
Whole Sequencing and Detailed Analysis of SARS-CoV-2 Genomes in Southeast Spain: Identification of Recurrent Mutations in the 20E (EU1) Variant with Some Clinical Implications

 , , , ,
, , , ,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Sample Collection
2.2. RNA Extraction and RT-PCR
2.3. DNA Library Construction and Sequencing for SARS-CoV-2
2.4. Sequencing Data Analysis
2.5. Statistical Analysis of the Data
3. Results
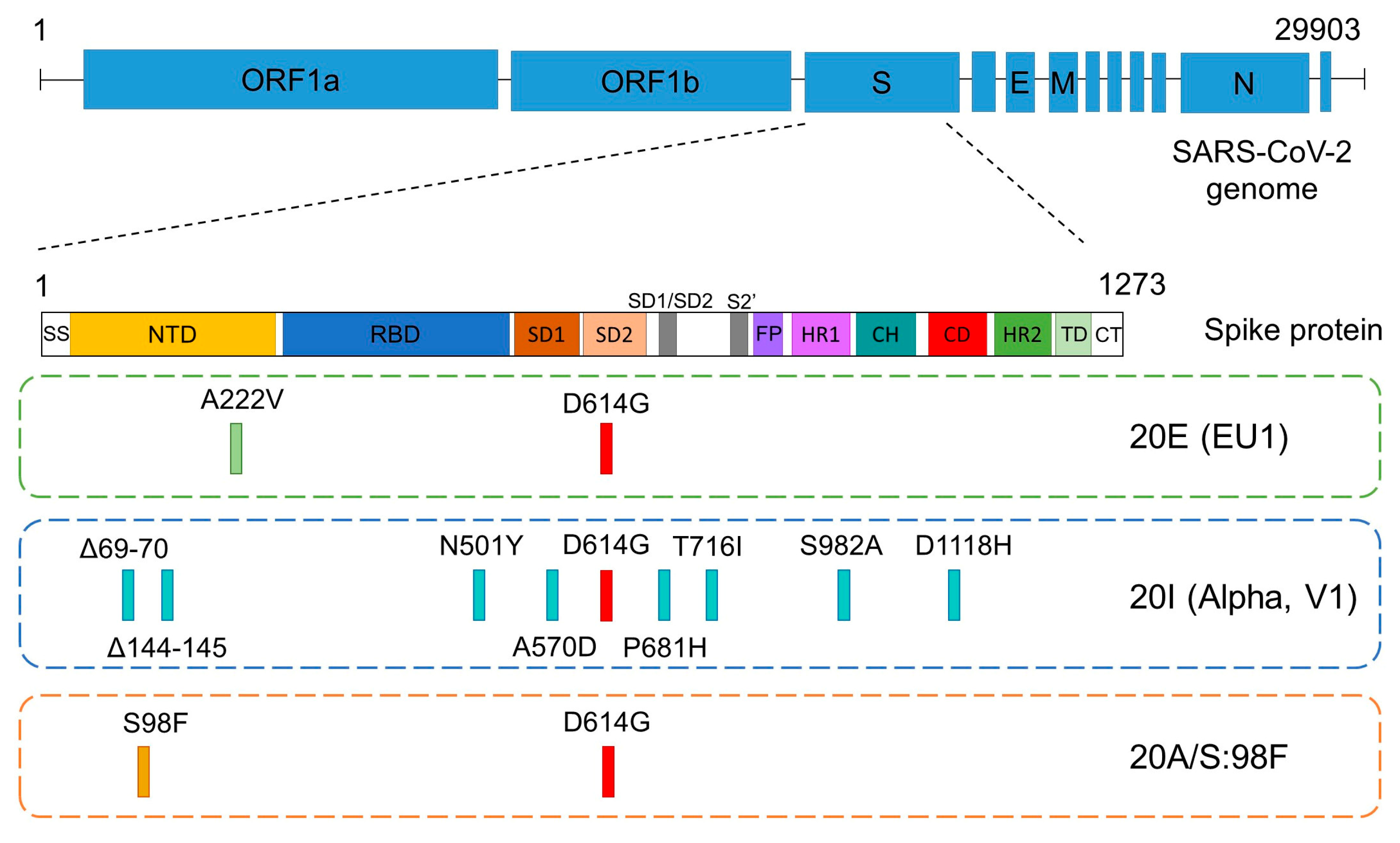
3.1. Description of the SARS-CoV-2 Variants Detected
3.2. Considerations of Concern on the Variant 20E (EU1) Background
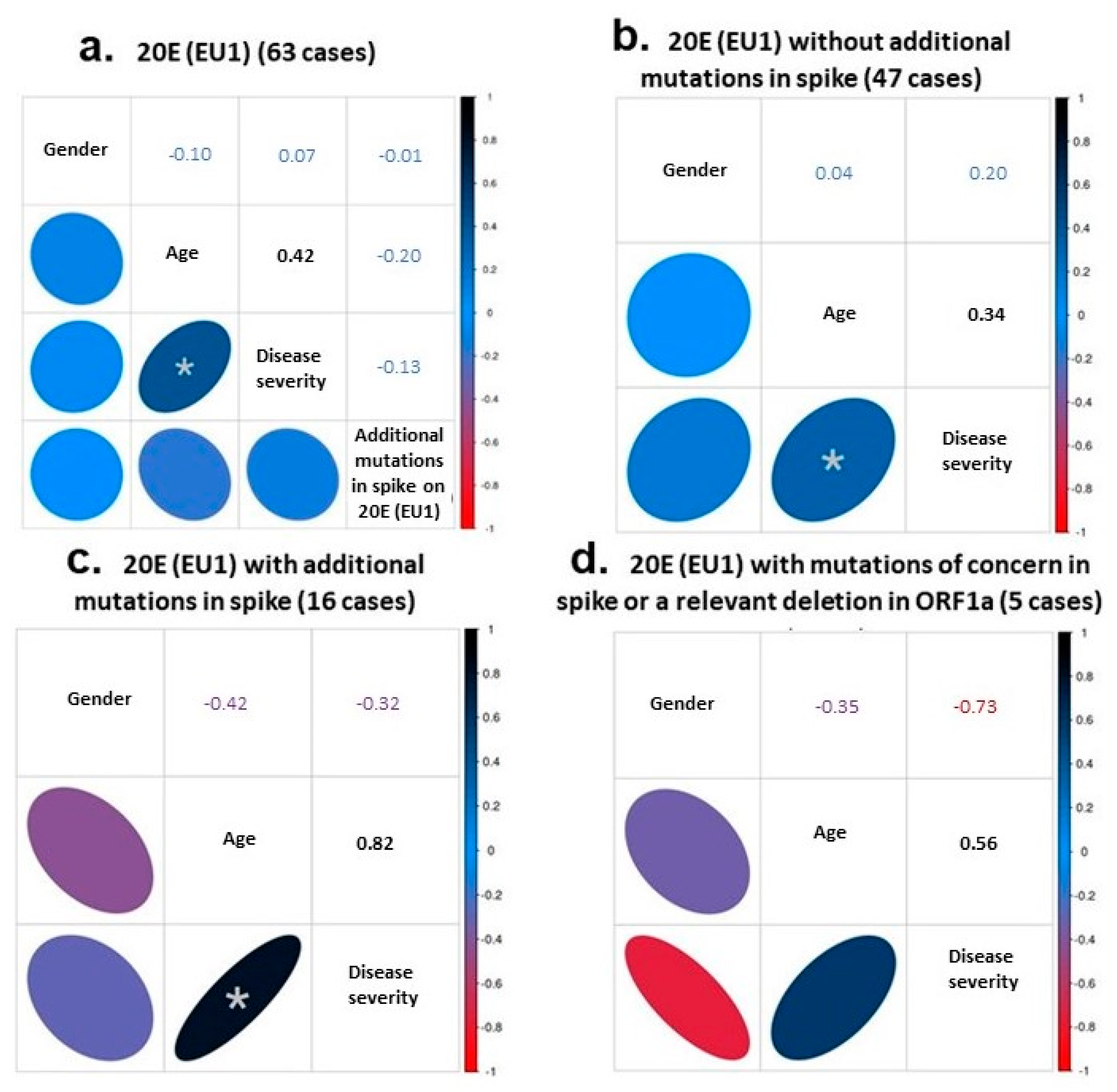
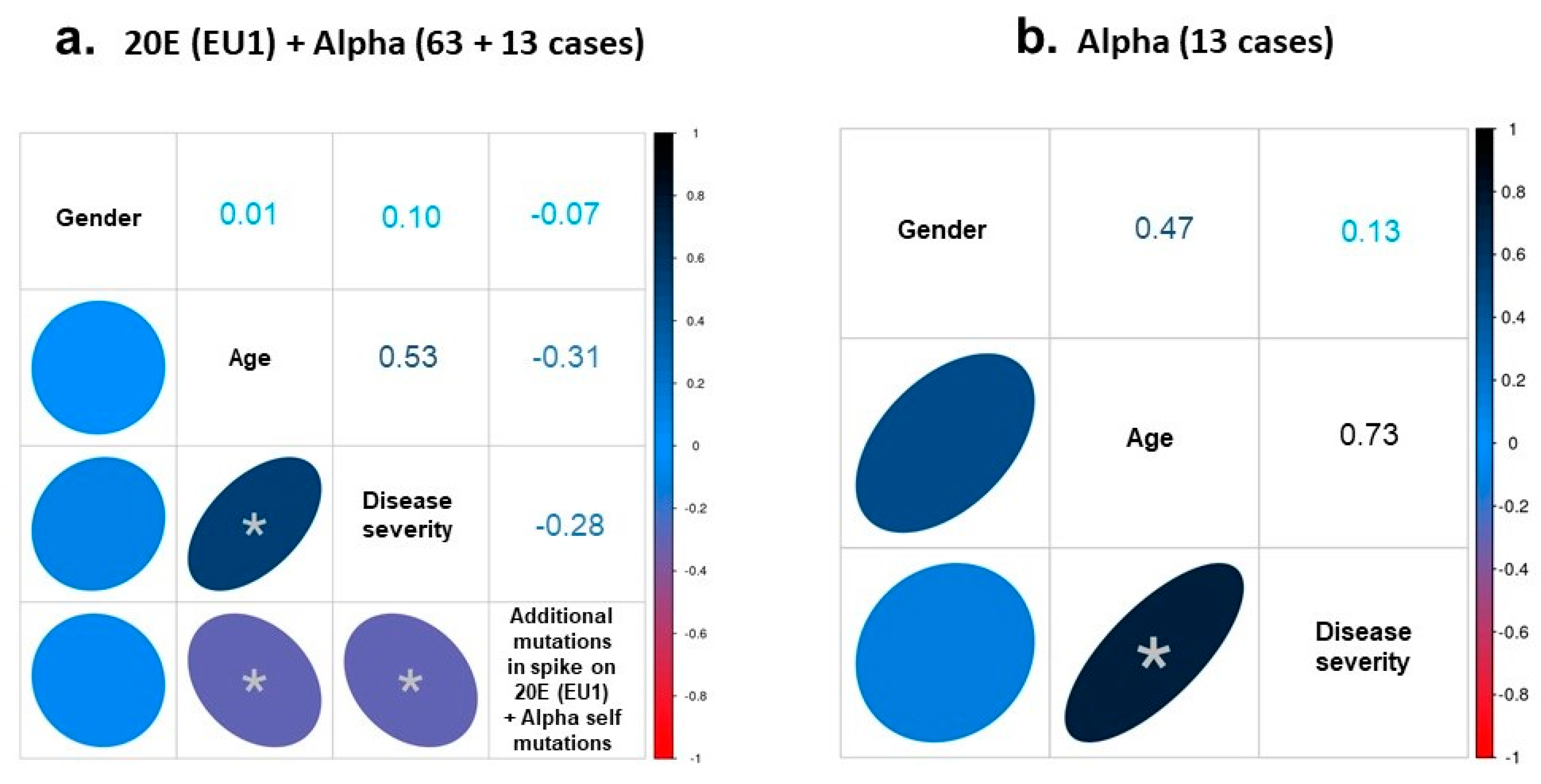
3.3. Correlations between Clinical Data and Genome of SARS-CoV-2 Variants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Okada, P.; Buathong, R.; Phuygun, S.; Thanadachakul, T.; Parnmen, S.; Wongboot, W.; Waicharoen, S.; Wacharapluesadee, S.; Uttayamakul, S.; Vachiraphan, A.; et al. Early transmission patterns of coronavirus disease 2019 (COVID-19) in travellers from Wuhan to Thailand, January 2020. Eurosurveillance 2020, 25, 2000097. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Perez-Gomez, R. The Development of SARS-CoV-2 Variants: The Gene Makes the Disease. J. Dev. Biol. 2021, 9, 58. [Google Scholar] [CrossRef]
- Obermeyer, F.; Jankowiak, M.; Barkas, N.; Schaffner, S.F.; Pyle, J.D.; Yurkovetskiy, L.; Bosso, M.; Park, D.J.; Babadi, M.; MacInnis, B.L.; et al. Analysis of 6.4 million SARS-CoV-2 genomes identifies mutations associated with fitness. Science 2022, 376, 1327–1332. [Google Scholar] [CrossRef] [PubMed]
- Wise, J. COVID-19: The E484K mutation and the risks it poses. BMJ 2021, 372, n359. [Google Scholar] [CrossRef] [PubMed]
