Aryl Hydrocarbon Receptor Role in Co-Ordinating SARS-CoV-2 Entry and Symptomatology: Linking Cytotoxicity Changes in COVID-19 and Cancers; Modulation by Racial Discrimination Stress




Abstract
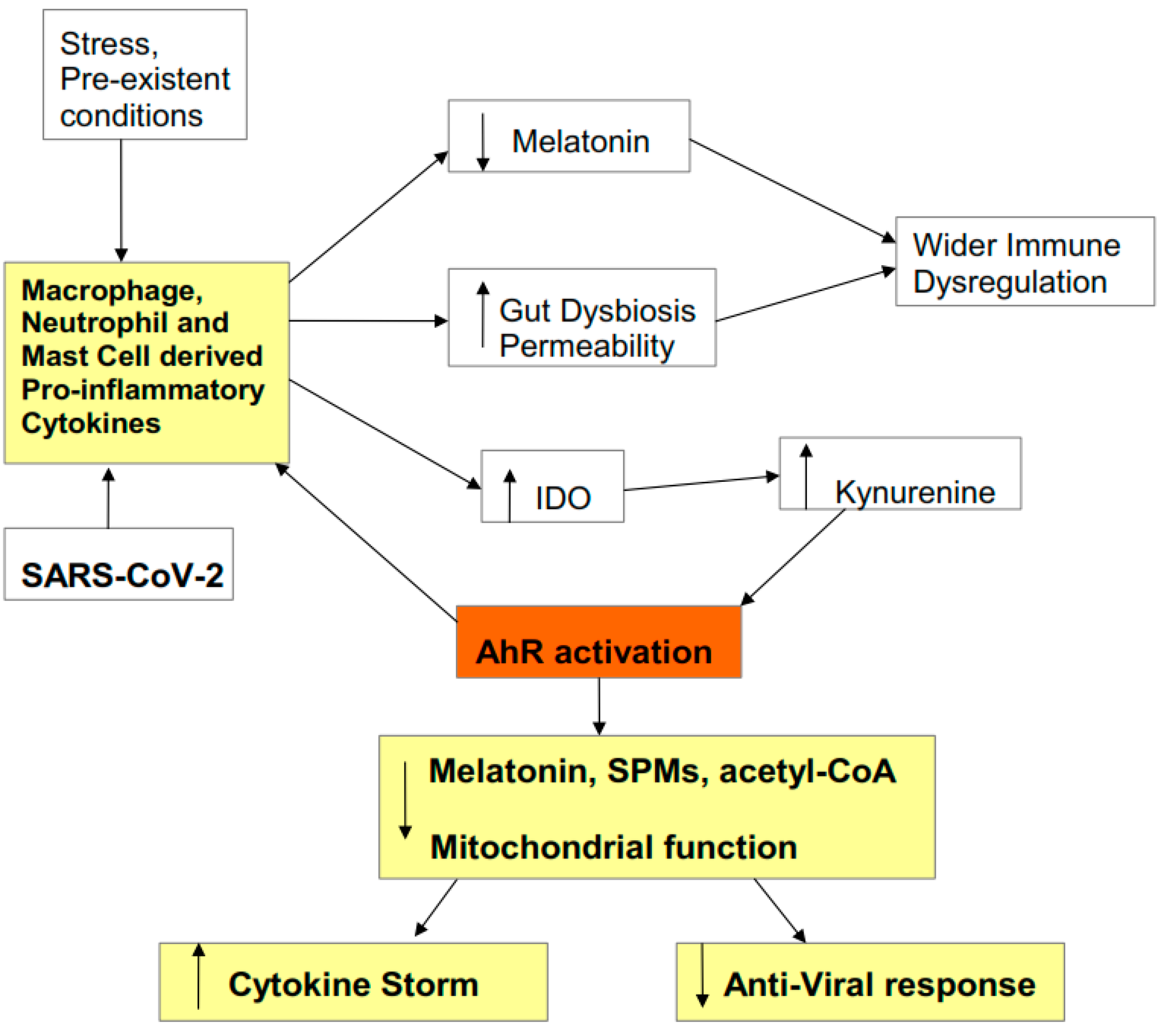
:1. Introduction
The Aryl Hydrocarbon Receptor
2. SARS-CoV-2 Entry and Pathophysiology
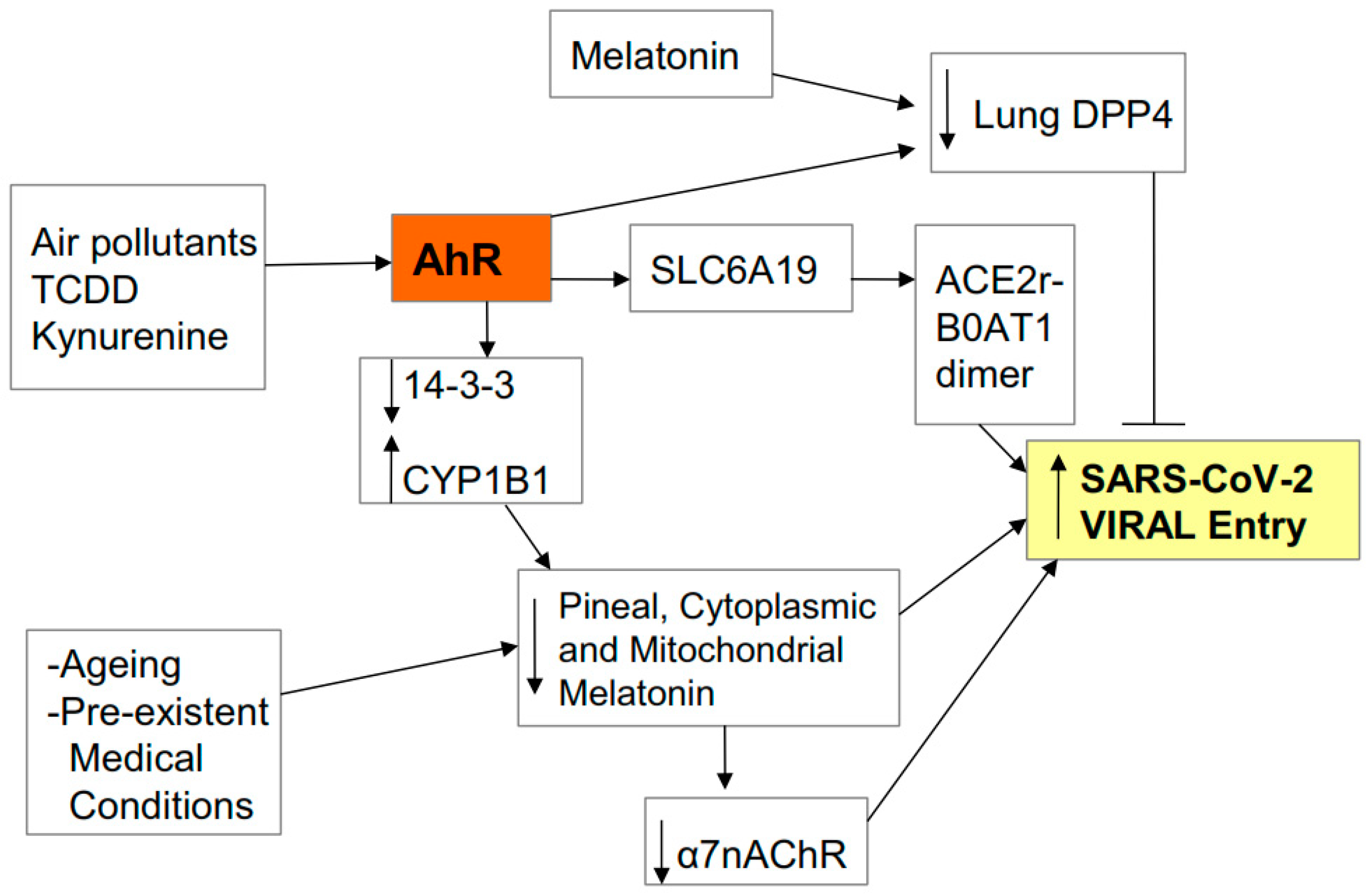
2.1. Entry
2.2. Pathophysiology
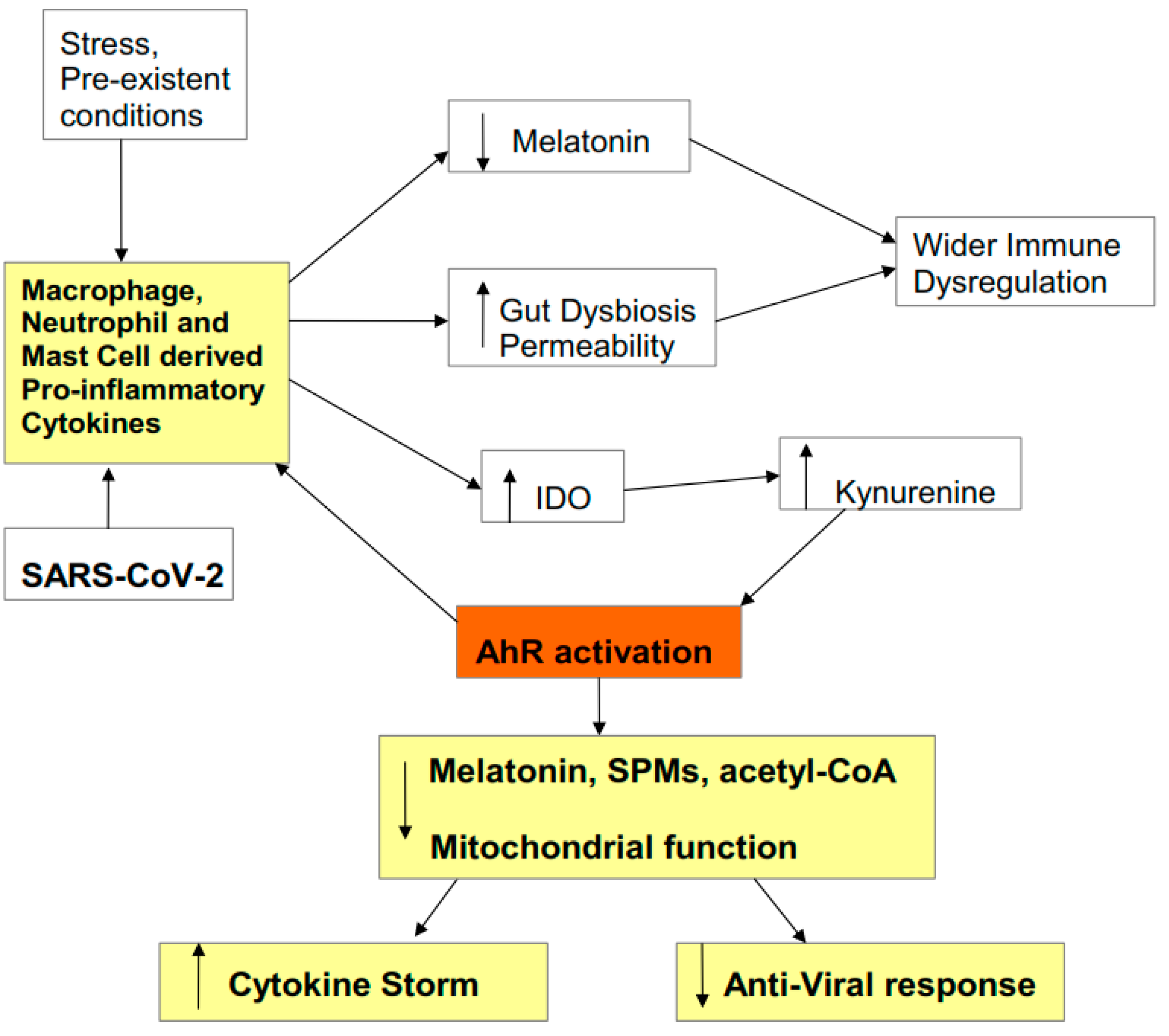
2.3. Cytokine Storm Consequences
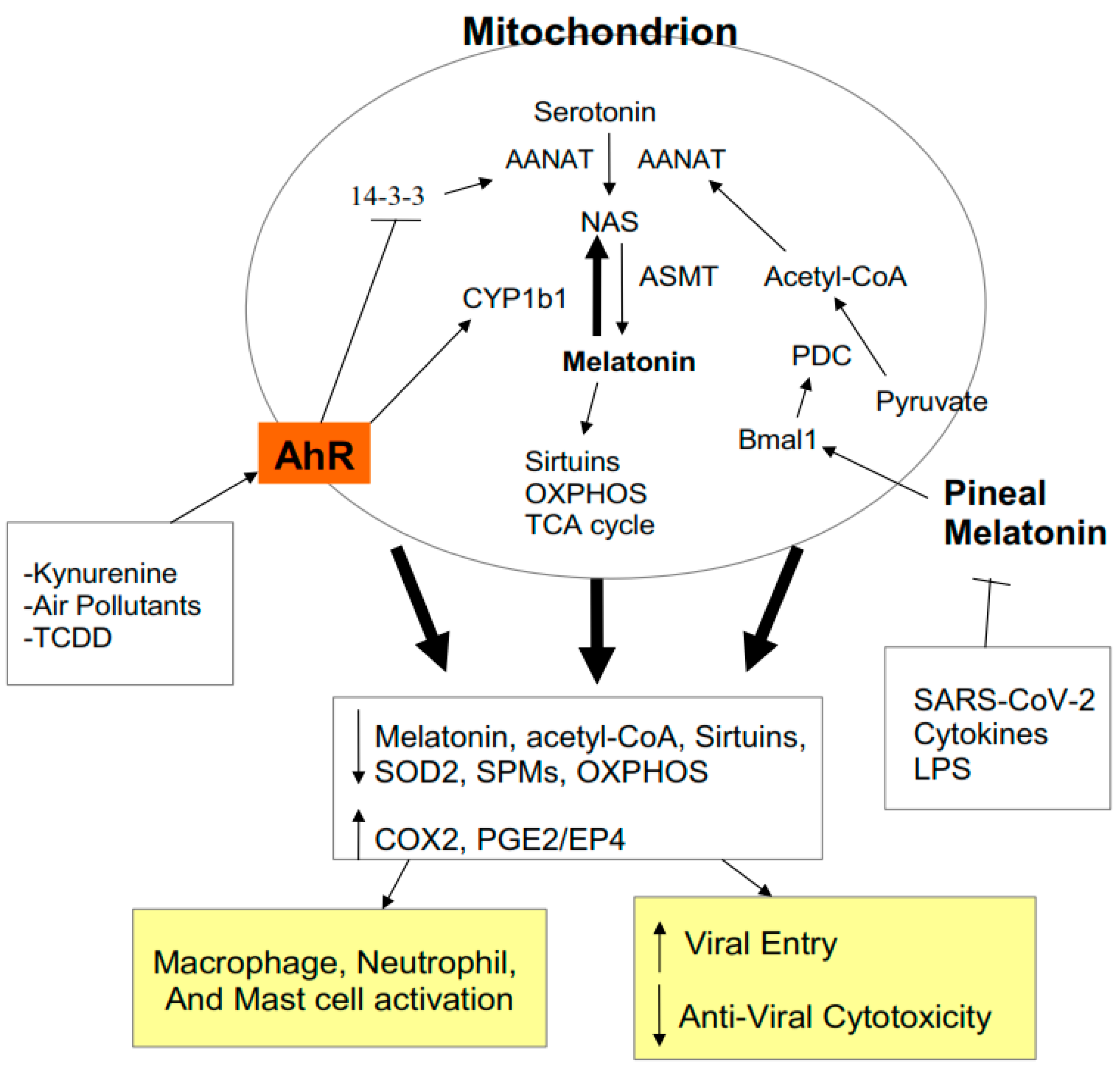
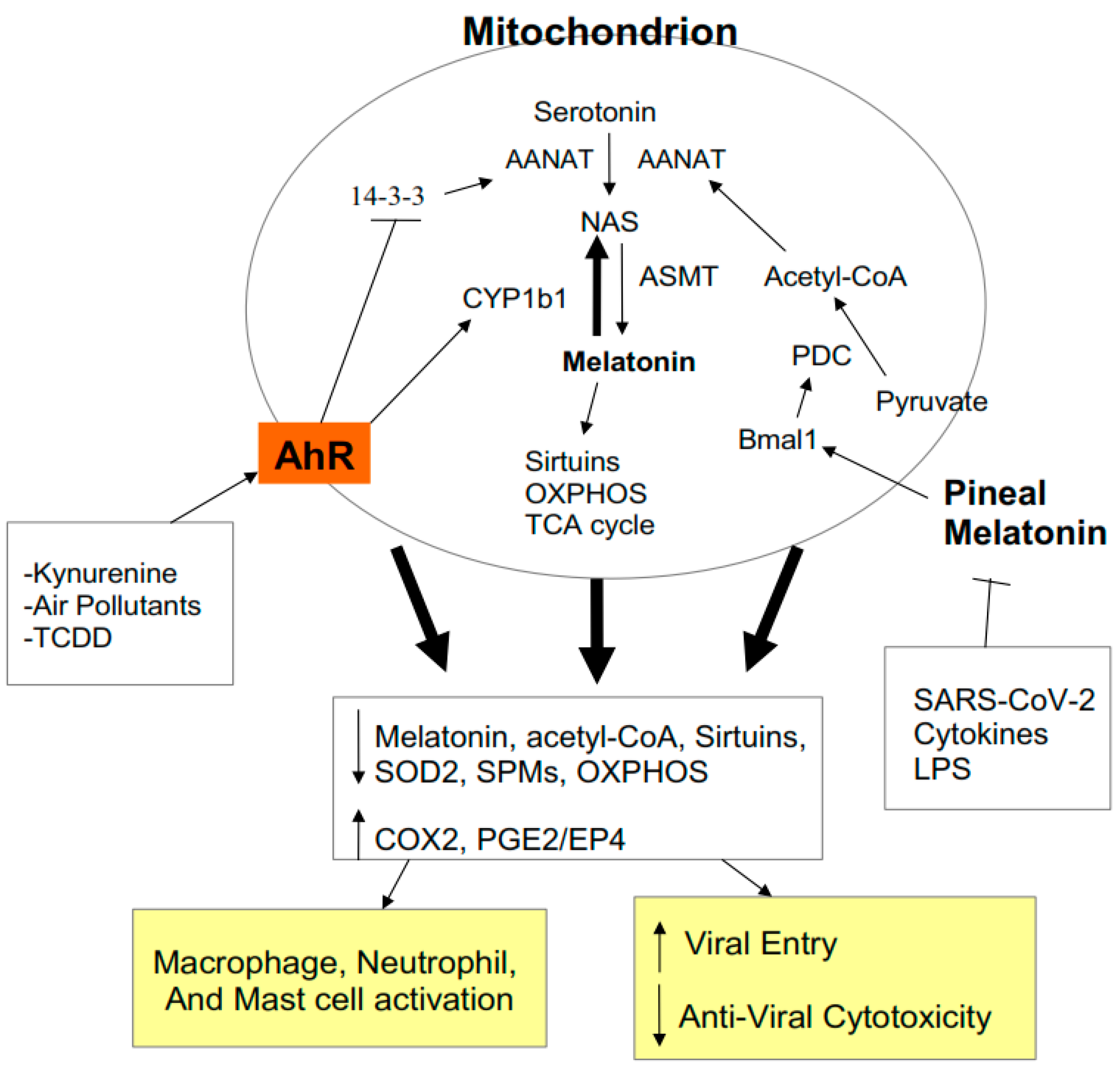
2.4. AhR Regulation of Mitochondrial Metabolism
2.5. AhR and Pre-Existing High-Risk COVID-19 Medical Conditions
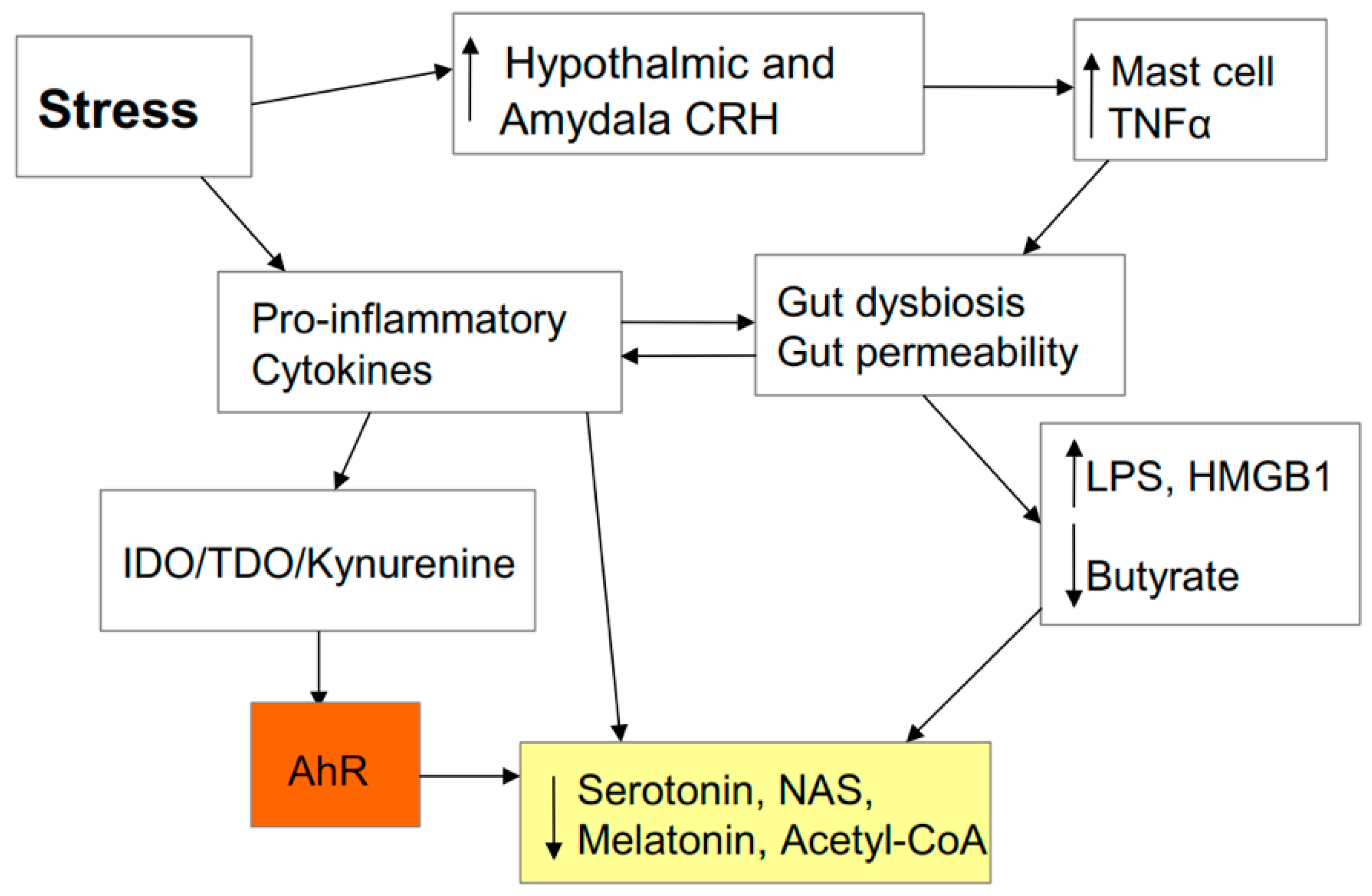
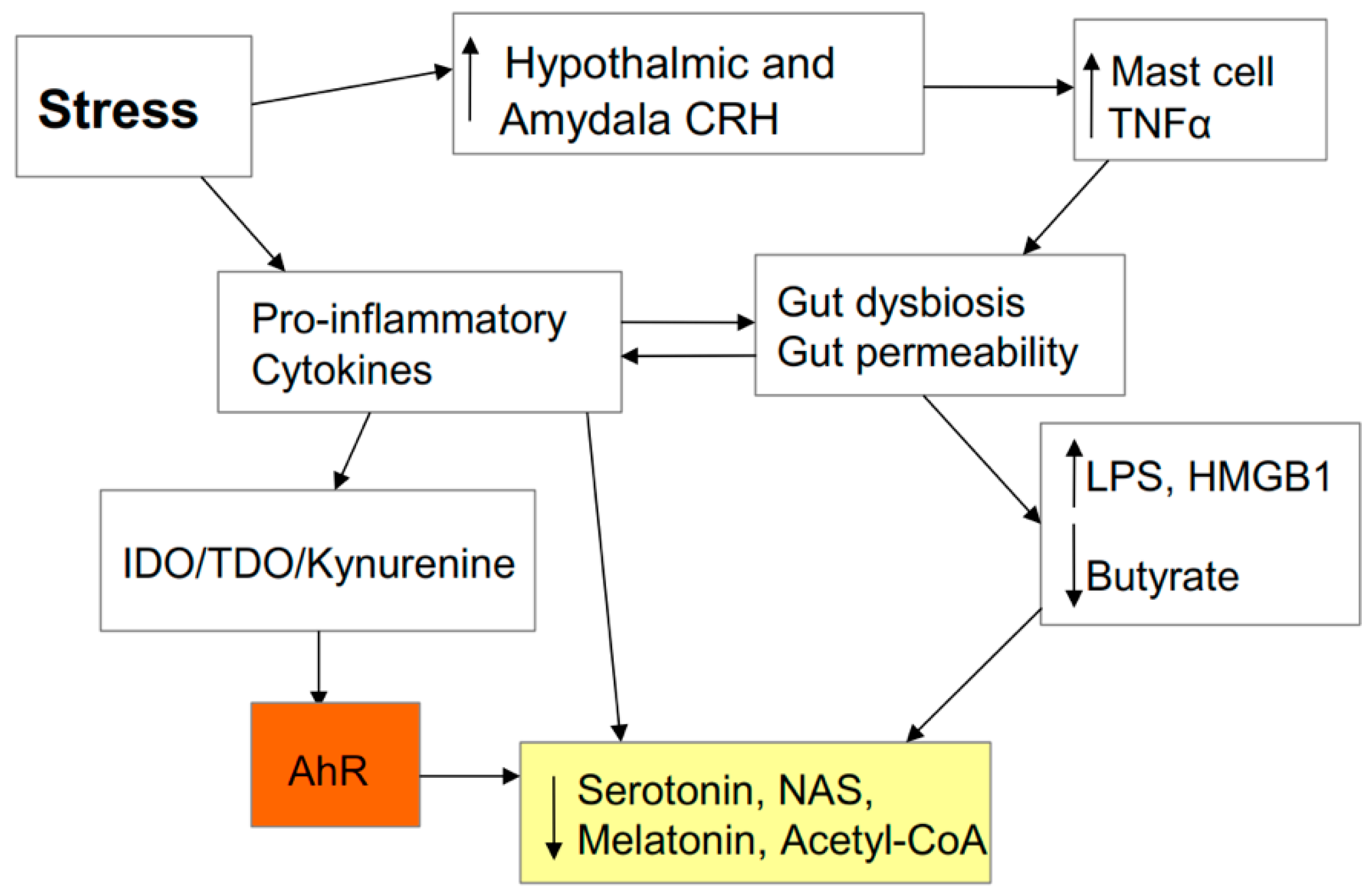
2.6. AhR and Stress
2.7. Stress and the Gut
3. AhR and Wider COVID-19 Pathophysiology
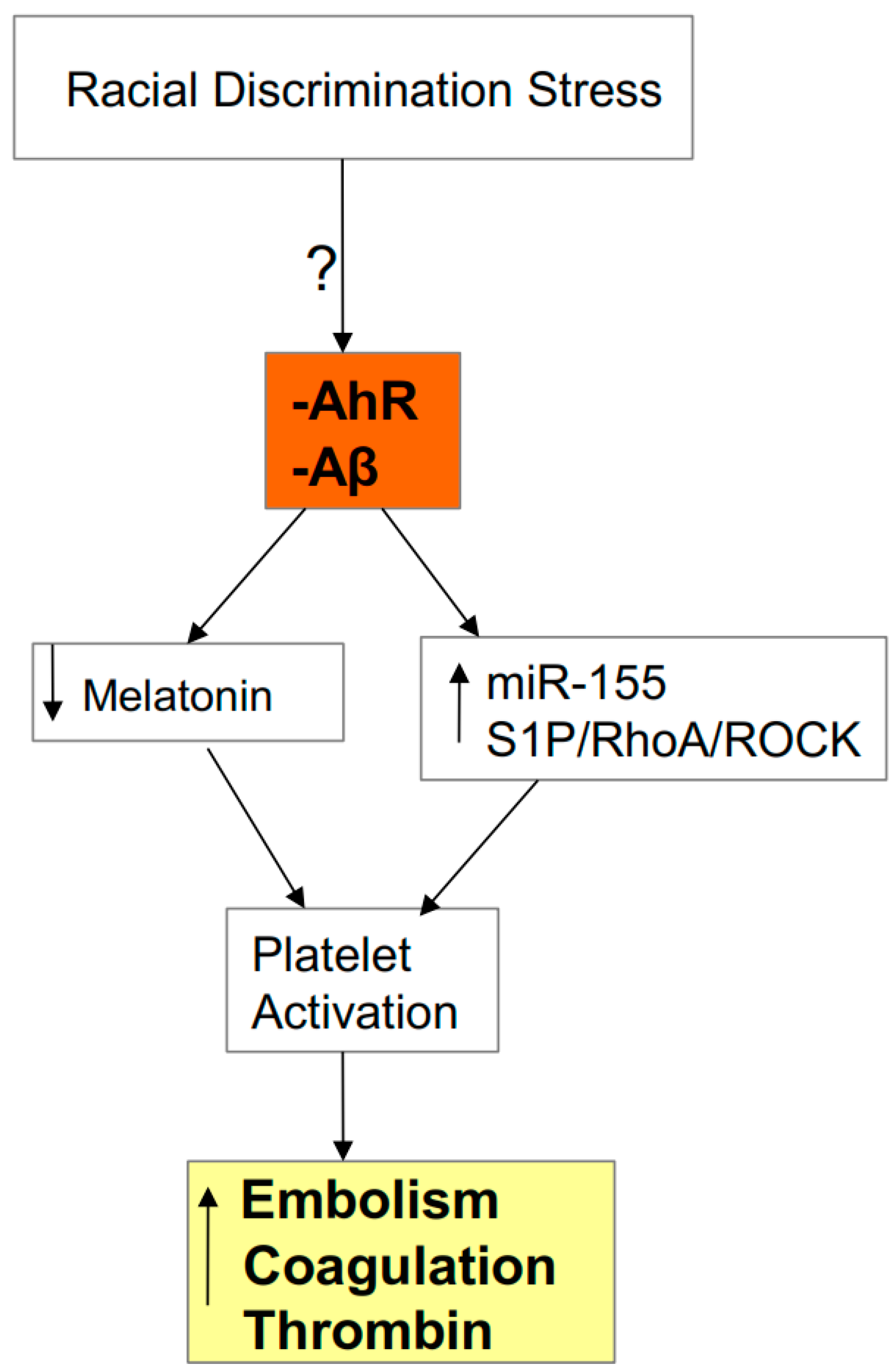
3.1. AhR, Platelets, ROCK, and SARS-CoV-2 Severity/Fatality
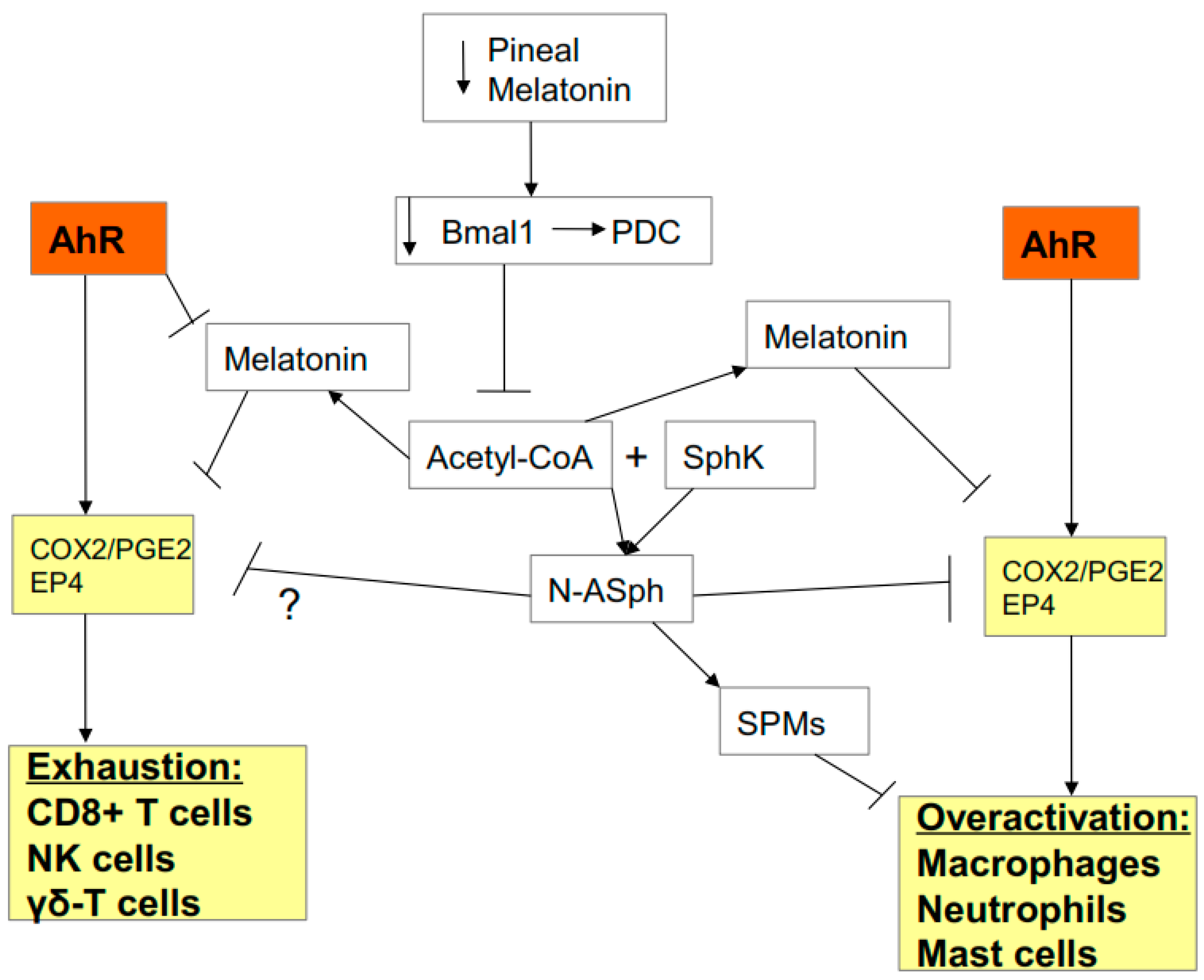
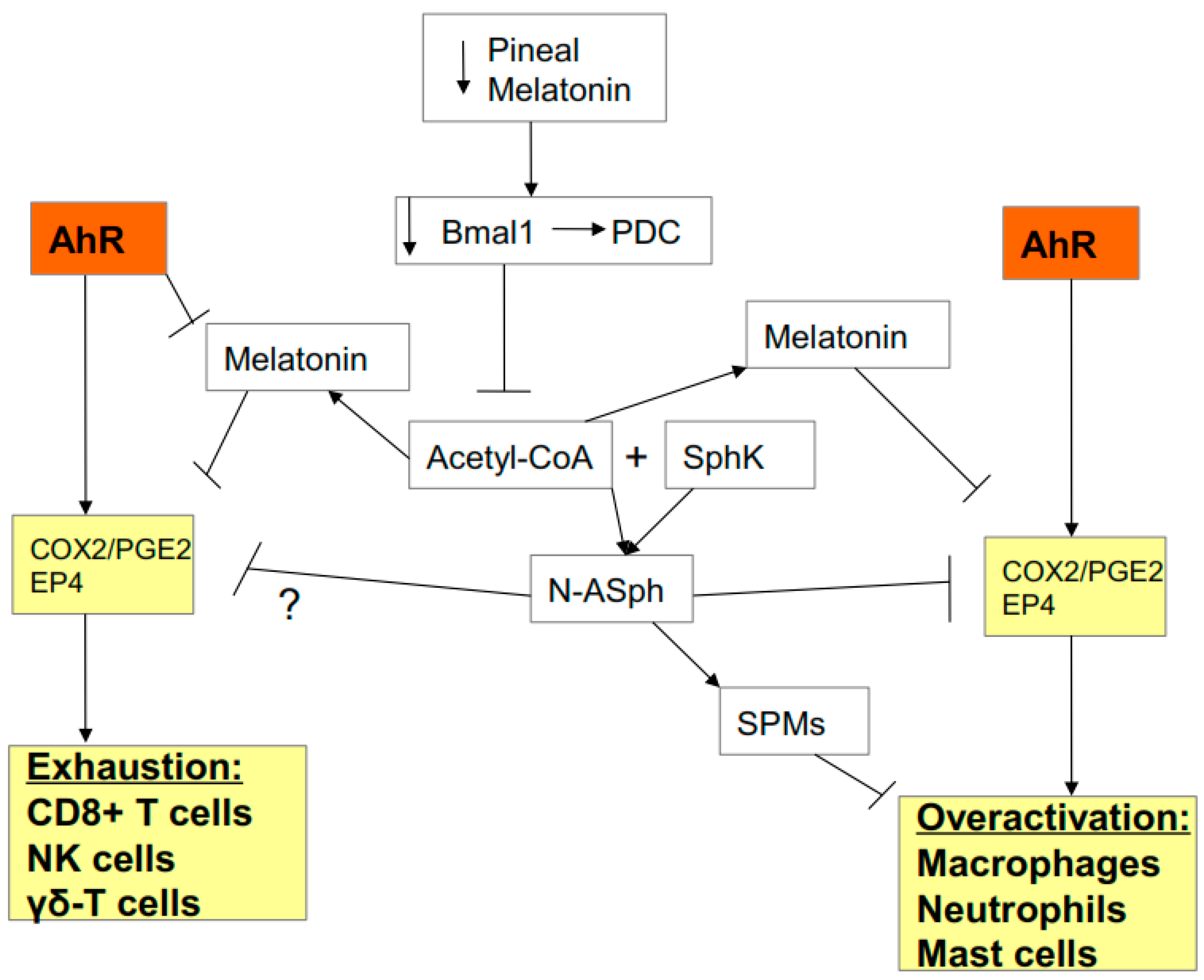
3.2. AhR, Acetyl-CoA, COX2, and Specialized Pro-Resolving Mediators (SPMs)
3.3. AhR, COX2, SPMs, Acetyl-CoA, and miR-155
3.4. Gut Dysbiosis: Interactions with Acetyl-CoA, COX2, SPMs, and AhR
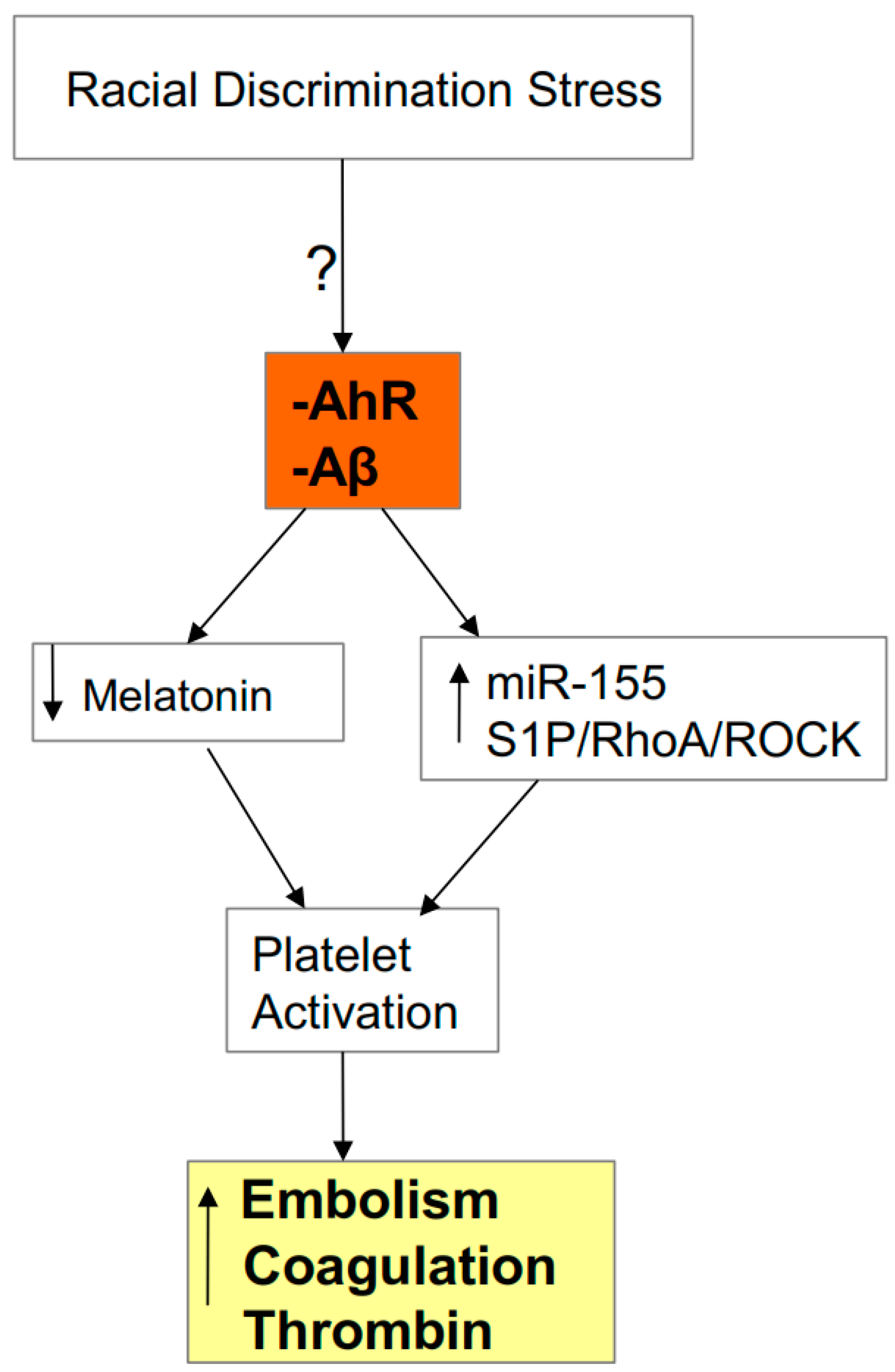
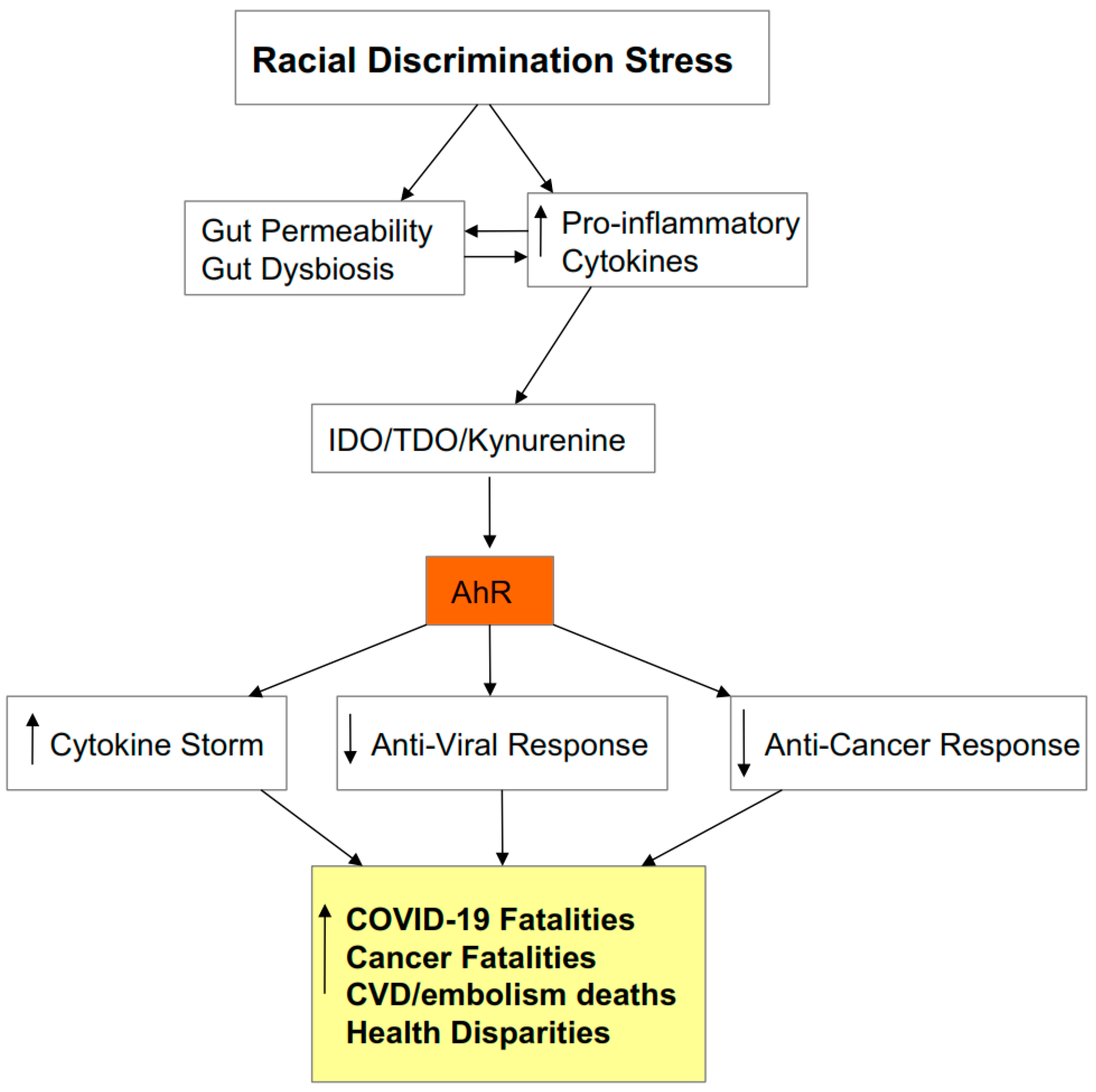
4. Integrating Racial Discrimination into SARS-CoV-2 Pathophysiology
5. Treatment Implications
5.1. AhR Antagonists
5.1.1. Vitamin B12 and Folic Acid
5.1.2. Green Tea Polyphenols
5.2. Melatonin
5.3. ROCK Inhibitors
5.4. IDO Inhibitors
5.5. Nimesulide
5.6. Prophylaxis
5.7. Treatment
6. Future Research
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| α7nAChR | alpha 7 nicotinic acetylcholine receptor |
| Aβ | amyloid-beta |
| AANAT | aralkylamine N-acetyltransferase |
| ACC | acetyl-CoA carboxylase |
| ACE2r | angiotensin-converting enzyme 2 receptor |
| acetyl-CoA | acetyl-coenzyme A |
| AhR | aryl hydrocarbon receptor |
| BAME | Black Asian and Minority Ethnic |
| CD8+ | cluster of differentiation 8 |
| COVID-19 | coronavirus disease-19 |
| COX2 | cyclooxygenase 2 |
| CRH | corticotropin-releasing hormone |
| CVD | cardiovascular disease |
| CYP | cytochdrome P450 |
| DPP-4 | Dipeptidyl peptidase 4 |
| EGCG | epigallocatechin gallate |
| EP4 | prostaglandin E2 receptor 4 |
| HMGB | high-mobility group box |
| IDO | indoleamine 2,3-dioxygenase |
| IFN | interferons |
| IL | interleukin |
| LPS | lipopolysaccharide |
| MDSC | myeloid-derived suppressor cells |
| N-ASph | N-acetylsphingosine |
| NAS | N-acetylserotonin |
| NK | natural killer |
| OXPHOS | oxidative phosphorylation |
| PDC | pyruvate dyhydroganse complex |
| PDK | pyruvate dehydrogenase kinase |
| PGE2 | prostaglandin E2 |
| ROCK | RhoA-associated kinase |
| S1P | sphingosine-1-phosphate |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome associatedcoronavirus |
| SNP | single nucleotide polymorphism |
| SOD | superoxide dismutase |
| SphK | sphingosine kinase |
| SPM | specialized pro-resolving mediators |
| TCA | tricarboxylic acid |
| TCDD | 2,3,7,8-tetrachlorodibenzo-p-dioxin |
| TDO | tryptophan 2,3-dioxygenase |
| TLR | toll-like receptor |
| TNF | tumor necrosis factor |
References
- Kloc, M.; Ghobrial, R.M.; Kubiak, J.Z. How nicotine can inhibit cytokine storm in the lungs and prevent or lessen the severity of COVID-19 infection? Immunol. Lett. 2020, 224, 28–29. [Google Scholar] [CrossRef] [PubMed]
