
A Novel Compound Nonsense Variant in CYP27B1 Causes an Atypical Form of Vitamin D-Dependent Rickets Type 1A: A Case Report of Two Siblings in a Mexican Family

 ,
,
Abstract
1. Introduction
2. Case Reports
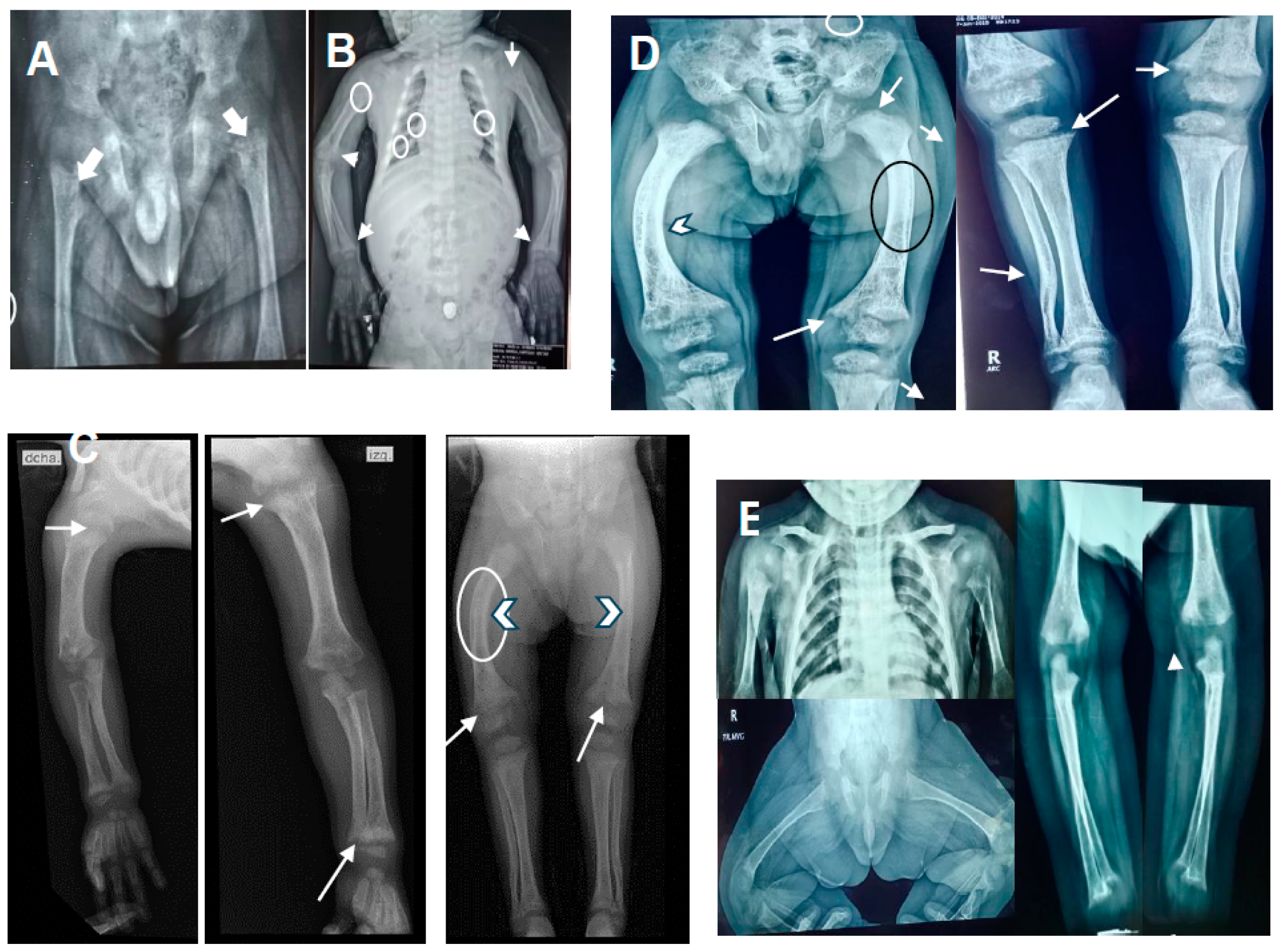
2.1. Patient 1
2.2. Patient 2
3. DNA Samples and Next-Generation Sequencing
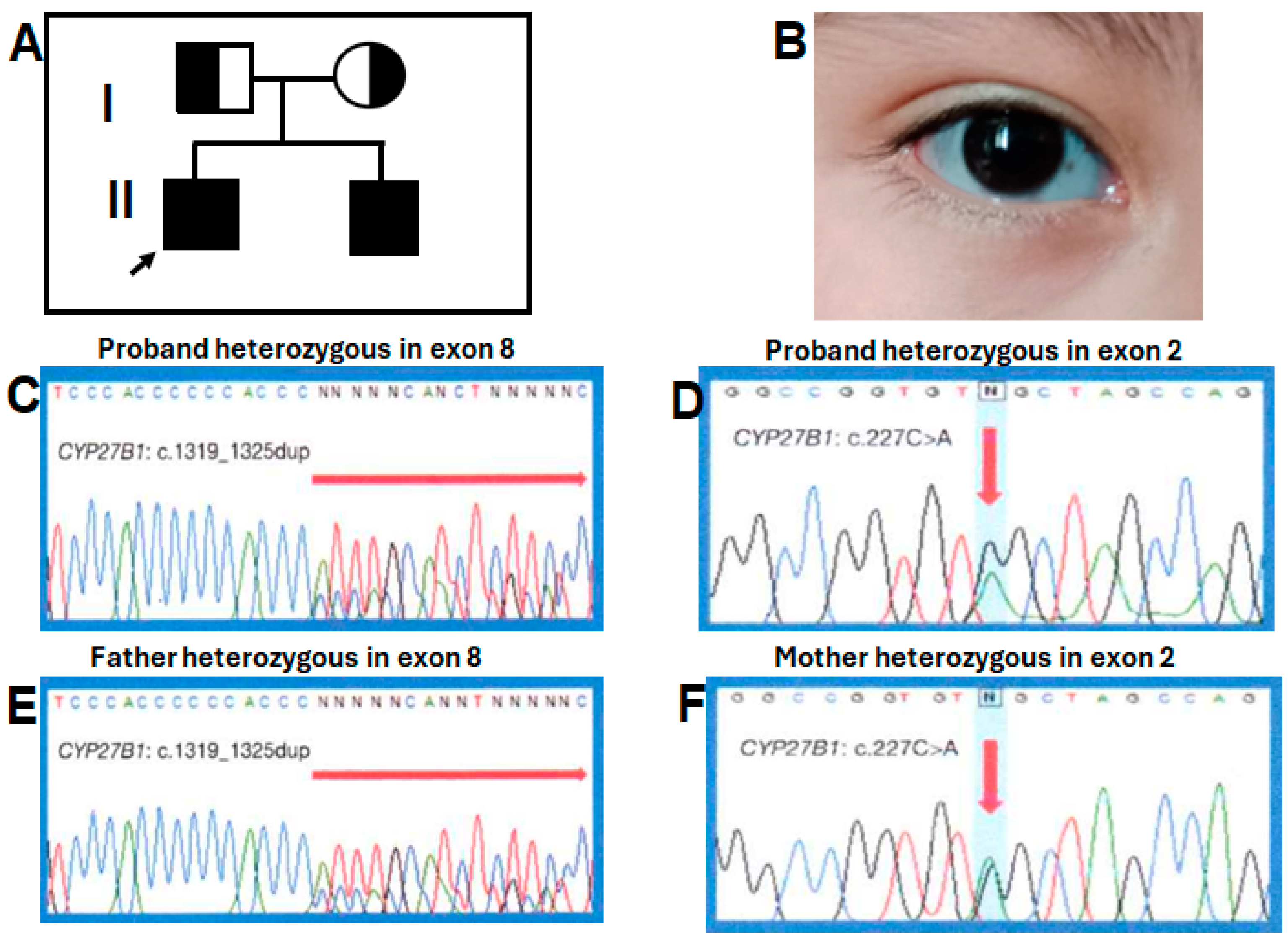
4. Variant’s Validation
5. Statistical Analysis
6. Genetic and Biochemical Results
7. Discussion
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fraser, D.; Kooh, S.W.; Kind, H.P.; Holick, M.F.; Tanaka, Y.; DeLuca, H.F. Pathogenesis of hereditary vitamin-D-dependent rickets. An inborn error of vitamin D metabolism involving defective conversion of 25-hydroxyvitamin D to 1 alpha,25-dihydroxyvitamin D. N. Engl. J. Med. 1973, 289, 817–822. [Google Scholar] [CrossRef]
- Labuda, M.; Morgan, K.; Glorieux, F.H. Mapping autosomal recessive vitamin D dependency type I to chromosome 12q14 by linkage analysis. Am. J. Hum. Genet. 1990, 47, 28–36. [Google Scholar]
- Dodamani, M.H.; Sehemby, M.; Memon, S.S.; Sarathi, V.; Lila, A.R.; Chapla, A.; Bhandare, V.V.; Patil, V.A.; Shah, N.S.; Thomas, N.; et al. Genotype and phenotypic spectrum of vitamin D dependent rickets type 1A: Our experience and systematic review. J. Pediatr. Endocrinol. Metab. 2021, 34, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Kitanaka, S.; Takeyama, K.; Murayama, A.; Sato, T.; Okumura, K.; Nogami, M.; Hasegawa, Y.; Niimi, H.; Yanagisawa, J.; Tanaka, T.; et al. Inactivating mutations in the 25-hydroxyvitamin D3 1alpha-hydroxylase gene in patients with pseudovitamin D-deficiency rickets. N. Engl. J. Med. 1998, 338, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.T.; Lin, C.J.; Burridge, S.M.; Fu, G.K.; Labuda, M.; Portale, A.A.; Miller, W.L. Genetics of vitamin D 1alpha-hydroxylase deficiency in 17 families. Am. J. Hum. Genet. 1998, 63, 1694–1702. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L.; Portale, A.A. Vitamin D 1 alpha-hydroxylase. Trends Endocrinol. Metab. 2000, 11, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Babiker, A.M.; Al Gadi, I.; Al-Jurayyan, N.A.; Al Nemri, A.M.; Al Haboob, A.A.; Al Boukai, A.A.; Al Zahrani, A.; Habib, H.A. A novel pathogenic mutation of the CYP27B1 gene in a patient with vitamin D-dependent rickets type 1: A case report. BMC Res. Notes 2014, 7, 783. [Google Scholar] [CrossRef] [PubMed]