- Meng, B.; Kemp, S.A.; Papa, G.; Datir, R.; Ferreira, I.A.; Marelli, S.; Harvey, W.T.; Lytras, S.; Mohamed, A.; Gallo, G.; et al. Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B.1.1.7. Cell Rep. 2021, 35, 109292. [Google Scholar] [CrossRef]
- Collier, D.A.; De Marco, A.; Ferreira, I.A.T.M.; Meng, B.; Datir, R.P.; Walls, A.C.; Kemp, S.A.; Bassi, J.; Pinto, D.; Silacci-Fregni, C.; et al. Sensitivity of SARS-CoV-2 B.1.1.7 to mRNA vaccine-elicited antibodies. Nature 2021, 593, 136–141. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e889. [Google Scholar] [CrossRef]
- Salleh, M.; Derrick, J.; Deris, Z. Structural Evaluation of the Spike Glycoprotein Variants on SARS-CoV-2 Transmission and Immune Evasion. Int. J. Mol. Sci. 2021, 22, 7425. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Assmann, J.L.; Kolijn, P.M.; Schrijver, B.; van Gammeren, A.J.; Loth, D.W.; Ermens, T.A.; Dik, W.A.; van der Velden, V.H.; Langerak, A.W. TRB sequences targeting ORF1a/b are associated with disease severity in hospitalized COVID-19 patients. J. Leukoc. Biol. 2021, 111, 283–289. [Google Scholar] [CrossRef]
- Christensen, P.A.; Olsen, R.J.; Long, S.W.; Snehal, R.; Davis, J.J.; Saavedra, M.O.; Reppond, K.; Shyer, M.N.; Cambric, J.; Gadd, R.; et al. Signals of Significantly Increased Vaccine Breakthrough, Decreased Hospitalization Rates, and Less Severe Disease in Patients with Coronavirus Disease 2019 Caused by the Omicron Variant of Severe Acute Respiratory Syndrome Coronavirus 2 in Houston, Texas. Am. J. Pathol. 2022, 192, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Kidd, M.; Richter, A.; Best, A.; Cumley, N.; Mirza, J.; Percival, B.; Mayhew, M.; Megram, O.; Ashford, F.; White, T.; et al. S-Variant SARS-CoV-2 Lineage B1.1.7 Is Associated With Significantly Higher Viral Load in Samples Tested by TaqPath Polymerase Chain Reaction. J. Infect. Dis. 2021, 223, 1666–1670. [Google Scholar] [CrossRef] [PubMed]