- Potter, H.; Boyd, T.D.; Clarke, P.; Pelak, V.S.; Tyler, K.L. Recruiting the innate immune system with GM-CSF to fight viral diseases, including West Nile Virus encephalitis and COVID-19. F1000Research 2020, 9, 345. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S. Focusing On a Unique Innate Memory Cell Population of Natural Killer Cells in the Fight Against COVID-19: Harnessing The Ubiquity Of Cytomegalovirus Exposure. Mediterr. J. Hematol. Infect. Dis. 2020, 12, e2020047. [Google Scholar] [CrossRef] [PubMed]
- Fierabracci, A.; Arena, A.; Rossi, P. COVID-19: A Review on Diagnosis, Treatment, and Prophylaxis. Int. J. Mol. Sci. 2020, 21, 5145. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.; Reiter, R.J. COVID-19 pathophysiology: Interactions of gut microbiome, melatonin, vitamin D, stress, kynurenine and the alpha 7 nicotinic receptor: Treatment implications. Melatonin Res. 2020, 3, 322–345. [Google Scholar] [CrossRef]
- Küster, O.C.; Laptinskaya, D.; Fissler, P.; Schnack, C.; Zügel, M.; Nold, V.; Thurm, F.; Pleiner, S.; Karabatsiakis, A.; Von Einem, B.; et al. Novel Blood-Based Biomarkers of Cognition, Stress, and Physical or Cognitive Training in Older Adults at Risk of Dementia: Preliminary Evidence for a Role of BDNF, Irisin, and the Kynurenine Pathway. J. Alzheimer’s Dis. 2017, 59, 1097–1111. [Google Scholar] [CrossRef]
- Nduhirabandi, F.; Du Toit, E.F.; Lochner, A. Melatonin and the metabolic syndrome: A tool for effective therapy in obesity-associated abnormalities? Acta Physiol. 2012, 205, 209–223. [Google Scholar] [CrossRef]
- Zhao, Y.; Fu, Y.; Sun, Y.; Zou, M.; Peng, X. Transcriptional Regulation of gga-miR-451 by AhR: Arnt in Mycoplasma gallisepticum (HS Strain) Infection. Int. J. Mol. Sci. 2019, 20, 3087. [Google Scholar] [CrossRef] [Green Version]
- Raisi-Estabragh, Z.; McCracken, C.; Bethell, M.S.; Cooper, J.; Cooper, C.; Caulfield, M.J.; Munroe, P.B.; Harvey, N.C.; Petersen, S.E. Greater risk of severe COVID-19 in Black, Asian and Minority Ethnic populations is not explained by cardiometabolic, socioeconomic or behavioural factors, or by 25(OH)-vitamin D status: Study of 1326 cases from the UK Biobank. J. Public Health 2020, 42. [Google Scholar] [CrossRef]
- Yan, C.; Luo, Z.; Li, W.; Li, X.; Dallmann, R.; Kurihara, H.; Li, Y.; He, R.-R. Disturbed Yin–Yang balance: Stress increases the susceptibility to primary and recurrent infections of herpes simplex virus type 1. Acta Pharm. Sin. B 2020, 10, 383–398. [Google Scholar] [CrossRef]
- Cohen, S.; Tyrrell, D.A.; Smith, A.P. Psychological Stress and Susceptibility to the Common Cold. N. Eng. J. Med. 1991, 325, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Scalise, M.; Indiveri, C. Repurposing Nimesulide, a Potent Inhibitor of the B0AT1 Subunit of the SARS-CoV-2 Receptor, as a Therapeutic Adjuvant of COVID-19. SLAS Discov. Adv. Sci. Drug Discov. 2020, 2472555220934421. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javed, K.; Cheng, Q.; Carroll, A.J.; Truong, T.T.; Bröer, S. Development of Biomarkers for Inhibition of SLC6A19 (B0AT1)—A Potential Target to Treat Metabolic Disorders. Int. J. Mol. Sci. 2018, 19, 3597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, W.; Fu, H.; Xu, T.; Xu, S.L.; Guo, Z.; Tian, J.; Tao, W.; Xie, H.Q.; Zhao, B. SLC6A19 is a novel putative gene, induced by dioxins via AhR in human hepatoma HepG2 cells. Environ. Pollut. 2018, 237, 508–514. [Google Scholar] [CrossRef]