- Dhull, R.S.; Jain, R.; Deepthi, B.; Cheong, H.I.; Saha, A.; Mehndiratta, M.; Basu, S. Vitamin D-dependent rickets (VDDR) type 1: Case series of two siblings with a CYP27B1 mutation and review of the literature. Braz. J. Nephrol. 2020, 42, 494–497. [Google Scholar] [CrossRef] [PubMed]
- Velásquez-Jones, L.; Medeiros, M.; Valverde-Rosas, S.; Jiménez-Triana, C.; Del Moral-Espinosa, I.; Romo-Vázquez, J.C.; Franco-Alvarez, I. Seguimiento a largo plazo de un paciente con raquitismo dependiente de vitamina D tipo I [Long term follow up of a patient with type I vitamin D-dependent rickets]. Boletín Médico Hosp. Infant. México 2015, 72, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Dursun, F.; Özgürhan, G.; Kırmızıbekmez, H.; Keskin, E.; Hacıhamdioğlu, B. Genetic and Clinical Characteristics of Patients with Vitamin D Dependent Rickets Type 1A. J. Clin. Res. Pediatr. Endocrinol. 2019, 11, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Edouard, T.; Alos, N.; Chabot, G.; Roughley, P.; Glorieux, F.H.; Rauch, F. Short- and long-term outcome of patients with pseudo-vitamin D deficiency rickets treated with calcitriol. J. Clin. Endocrinol. Metab. 2011, 96, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Tahir, S.; Demirbilek, H.; Ozbek, M.N.; Baran, R.T.; Tanriverdi, S.; Hussain, K. Genotype and Phenotype Characteristics in 22 Patients with Vitamin D-Dependent Rickets Type I. Horm. Res. Paediatr. 2016, 85, 309–317. [Google Scholar] [CrossRef]
- Zou, M.; Guven, A.; BinEssa, H.A.; Al-Rijjal, R.A.; Meyer, B.F.; Alzahrani, A.S.; Shi, Y. Molecular Analysis of CYP27B1 Mutations in Vitamin D-Dependent Rickets Type 1A: C.590G > A (p.G197D) Missense Mutation Causes a RNA Splicing Error. Front. Genet. 2020, 11, 607517. [Google Scholar] [CrossRef]
- Yamazaki, M.; Michigami, T. Osteocytes and the pathogenesis of hypophosphatemic rickets. Front. Endocrinol. 2022, 13, 1005189. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yuan, X.; Chen, R.; Lin, X.; Shangguan, H.; Yang, X.; Zhang, Y. Clinical and genetic analysis of two Chinese families with vitamin D-dependent rickets type IA and follow-up. Orphanet J. Rare Dis. 2020, 15, 273. [Google Scholar] [CrossRef]