- Pillay, S.; Giandhari, J.; Tegally, H.; Wilkinson, E.; Chimukangara, B.; Lessells, R.; Moosa, Y.; Gazy, I.; Fish, M.; Singh, L.; et al. Illumina Nextera DNA Flex library construction and sequencing for SARS-CoV-2: Adapting COVID-19 ARTIC protocol V.1. Antivir. Res. 2020, 200, 10-17504. [Google Scholar] [CrossRef]
- Pillay, S.; Giandhari, J.; Tegally, H.; Wilkinson, E.; Chimukangara, B.; Lessells, R.; Moosa, Y.; Mattison, S.; Gazy, I.; Fish, M.; et al. Whole Genome Sequencing of SARS-CoV-2: Adapting Illumina Protocols for Quick and Accurate Outbreak Investigation during a Pandemic. Genes 2020, 11, 949. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019; Available online: https://www.R-project.org/ (accessed on 22 February 2023).
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Vaughan, T.G.; Crawford, K.H.D.; Althaus, C.L.; Reichmuth, M.L.; Bowen, J.E.; Walls, A.C.; Corti, D.; et al. Spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nature 2021, 595, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef]
- López, M.G.; Chiner-Oms, Á.; de Viedma, D.G.; Ruiz-Rodriguez, P.; Bracho, M.A.; Cancino-Muñoz, I.; D’Auria, G.; de Marco, G.; García-González, N.; Goig, G.A.; et al. The first wave of the COVID-19 epidemic in Spain was associated with early introductions and fast spread of a dominating genetic variant. Nat. Genet. 2021, 53, 1405–1414. [Google Scholar] [CrossRef]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef] [PubMed]