- Wang, Y.; Han, D.; Zhou, T.; Zhang, J.; Liu, C.; Cao, F.; Dong, N. Melatonin ameliorates aortic valve calcification via the regulation of circular RNA CircRIC3/miR-204-5p/DPP4 signaling in valvular interstitial cells. J. Pineal Res. 2020, 69, 12666. [Google Scholar] [CrossRef]
- Lv, D.; Xu, Y.; Cheng, H.; Ke, Y.; Zhang, X.; Ying, K. A novel cell-based assay for dynamically detecting neutrophil extracellular traps-induced lung epithelial injuries. Exp. Cell Res. 2020, 394, 112101. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, X.; Dong, W.; Fang, Y.; Lv, J.; Zhang, T.; Fiskesund, R.; Xie, J.; Liu, J.; Yin, X.; et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8+ T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell 2018, 33, 480–494. [Google Scholar] [CrossRef] [Green Version]
- Reiter, R.J.; Sharma, R.; Ma, Q.; Rorsales-Corral, S.; Chuffa, L.G.D.A. Melatonin inhibits Warburg-dependent cancer by redirecting glucose oxidation to the mitochondria: A mechanistic hypothesis. Cell. Mol. Life Sci. 2020, 77, 2527–2542. [Google Scholar] [CrossRef]
- Anderson, G.; Rodriguez, M.; Reiter, R.J. Multiple Sclerosis: Melatonin, Orexin, and Ceramide Interact with Platelet Activation Coagulation Factors and Gut-Microbiome-Derived Butyrate in the Circadian Dysregulation of Mitochondria in Glia and Immune Cells. Int. J. Mol. Sci. 2019, 20, 5500. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.; Wang, H.; Xu, S.; Zhuang, Y.; An, J.; Su, C.; Xia, Y.; Chen, J.; Xu, Z.Z.; Liu, Q.; et al. Alteration in gut microbiota is associated with dysregulation of cytokines and glucocorticoid therapy in systemic lupus erythematosus. Gut Microbes 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Zhang, F.; Lui, G.C.; Yeoh, Y.K.; Li, A.Y.; Zhan, H.; Wan, Y.; Chung, A.; Cheung, C.P.; Chen, N.; et al. Alterations in Gut Microbiota of Patients With COVID-19 During Time of Hospitalization. Gastroenterology 2020. [Google Scholar] [CrossRef] [PubMed]
- Vitale, J.A.; Perazzo, P.; Silingardi, M.; Biffi, M.; Banfi, G.; Negrini, F. Is disruption of sleep quality a consequence of severe Covid-19 infection? A case-series examination. Chronobiol. Int. 2020. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Stefanoni, D.; Reisz, J.A.; Nemkov, T.; Bertolone, L.; Francis, R.O.; Hudson, K.E.; Zimring, J.C.; Hansen, K.C.; Hod, E.A.; et al. COVID-19 infection alters kynurenine and fatty acid metabolism, correlating with IL-6 levels and renal status. JCI Insight 2020, 5, 140327. [Google Scholar] [CrossRef]
- Anderson, G.; Maes, M. Mitochondria and immunity in chronic fatigue syndrome. Prog. Neuro-Psychopharmacol. Boil. Psychiatry 2020, 103, 109976. [Google Scholar] [CrossRef]
- Anderson, G.; Mazzoccoli, G. Left Ventricular Hypertrophy: Roles of Mitochondria CYP1B1 and Melatonergic Pathways in Co-Ordinating Wider Pathophysiology. Int. J. Mol. Sci. 2019, 20, 4068. [Google Scholar] [CrossRef] [Green Version]