- Durmaz, E.; Zou, M.; Al-Rijjal, R.A.; Bircan, I.; Akçurin, S.; Meyer, B.; Shi, Y. Clinical and genetic analysis of patients with vitamin D-dependent rickets type 1A. Clin. Endocrinol. 2012, 77, 363–369. [Google Scholar] [CrossRef]
- Yamamoto, K.; Masuno, H.; Sawada, N.; Sakaki, T.; Inouye, K.; Ishiguro, M.; Yamada, S. Homology modeling of human 25-hydroxyvitamin D3 1alpha-hydroxylase (CYP27B1) based on the crystal structure of rabbit CYP2C5. J. Steroid Biochem. Mol. Biol. 2004, 89–90, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Koek, W.N.; Zillikens, M.C.; van der Eerden, B.C.; van Leeuwen, J.P. Novel Compound Heterozygous Mutations in the CYP27B1 Gene Lead to Pseudovitamin D-Deficient Rickets. Calcif. Tissue Int. 2016, 99, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Guan, Z.; Mei, H.; Zhang, W.; Zhou, Z.; Su, L.; Cheng, J.; Zheng, R.; Liang, C.; Cai, Y.; et al. Clinical characteristics and long-term outcomes of 12 children with vitamin D-dependent rickets type 1A: A retrospective study. Front. Pediatr. 2022, 10, 1007219. [Google Scholar] [CrossRef] [PubMed]
- Özcabı, B.; Bucak, F.T.; Jaferova, S.; Oruç, Ç.; Adrovic, A.; Ceylaner, S.; Ercan, O.; Evliyaoğlu, O. A Case of Vitamin D-Dependent Rickets Type 1A with a Novel Mutation in the Uzbek Population. J. Clin. Res. Pediatr. Endocrinol. 2016, 8, 484–489. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Proband (Patient 1) | Proband’s Brother (Patient 2) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Features | Reference Ranges | Level Pre-Treatment | After 2 at 18 Months of Treatment | After 18 at 53 Months of Treatment | After 53 Months of Treatment | F of ANOVA Test, p Value | Pre-Treatment | 16 mo Post-Treatment | Student’s t-Test, p Value |
| Age, y (year), mo (months) | 1y-1 mo | 1y-4 mo at 2y-8 mo | 2y-8 mo at 5y-7 mo | 5y-7 mo to 6y-10 mo | <3 mo | 3 to 1y-7 mo | |||
| Calcium, mg/dL, mean ± SD | 8.3–10.6 | 8.3 | 8.14 ± 0.6 | 8.6.0 ± 0.2 | 9.5 ± 0.4 | <0.001 * | 8.0 | 9.0 | 0.02 ** |
| Phosphorus, mg/dL, mean ± SD | 2.4–5.1 | 2.1 | 3.1 ± 1.1 | 3.2 ± 0.5 | 4.8 ± 0.7 | <0.006 * | 3.8 | 4.2 | 0.59 |