- Follis, K.E.; York, J.; Nunberg, J.H. Furin cleavage of the SARS coronavirus spike glycoprotein enhances cell–cell fusion but does not affect virion entry. Virology 2006, 350, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Aleem, A.; Samad, A.B.A.; Vaqar, S. Emerging Variants of SARS-CoV-2 and Novel Therapeutics against Coronavirus (COVID-19); StatPearls, Lehigh Valley Health Network: Allentown, PA, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK570580/ (accessed on 22 February 2023).
- Lasek-Nesselquist, E.; Lapierre, P.; Schneider, E.; George, K.S.; Pata, J. The localized rise of a B.1.526 SARS-CoV-2 variant containing an E484K mutation in New York State. medRxiv 2021. [Google Scholar] [CrossRef]
- Ku, Z.; Xie, X.; Davidson, E.; Ye, X.; Su, H.; Menachery, V.D.; Li, Y.; Yuan, Z.; Zhang, X.; Muruato, A.E.; et al. Molecular determinants and mechanism for antibody cocktail preventing SARS-CoV-2 escape. Nat. Commun. 2021, 12, 469. [Google Scholar] [CrossRef]
- McCarthy, K.R.; Rennick, L.J.; Nambulli, S.; Robinson-McCarthy, L.R.; Bain, W.G.; Haidar, G.; Duprex, W.P. Recurrent deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape. Science 2021, 371, 1139–1142. [Google Scholar] [CrossRef]
- Kemp, S.A.; Collier, D.A.; Datir, R.P.; Ferreira, I.A.T.M.; Gayed, S.; Jahun, A.; Hosmillo, M.; Rees-Spear, C.; Mlcochova, P.; Lumb, I.U.; et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature 2021, 592, 277–282. [Google Scholar] [CrossRef]
- Gong, S.Y.; Chatterjee, D.; Richard, J.; Prévost, J.; Tauzin, A.; Gasser, R.; Bo, Y.; Vézina, D.; Goyette, G.; Gendron-Lepage, G.; et al. Contribution of single mutations to selected SARS-CoV-2 emerging variants spike antigenicity. Virology 2021, 563, 134–145. [Google Scholar] [CrossRef]
- Callaway, E. The mutation that helps Delta spread like wildfire. Nature 2021, 596, 472–473. [Google Scholar] [CrossRef]
