Y.A.L.; Van Der Vlies, A.E.; Koedam, E.L.G.E.; Bouwman, F.H.; Reuling, I.E.W.; Scheltens, P.; Van Der Flier, W.M. Early Onset APOE E4-Negative Alzheimer’s Disease Patients Show Faster Cognitive Decline on Non-Memory Domains. Eur. Neuropsychopharmacol. 2015, 25, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Marra, C.; Bizzarro, A.; Daniele, A.; De Luca, L.; Ferraccioli, M.; Valenza, A.; Brahe, C.; Tiziano, F.D.; Gainotti, G.; Masullo, C. Apolipoprotein E Ε4 Allele Differently Affects the Patterns of Neuropsychological Presentation in Early- and Late-Onset Alzheimer’s Disease Patients. Dement. Geriatr. Cogn. Disord. 2004, 18, 125–131. [Google Scholar] [CrossRef]
- Lehtovirta, M.; Soininen, H.; Helisalmi, S.; Mannermaa, A.; Helkala, E.-L.; Hartikainen, P.; Hanninen, T.; Ryynanen, M.; Riekkinen, P.J. Clinical and Neuropsychological Characteristics in Familial and Sporadic Alzheimer’s Disease: Relation to Apolipoprotein E Polymorphism. Neurology 1996, 46, 413–419. [Google Scholar] [CrossRef]
- Almkvist, O.; Johansson, C.; Laffita-Mesa, J.; Thordardottir, S.; Graff, C. APOE Ε4 Influences Cognitive Decline Positively in APP and Negatively in PSEN1 Mutation Carriers with Autosomal-dominant Alzheimer’s Disease. Eur. J. Neurol. 2022, 29, 3580–3589. [Google Scholar] [CrossRef]
- Almkvist, O.; Graff, C. The APOE Ε4 Allele Affects Cognitive Functions Differently in Carriers of APP Mutations Compared to Carriers of PSEN1 Mutations in Autosomal-Dominant Alzheimer’s Disease. Genes 2021, 12, 1954. [Google Scholar] [CrossRef] [PubMed]
- Gharbi-Meliani, A.; Dugravot, A.; Sabia, S.; Regy, M.; Fayosse, A.; Schnitzler, A.; Kivimäki, M.; Singh-Manoux, A.; Dumurgier, J. The Association of APOE Ε4 with Cognitive Function over the Adult Life Course and Incidence of Dementia: 20 Years Follow-up of the Whitehall II Study. Alzheimers Res. Ther. 2021, 13, 5. [Google Scholar] [CrossRef] [PubMed]
- Vélez, J.I.; Lopera, F.; Sepulveda-Falla, D.; Patel, H.R.; Johar, A.S.; Chuah, A.; Tobón, C.; Rivera, D.; Villegas, A.; Cai, Y.; et al. APOE*E2 Allele Delays Age of Onset in PSEN1 E280A Alzheimer’s Disease. Mol. Psychiatry 2016, 21, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Raulin, A.-C.; Doss, S.V.; Trottier, Z.A.; Ikezu, T.C.; Bu, G.; Liu, C.-C. ApoE in Alzheimer’s Disease: Pathophysiology and Therapeutic Strategies. Mol. Neurodegener. 2022, 17, 72. [Google Scholar] [CrossRef] [PubMed]
- Korczyn, A.D.; Grinberg, L.T. Is Alzheimer Disease a Disease? Nat. Rev. Neurol. 2024, 20, 245–251. [Google Scholar] [CrossRef]
- Ellouze, I.; Sheffler, J.; Nagpal, R.; Arjmandi, B. Dietary Patterns and Alzheimer’s Disease: An Updated Review Linking Nutrition to Neuroscience. Nutrients 2023, 15, 3204. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | EOAD | LOAD |
|---|---|---|
| Age at onset | <65 years | ≥65 years |
| Sex distribution | Female = male | Female > male |
| Atypical signs and symptoms | Higher prevalence of dysexecutive symptoms and atypical presentations | Uncommon |
| Episodic memory loss | Early (late in atypical cases) | Early |
| Genetic contributions | Probably polygenic and autosomal dominant in some cases | Probably polygenic |
| Heritability | 92–100% | 60–80% |
| Neuropathological hallmarks | Plaques and tangles | Plaques and tangles |
| Initial PET signal evidence | Precuneus and mesial temporal region | Mesial temporal region |
| Initial Aβ accumulation | Neocortex (striatal in some autosomal dominant cases) | Neocortex |
| Rate of brain atrophy | Fast | Low |
| Tau burden | Higher in cortical and stratum areas | Higher in limbic areas |
| Effect of APOE genotype | APOEε4: Accelerates/diminishes cognitive decline depending on other factors [140,167,168,169,175,176,177]. APOEε2: Significantly delays the age of onset [178]. APOEε3: Further research is needed, although a rare variant has been described to exert a protective effect [133]. | APOEε4: Accelerates cognitive decline and especially promotes the appearance of amnesic phenotypes [129,130,131,132,134,135,136,137]. APOEε2: Slows general cognitive rate decline [132,142]. APOEε3: Is considered a neutral risk factor for AD development. |
| Pharmacological therapy | Therapies focus on the underlying pathological mechanisms of the disease. Patients may benefit from gene-based therapies aimed at reducing the production or aggregation of Aβ [179] | Therapies primarily focus on managing symptoms and improving cognitive function rather than targeting the underlying disease process as well as Gamma-Secretase modulators. In these patients, a gene-based therapy may not be very useful [100,102,103,104,105,106,107,108,109,110] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valdez-Gaxiola, C.A.; Rosales-Leycegui, F.; Gaxiola-Rubio, A.; Moreno-Ortiz, J.M.; Figuera, L.E. Early- and Late-Onset Alzheimer’s Disease: Two Sides of the Same Coin? Diseases 2024, 12, 110. https://doi.org/10.3390/diseases12060110
Valdez-Gaxiola CA, Rosales-Leycegui F, Gaxiola-Rubio A, Moreno-Ortiz JM, Figuera LE. Early- and Late-Onset Alzheimer’s Disease: Two Sides of the Same Coin? Diseases. 2024; 12(6):110. https://doi.org/10.3390/diseases12060110
Chicago/Turabian StyleValdez-Gaxiola, César A., Frida Rosales-Leycegui, Abigail Gaxiola-Rubio, José Miguel Moreno-Ortiz, and Luis E. Figuera. 2024. "Early- and Late-Onset Alzheimer’s Disease: Two Sides of the Same Coin?" Diseases 12, no. 6: 110. https://doi.org/10.3390/diseases12060110
APA StyleValdez-Gaxiola, C. A., Rosales-Leycegui, F., Gaxiola-Rubio, A., Moreno-Ortiz, J. M., & Figuera, L. E. (2024). Early- and Late-Onset Alzheimer’s Disease: Two Sides of the Same Coin? Diseases, 12(6), 110. https://doi.org/10.3390/diseases12060110