- Muxel, S.M.; Lapa, M.; Monteiro, A.W.A.; Cecon, E.; Tamura, E.K.P.; Flöeter-Winter, L.M.; Markus, R.P. NF-κB Drives the Synthesis of Melatonin in RAW 264.7 Macrophages by Inducing the Transcription of the Arylalkylamine-N-Acetyltransferase (AA-NAT) Gene. PLoS ONE 2012, 7, e52010. [Google Scholar] [CrossRef] [Green Version]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 10. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, C.; Highton, A.J.; Peine, S.; Sauter, J.; Schmidt, A.H.; Bunders, M.J.; Altfeld, M.; Körner, C. Natural Killer Cell Education Is Associated With a Distinct Glycolytic Profile. Front. Immunol. 2018, 9, 3020. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Jiang, M.; Chen, X.; Montaner, L.J. Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: Review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J. Leukoc. Boil. 2020, 108, 17–41. [Google Scholar] [CrossRef]
- Reiter, R.J.; Sharma, R.; Ma, Q.; Rosales-Corral, S.; Acuna-Castroviejo, D.; Escames, G. Inhibition of mitochondrial pyruvate dehydrogenase kinase: A proposed mechanism by which melatonin causes cancer cells to overcome cytosolic glycolysis, reduce tumor biomass and reverse insensitivity to chemotherapy. Melatonin Res. 2019, 2, 105–119. [Google Scholar] [CrossRef]
- Anderson, G. Daytime orexin and night-time melatonin regulation of mitochondria melatonin: Roles in circadian oscillations systemically and centrally in breast cancer symptomatology. Melatonin Res. 2019, 2, 1–8. [Google Scholar] [CrossRef]
- Acuña-Castroviejo, D.; Rahim, I.; Acuña-Fernández, C.; Ortiz, F.; Solera-Marín, J.; Sayed, R.K.A.; Díaz-Casado, M.E.; Rusanova, I.; López, L.C.; Escames, G. Melatonin, clock genes and mitochondria in sepsis. Cell. Mol. Life Sci. 2017, 74, 3965–3987. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Correia, M.A. Heme: A Regulator of Rat Hepatic Tryptophan 2,3-Dioxygenase? Arch. Biochem. Biophys. 2000, 377, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Michels, N.; Clarke, G.; Olavarría-Ramírez, L.; Gomez-Martinez, S.; Díaz-Prieto, L.; Marcos, A.; Widhalm, K.; Carvalho, L.A. Psychosocial stress and inflammation driving tryptophan breakdown in children and adolescents: A cross-sectional analysis of two cohorts. Psychoneuroendocrinology 2018, 94, 104–111. [Google Scholar] [CrossRef]
- Chiappelli, J.; Rowland, L.M.; Notarangelo, F.M.; Wijtenburg, S.A.; Thomas, M.A.R.; Pocivavsek, A.; Jones, A.; Wisner, K.; Kochunov, P.; Schwarcz, R.; et al. Salivary kynurenic acid response to psychological stress: Inverse relationship to cortical glutamate in schizophrenia. Neuropsychopharmacology 2018, 43, 1706–1711. [Google Scholar] [CrossRef]
- Bettison, T.M.; Nahm, C.B.; Gill, A.J.; Mittal, A.; Malhi, G.S.; Samra, J.S. Understanding the Pathophysiology of Psychological Distress and Pancreatic Cancer. Pancreas 2018, 47, 376–381. [Google Scholar] [CrossRef]
- Anderson, G.; Maes, M. Interactions of Tryptophan and Its Catabolites with Melatonin and the Alpha 7 Nicotinic Receptor in Central Nervous System and Psychiatric Disorders: Role of the Aryl Hydrocarbon Receptor and Direct Mitochondria Regulation. Int. J. Tryptophan Res. 2017, 10. [Google Scholar] [CrossRef]
- Sakurai, M.; Yamamoto, Y.; Kanayama, N.; Hasegawa, M.; Mouri, A.; Takemura, M.; Matsunami, H.; Miyauchi, T.; Tokura, T.; Kimura, H.; et al. Serum Metabolic Profiles of the Tryptophan-Kynurenine Pathway in the high risk subjects of major depressive disorder. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Anderson, G. Linking the biological underpinnings of depression: Role of mitochondria interactions with melatonin, inflammation, sirtuins, tryptophan catabolites, DNA repair and oxidative and nitrosative stress, with consequences for classification and cognition. Prog. Neuro-Psychopharmacol. Boil. Psychiatry 2018, 80, 255–266. [Google Scholar] [CrossRef]