- Hodcroft, E.B.; Domman, D.B.; Snyder, D.J.; Oguntuyo, K.Y.; Diest, M.V.; Densmore, K.H.; Schwalm, K.C.; Femling, J.; Carroll, J.L.; Scott, R.S.; et al. Emergence in late 2020 of multiple lineages of SARS-CoV-2 Spike protein variants affecting amino acid position 677. medRxiv 2021. [Google Scholar] [CrossRef]
- Pater, A.A.; Bosmeny, M.S.; Barkau, C.L.; Ovington, K.N.; Chilamkurthy, R.; Parasrampuria, M.; Eddington, S.B.; Yinusa, A.O.; White, A.A.; Metz, P.E.; et al. Emergence and evolution of a prevalent new SARS-CoV-2 variant in the United States. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wang, P.; Casner, R.G.; Nair, M.S.; Wang, M.; Yu, J.; Cerutti, G.; Liu, L.; Kwong, P.D.; Huang, Y.; Shapiro, L.; et al. Increased resistance of SARS-CoV-2 variant P.1 to antibody neutralization. Cell Host Microbe 2021, 29, 747–751. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.; De Marco, A.; Lempp, F.A.; Tortorici, M.A.; Pinto, D.; Walls, A.C.; Beltramello, M.; Chen, A.; Liu, Z.; Zatta, F.; et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell 2021, 184, 2332–2347.e16. [Google Scholar] [CrossRef] [PubMed]
- Benvenuto, D.; Angeletti, S.; Giovanetti, M.; Bianchi, M.; Pascarella, S.; Cauda, R.; Ciccozzi, M.; Cassone, A. Evolutionary analysis of SARS-CoV-2: How mutation of Non-Structural Protein 6 (NSP6) could affect viral autophagy. J. Infect. 2020, 81, e24–e27. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Nextstrain Clade | Pango Lineage | GISAID * Clade | WHO ** Name/ Status | % | Nonsynonymous Defining Mutations in Spike |
|---|---|---|---|---|---|
| 20E (EU1) | B.1.177 | GV | - | 63/88: 71.6% | A222V, D614G |
| 20I (Alpha, V1) | B.1.1.7 | GRY | Alpha/ VOC | 13/88: 14.8% | Δ69–70, Δ144–145, N501Y, A570D, D614G, P681H, T716I, S982A, D1118H |