| AP, UI/L, mean ± SD | 45–129 | 9200 | 5098 ± 2108 | 2635 ± 300 | 171 ± 62 | 0.02 * | 1336 | 682 | 0.05 |
| PTH, pg/mL, mean ± SD | 10–88 | 1350 | 702 ± 185 | 289 ± 137 | 60 ± 27 | <0.001 * | 314 | 67 | <0.001 ** |
| 25(OH)D3, ng/mL, mean ± SD | 30–100 | 32.9 | 73.9 ± 13 | 51.9 ± 12 | 62 ± 15 | 0.06 | 81 | 91 | 0.62 |
| 1,25(OH)2D3 pg/mL, mean ± SD | 19.6–54.3 | 6 | 36.7 ± 14 | 34.8 ± 12 | 45.3 ± 4 | 0.12 (<0.001 **) | 9 | 43 | 0.01 ** |
| Height, cm | 67 | 67 at 77 | 77 at 92 | 92 at 103 | NA | 59 | 79 | ||
| Height, SD | −3.2 | −4.3 to −5.3 | −5.3 to −6.6 | −6.6 to −5.6 | NA | −0.6 | −1.3 | ||
| Calcitriol dose, μg/day | NA | 0.5 | 1.5 | 1.5 | NA | NA | 1.0 | ||
| Cases/Captation Age | DNA Mutation | Exon | Amino Acid Change | Phenotype | Clinical Response at Calcitriol Treatment | Author |
|---|---|---|---|---|---|---|
| 2 cases (4 mo). 2 cases (18 and 19 mo). 1 case (INR) | c.1319_1325dupCCCACCC. c.1319_1325dupCCCACC. c.1166G>A; c.1079 C>A | 8 8 7 6 | p.Phe443Profs*24. p.Phe443Profs*24. p.Arg389His; p.Ser360* | Severe hypocalcemia and seizure. Delay in walking and mild hypocalcemia. Bowed legs. | INR | [16] |
| 1 case (13 mo) | c.1510C > T. | 9 | p.Q504*. | Multiple fractures, bossing frontal, and classic VDDR1A. | Good biochemical response at 10 mo post-treatment. Growth and deformity NR | [7] |
| Case 8 (INR) Case 7 (INR) | c.574A>G; c.1319_1325dupCCCACCC. c.1319_1325dupCCCACCC | 3 8 8 | p.K192E; p.Phe443Profs*24. p.Phe443Profs*24. | Mild phenotype. Severe phenotype. | INR | [12] |
| Case 5 (14 mo) Case 8 (24 mo) | c.1319_1325dupCCCACCC. c.1319_1325dupCCCACCC | 8 8 | p.Phe443Profs*24. p.Phe443Profs*24. | Growth retardation and hypocalcemia. Inability to walk and mild hypocalcemia. | INR | [10] |
| 2 cases (INR) | c.1319_1325dupCCCACCC. | 8 | p.Phe443Profs*24. | Hypocalcemic seizure, severe growth retardation, walking difficulty, and skeletal deformities. | INR | [15] |
| 3 cases (4–18 mo) | c.1319_1325dupCCCACCC. | 8 | p.Phe443Profs*24. | Hypocalcemic seizures in infancy, rickets, dental anomalies, and fractures. | Good biochemical response, two patients > 12 years persisted deformity | [3] |