- Evrensel, A.; Ünsalver, B.Ö.; Ceylan, M.E. Immune-Kynurenine Pathways and the Gut Microbiota-Brain Axis in Anxiety Disorders. Adv. Exp. Med. Biol. 2020, 1191, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Petrozzi, B.P.; ElYamany, O.; Rummel, C.; Mulert, C. Effects of inflammation on the kynurenine pathway in schizophrenia—A systematic review. J. Neuroinflamm. 2020, 17, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G. Neuronal–immune interactions in mediating stress effects in the etiology and course of schizophrenia: Role of the amygdala in developmental co-ordination. Med. Hypotheses 2011, 76, 54–60. [Google Scholar] [CrossRef]
- Anderson, G.; Maes, M. Gut Dysbiosis Dysregulates Central and Systemic Homeostasis via Suboptimal Mitochondrial Function: Assessment, Treatment and Classification Implications. Curr. Top. Med. Chem. 2020, 20, 524–539. [Google Scholar] [CrossRef] [PubMed]
- Vanuytsel, T.; Van Wanrooy, S.; Vanheel, H.; Vanormelingen, C.; Verschueren, S.; Houben, E.; Rasoel, S.S.; Toth, J.; Holvoet, L.; Farré, R.; et al. Psychological stress and corticotropin-releasing hormone increase intestinal permeability in humans by a mast cell-dependent mechanism. Gut 2014, 63, 1293–1299. [Google Scholar] [CrossRef] [PubMed]
- Martín-Hernández, D.; Tendilla-Beltran, H.; Madrigal, J.L.M.; García-Bueno, B.; Leza, J.C.; Caso, J.R. Chronic Mild Stress Alters Kynurenine Pathways Changing the Glutamate Neurotransmission in Frontal Cortex of Rats. Mol. Neurobiol. 2019, 56, 490–501. [Google Scholar] [CrossRef]
- Fuertig, R.; Azzinnari, D.; Bergamini, G.; Cathomas, F.; Sigrist, H.; Seifritz, E.; Vavassori, S.; Luippold, A.; Hengerer, B.; Ceci, A.; et al. Mouse chronic social stress increases blood and brain kynurenine pathway activity and fear behaviour: Both effects are reversed by inhibition of indoleamine 2,3-dioxygenase. Brain Behav. Immun. 2016, 54, 59–72. [Google Scholar] [CrossRef] [Green Version]
- Nold, V.; Sweatman, C.; Karabatsiakis, A.; Böck, C.; Bretschneider, T.; Lawless, N.; Fundel-Clemens, K.; Kolassa, I.-T.; Allers, K.; Catherine, S. Activation of the kynurenine pathway and mitochondrial respiration to face allostatic load in a double-hit model of stress. Psychoneuroendocrinology 2019, 107, 148–159. [Google Scholar] [CrossRef]
- Wang, B.; Lian, Y.-J.; Su, W.-J.; Peng, W.; Dong, X.; Liu, L.-L.; Gong, H.-Y.; Zhang, T.; Jiang, C.-L.; Wang, Y.-X. HMGB1 mediates depressive behavior induced by chronic stress through activating the kynurenine pathway. Brain Behav. Immun. 2018, 72, 51–60. [Google Scholar] [CrossRef]
- Marin, I.A.; Goertz, J.; Ren, T.; Rich, S.S.; Onengut-Gumuscu, S.; Farber, E.; Wu, M.; Overall, C.C.; Kipnis, J.; Gaultier, A. Microbiota alteration is associated with the development of stress-induced despair behavior. Sci. Rep. 2017, 7, 43859. [Google Scholar] [CrossRef]
- Bompard, F.; Monnier, H.; Saab, I.; Tordjman, M.; Abdoul, H.; Fournier, L.; Sanchez, O.; Lorut, C.; Chassagnon, G.; Revel, M.-P. Pulmonary embolism in patients with COVID-19 pneumonia. Eur. Respir. J. 2020, 56, 2001365. [Google Scholar] [CrossRef] [PubMed]