| 20A/S:98F | B.1.221 | G | - | 6/88: 6.8% | S98F, D614G |
| 20A or 20C (miscellaneous group) | B.1, B.1.36.10 or B.1.499 | GH | - | 4/88: 4.56% | D614G |
| 20J (Gamma, V3) | P.1 or B.1.1.28.1 | GR | Gamma/ VOC | 1/88: 1.14% | L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, D614G, H655Y, T1027I, V1176F |
| 21H | B.1.621 | GH | Mu/ VOI | 1/88: 1.14% | T95I, Y144S, Y145N, R346K, E484K, N501Y, D614G, P681H, D950N |
| Sample Name | GISAID Accession ID | Mutations in Spike | Presence of ORF1a: S3675-, G3676- and F3677-Deletion |
|---|---|---|---|
| RS23 | EPI_ISL_11623867 | A222V, D614G, Q675H | No |
| RS25 | EPI_ISL_11634150 | T19R, A222V, D614G | No |
| RS26 | EPI_ISL_11634151 | S98F, A222V, D614G | No |
| RS34 | EPI_ISL_11635219 | A222V, D614G, P681H, T716I | No |
| RS38 | EPI_ISL_11646217 | A222V, S254P, A262S, D614G, P809S | No |
| RS40 | EPI_ISL_11685202 | Δ69–70, A222V, E484K, D614G, A701G, P1069S | Yes |
| RS48 | EPI_ISL_11696722 | T95I, A222V, D614G, A1020S | No |
| RS50 | EPI_ISL_11696733 | T20N, A222V, D614G | No |
| RS52 | EPI_ISL_11696749 | A222V, S254P, A262S, D614G, P809S | No |
| RS56 | EPI_ISL_11728097 | T95I, A222V, D614G, A1020S | No |
| RS62 | EPI_ISL_11755017 | A222V, P251H, D614G | No |
| RS63 | EPI_ISL_11755804 | T95I, A222V, D614G, A1020S | No |
| RS67 | EPI_ISL_11760681 | F192L, A222V, D614G | Yes |
| RS69 | EPI_ISL_11765429 | A222V, D614G, N658D | No |
| RS73 | EPI_ISL_11767045 | A222V, E484K, D614G, A701G, P1069S | Yes |