| 12 cases | c.1319_1325dupCCCACCC (Eight cases involving this variant) | 8 | p.Phe443Profs*24. | Delayed walking and severe growth retardation. | Good rickets and biochemical response (6 mo to 15.6y of follow-up), and 58% of patients remained with short stature | [19] |
| Case 1 (proband; 13 mo) Case 2 (Brother; 2 mo) | c.227G>A; c.1319_1325dupCCCACCC. c.227G>A; c.1319_1325dupCCCACCC. | 2 8 2 8 | p.Trp76*; p.Phe443Profs*24. p.Trp76*; p.Phe443Profs*24. | Low-normal calcemia, no seizures, fractures, severe growth retardation, sclera gray, café-au-lait spots, frontal bossing, mild medial facial hypoplasia, and pectum carinatum. Gray sclera. | Good biochemical response, partial to rickets, and bad growth deformity prevention Good biochemical, rickets, growth response, and prevention of deformities | This study |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toral López, J.; Candia Tenopala, C.; Reyes Mosqueda, A.D.; Fonseca Sánchez, M.Á.; González Huerta, L.M. A Novel Compound Nonsense Variant in CYP27B1 Causes an Atypical Form of Vitamin D-Dependent Rickets Type 1A: A Case Report of Two Siblings in a Mexican Family. Diseases 2024, 12, 248. https://doi.org/10.3390/diseases12100248
Toral López J, Candia Tenopala C, Reyes Mosqueda AD, Fonseca Sánchez MÁ, González Huerta LM. A Novel Compound Nonsense Variant in CYP27B1 Causes an Atypical Form of Vitamin D-Dependent Rickets Type 1A: A Case Report of Two Siblings in a Mexican Family. Diseases. 2024; 12(10):248. https://doi.org/10.3390/diseases12100248
Chicago/Turabian StyleToral López, Jaime, Cesar Candia Tenopala, Alix Daniela Reyes Mosqueda, Miguel Ángel Fonseca Sánchez, and Luz María González Huerta. 2024. "A Novel Compound Nonsense Variant in CYP27B1 Causes an Atypical Form of Vitamin D-Dependent Rickets Type 1A: A Case Report of Two Siblings in a Mexican Family" Diseases 12, no. 10: 248. https://doi.org/10.3390/diseases12100248
APA StyleToral López, J., Candia Tenopala, C., Reyes Mosqueda, A. D., Fonseca Sánchez, M. Á., & González Huerta, L. M. (2024). A Novel Compound Nonsense Variant in CYP27B1 Causes an Atypical Form of Vitamin D-Dependent Rickets Type 1A: A Case Report of Two Siblings in a Mexican Family. Diseases, 12(10), 248. https://doi.org/10.3390/diseases12100248