- Pombo, M.; Lame, M.W.; Walker, N.J.; Huynh, D.H.; Tablin, F. TCDD and omeprazole prime platelets through the aryl hydrocarbon receptor (AhR) non-genomic pathway. Toxicol. Lett. 2015, 235, 28–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-C.; Tsai, C.-F.; Chuang, H.-L.; Chang, Y.-C.; Chen, H.-S.; Lee, J.-N.; Tsai, E.-M. Benzyl butyl phthalate promotes breast cancer stem cell expansion via SPHK1/S1P/S1PR3 signaling. Oncotarget 2016, 7, 29563–29576. [Google Scholar] [CrossRef]
- Wang, H.-C.; Wong, T.-H.; Wang, L.-T.; Su, H.-H.; Yu, H.-Y.; Wu, A.-H.; Lin, Y.-C.; Chen, H.-L.; Suen, J.-L.; Hsu, S.-H.; et al. Aryl hydrocarbon receptor signaling promotes ORMDL3-dependent generation of sphingosine-1-phosphate by inhibiting sphingosine-1-phosphate lyase. Cell. Mol. Immunol. 2019, 16, 783–790. [Google Scholar] [CrossRef]
- Punsawad, C.; Viriyavejakul, P. Expression of sphingosine kinase 1 and sphingosine 1-phosphate receptor 3 in malaria-associated acute lung injury/acute respiratory distress syndrome in a mouse model. PLoS ONE 2019, 14, e0222098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Shi, L.; Ma, X.; Su, H.; Ma, G.; Wu, X.; Ying, K.; Zhang, R. RhoA-Rho associated kinase signaling leads to renin-angiotensin system imbalance and angiotensin converting enzyme 2 has a protective role in acute pulmonary embolism. Thromb. Res. 2019, 176, 85–94. [Google Scholar] [CrossRef]
- Lu, Y.; Lian, Z.; Yang, H.; Jiang, Q.; Zou, Y.; Zhu, Y.; Ling, W.; Yuan, L.; Jiang, X.; Chen, S. TNF-α activates RhoA/ROCK signaling pathway and increases permeability of endothelial cells infected with Listeria monocytogenes. Chin. J. Cell. Infect. Mol. Immunol. 2020, 36, 193–197. [Google Scholar]
- Sonkar, V.K.; Kulkarni, P.; Dash, D. Amyloid β peptide stimulates platelet activation through RhoA-dependent modulation of actomyosin organization. FASEB J. 2014, 28, 1819–1829. [Google Scholar] [CrossRef]
- Lee, J.Y.; Han, S.H.; Park, M.H.; Song, I.-S.; Choi, M.-K.; Yu, E.; Park, C.-M.; Kim, H.J.; Kim, S.H.; Schuchman, E.H.; et al. N-AS-triggered SPMs are direct regulators of microglia in a model of Alzheimer’s disease. Nat. Commun. 2020, 11, 1–19. [Google Scholar] [CrossRef]
- Zhu, P.; Yu, H.; Zhou, K.; Bai, Y.; Qi, R.-Q.; Zhang, S.-G. 3,3′-Diindolylmethane modulates aryl hydrocarbon receptor of esophageal squamous cell carcinoma to reverse epithelial-mesenchymal transition through repressing RhoA/ROCK1-mediated COX2/PGE2 pathway. J. Exp. Clin. Cancer Res. 2020, 39, 1–18. [Google Scholar] [CrossRef]
- Cheng, C.-I.; Chen, P.-H.; Lin, Y.-C.; Kao, Y.-H. High glucose activates Raw264.7 macrophages through RhoA kinase-mediated signaling pathway. Cell. Signal. 2015, 27, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Gdula-Argasińska, J.; Czepiel, J.; Totoń-Żurańska, J.; Jurczyszyn, A.; Wołkow, P.P.; Librowski, T.; Perucki, W. Resolvin D1 down-regulates CYP1A1 and PTGS2 gene in the HUVEC cells treated with benzo(a)pyrene. Pharmacol. Rep. 2016, 68, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.A.; Kado, S.Y.; Kobayashi, R.; Liu, X.; Wong, P.; Na, K.; Durbin, T.; Okamoto, R.A.; Kado, N.Y. Inflammatory marker and aryl hydrocarbon receptor-dependent responses in human macrophages exposed to emissions from biodiesel fuels. Chemosphere 2019, 220, 993–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castañeda, A.R.; Pinkerton, K.E.; Bein, K.J.; Magaña-Méndez, A.; Yang, H.T.; Ashwood, P.; Vogel, C.F. Ambient particulate matter activates the aryl hydrocarbon receptor in dendritic cells and enhances Th17 polarization. Toxicol. Lett. 2018, 292, 85–96. [Google Scholar] [CrossRef]