| RS101 | EPI_ISL_11794379 | M153I, A222V, D614G | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Andreo, M.J.; Vicente-Romero, M.R.; Bernal, E.; Navarro-González, I.; Salazar-Martínez, F.; Cánovas-Cánovas, V.; Gil-Ortuño, C.; Riquelme-Rocamora, M.G.; Solano, F.; Ibáñez-López, F.J.; et al. Whole Sequencing and Detailed Analysis of SARS-CoV-2 Genomes in Southeast Spain: Identification of Recurrent Mutations in the 20E (EU1) Variant with Some Clinical Implications. Diseases 2023, 11, 54. https://doi.org/10.3390/diseases11020054
López-Andreo MJ, Vicente-Romero MR, Bernal E, Navarro-González I, Salazar-Martínez F, Cánovas-Cánovas V, Gil-Ortuño C, Riquelme-Rocamora MG, Solano F, Ibáñez-López FJ, et al. Whole Sequencing and Detailed Analysis of SARS-CoV-2 Genomes in Southeast Spain: Identification of Recurrent Mutations in the 20E (EU1) Variant with Some Clinical Implications. Diseases. 2023; 11(2):54. https://doi.org/10.3390/diseases11020054
Chicago/Turabian StyleLópez-Andreo, María José, María Rosario Vicente-Romero, Enrique Bernal, Inmaculada Navarro-González, Francisco Salazar-Martínez, Vanesa Cánovas-Cánovas, Cristina Gil-Ortuño, María Gema Riquelme-Rocamora, Francisco Solano, Francisco Javier Ibáñez-López, and et al. 2023. "Whole Sequencing and Detailed Analysis of SARS-CoV-2 Genomes in Southeast Spain: Identification of Recurrent Mutations in the 20E (EU1) Variant with Some Clinical Implications" Diseases 11, no. 2: 54. https://doi.org/10.3390/diseases11020054
APA StyleLópez-Andreo, M. J., Vicente-Romero, M. R., Bernal, E., Navarro-González, I., Salazar-Martínez, F., Cánovas-Cánovas, V., Gil-Ortuño, C., Riquelme-Rocamora, M. G., Solano, F., Ibáñez-López, F. J., Tomás, C., Candel-Pérez, C., Pérez-Parra, S., & Flores-Flores, C. (2023). Whole Sequencing and Detailed Analysis of SARS-CoV-2 Genomes in Southeast Spain: Identification of Recurrent Mutations in the 20E (EU1) Variant with Some Clinical Implications. Diseases, 11(2), 54. https://doi.org/10.3390/diseases11020054