- Zavan, B.; De Almeida, E.M.; Salles, É.D.S.L.; Amarante-Paffaro, A.M.D.; Paffaro, V.A. COX-2 plays a role in angiogenic DBA + uNK cell subsets activation and pregnancy protection in LPS-exposed mice. Placenta 2016, 44, 34–45. [Google Scholar] [CrossRef]
- Mitchell, J.A.; Shala, F.; Elghazouli, Y.; Warner, T.D.; Gaston-Massuet, C.; Crescente, M.; Armstrong, P.C.; Herschman, H.R.; Kirkby, N.S. Cell-Specific Gene Deletion Reveals the Antithrombotic Function of COX1 and Explains the Vascular COX1/Prostacyclin Paradox. Circ. Res. 2019, 125, 847–854. [Google Scholar] [CrossRef]
- Ma, X.; Holt, D.; Kundu, N.; Reader, J.; Goloubeva, O.; Take, Y.; Fulton, A.M. A prostaglandin E (PGE) receptor EP4 antagonist protects natural killer cells from PGE2-mediated immunosuppression and inhibits breast cancer metastasis. OncoImmunology 2013, 2, e22647. [Google Scholar] [CrossRef] [Green Version]
- Holt, D.; Ma, X.; Kundu, N.; Fulton, A.M. Prostaglandin E2 (PGE2) suppresses natural killer cell function primarily through the PGE2 receptor EP4. Cancer Immunol. Immunother. 2011, 60, 1577–1586. [Google Scholar] [CrossRef] [Green Version]
- Eberstål, S.; Fritzell, S.; Sandén, E.; Visse, E.; Darabi, A.; Siesjö, P. Immunizations with unmodified tumor cells and simultaneous COX-2 inhibition eradicate malignant rat brain tumors and induce a long-lasting CD8+ T cell memory. J. Neuroimmunol. 2014, 274, 161–167. [Google Scholar] [CrossRef]
- Tawfik, D.; Groth, C.; Gundlach, J.-P.; Peipp, M.; Kabelitz, D.; Becker, T.; Oberg, H.-H.; Trauzold, A.; Wesch, D. TRAIL-Receptor 4 Modulates γδ T Cell-Cytotoxicity Toward Cancer Cells. Front. Immunol. 2019, 10, 10. [Google Scholar] [CrossRef]
- Inada, T.; Kubo, K.; Shingu, K. Promotion of interferon-gamma production by natural killer cells via suppression of murine peritoneal macrophage prostaglandin E2 production using intravenous anesthetic propofol. Int. Immunopharmacol. 2010, 10, 1200–1208. [Google Scholar] [CrossRef] [PubMed]
- Haworth, O.; Cernadas, M.; Levy, B.D. NK cells are effectors for resolvin E1 in the timely resolution of allergic airway inflammation. J. Immunol. 2011, 186, 6129–6135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chia, N.; Kumar, R.K.; Foster, P.S.; Herbert, C. Enhanced Pro-Inflammatory Response of Macrophages to Interleukin-33 in an Allergic Environment. Int. Arch. Allergy Immunol. 2018, 176, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Barden, A.; Shinde, S.; Tsai, I.-J.; Croft, K.D.; Beilin, L.; Puddey, I.; Mori, T. Effect of weight loss on neutrophil resolvins in the metabolic syndrome. Prostaglandins Leukot. Essent. Fat. Acids 2019, 148, 25–29. [Google Scholar] [CrossRef]
- Freire, M.; Dalli, J.; Serhan, C.N.; Van Dyke, T.E.; Charles, S.N. Neutrophil Resolvin E1 Receptor Expression and Function in Type 2 Diabetes. J. Immunol. 2017, 198, 718–728. [Google Scholar] [CrossRef] [Green Version]
- Kang, G.-J.; Lee, H.-J.; Kang, Y.P.; Kim, E.J.; Kim, H.J.; Byun, H.J.; Park, M.K.; Cho, H.; Kwon, S.W.; Lee, C.-H. High-mobility group box 1 suppresses resolvin D1-induced phagocytosis via induction of resolvin D1-inactivating enzyme, 15-hydroxyprostaglandin dehydrogenase. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 1981–1988. [Google Scholar] [CrossRef] [Green Version]
- Chiurchiù, V.; Leuti, A.; Saracini, S.; Fontana, D.; Finamore, P.; Giua, R.; Padovini, L.; Incalzi, R.A.; Maccarrone, M. Resolution of inflammation is altered in chronic heart failure and entails a dysfunctional responsiveness of T lymphocytes. FASEB J. 2018, 33, 909–916. [Google Scholar] [CrossRef]
- Li, T.; Liu, J.; Guo, G.; Ning, B.; Li, X.; Zhu, G.; Yang, D.; Moran, T.H.; Smith, W.W. Synphilin-1 Interacts with AMPK and Increases AMPK Phosphorylation. Int. J. Mol. Sci. 2020, 21, 4352. [Google Scholar] [CrossRef]
- Elesela, S.; Morris, S.B.; Narayanan, S.; Kumar, S.; Lombard, D.B.; Lukacs, N.W. Sirtuin 1 regulates mitochondrial function and immune homeostasis in respiratory syncytial virus infected dendritic cells. PLoS Pathog. 2020, 16, e1008319. [Google Scholar] [CrossRef]
- Mills, C.A.; Trub, A.G.; Hirschey, M.D. Sensing Mitochondrial Acetyl-CoA to Tune Respiration. Trends Endocrinol. Metab. 2018, 30, 1–3. [Google Scholar] [CrossRef]
- Currais, A.; Huang, L.; Goldberg, J.; Petrascheck, M.; Ates, G.; Pinto-Duarte, A.; Shokhirev, M.N.; Schubert, D.; Maher, P. Elevating acetyl-CoA levels reduces aspects of brain aging. Elife 2019, 8, e47866. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.-X.; Zeng, D.-Y.; Li, R.-T.; Pang, R.-P.; Yang, H.; Hu, Y.-L.; Zhang, Q.; Jiang, Y.; Huang, L.-Y.; Tang, Y.-B.; et al. Essential Role of MicroRNA-155 in Regulating Endothelium-Dependent Vasorelaxation by Targeting Endothelial Nitric Oxide Synthase. Hypertension 2012, 60, 1407–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, A.M.; Fagundes, C.T.; Yang, G.; Palsson-McDermott, E.M.; Wochal, P.; McGettrick, A.F.; Foley, N.H.; Early, J.O.; Chen, L.; Zhang, H.; et al. Circadian control of innate immunity in macrophages by miR-155 targeting Bmal1. Proc. Natl. Acad. Sci. USA 2015, 112, 7231–7236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tryggestad, J.B.; Teague, A.M.; Sparling, D.P.; Jiang, S.; Chernausek, S.D. Macrophage-Derived microRNA-155 Increases in Obesity and Influences Adipocyte Metabolism by Targeting Peroxisome Proliferator-Activated Receptor Gamma. Obesity 2019, 27, 1856–1864. [Google Scholar] [CrossRef]
- El Samaloty, N.M.; Hassan, Z.A.; Hefny, Z.; Abdelaziz, D.H.A. Circulating microRNA-155 is associated with insulin resistance in chronic hepatitis C patients. Arab. J. Gastroenterol. 2019, 20, 1–7. [Google Scholar] [CrossRef]
- Hu, J.; Huang, C.-X.; Rao, P.-P.; Cao, G.-Q.; Zhang, Y.; Zhou, J.-P.; Zhu, L.-Y.; Liu, M.-X.; Zhang, G. MicroRNA-155 inhibition attenuates endoplasmic reticulum stress-induced cardiomyocyte apoptosis following myocardial infarction via reducing macrophage inflammation. Eur. J. Pharmacol. 2019, 857, 172449. [Google Scholar] [CrossRef]
- Li, Y.; Tian, Z.; Tan, Y.; Lian, G.; Chen, S.; Chen, S.; Li, J.; Li, X.; Huang, K.; Chen, Y. Bmi-1-induced miR-27a and miR-155 promote tumor metastasis and chemoresistance by targeting RKIP in gastric cancer. Mol. Cancer 2020, 19, 1–14. [Google Scholar] [CrossRef]
- Sun, X.; Song, M.; Song, H.; Wang, Y.; Luo, M.; Yin, L. miR-155 mediates inflammatory injury of hippocampal neuronal cells via the activation of microglia. Mol. Med. Rep. 2019, 19, 2627–2635. [Google Scholar] [CrossRef]
- Ekiz, H.A.; Ramstead, A.G.; Lee, S.-H.; Nelson, M.C.; Bauer, K.M.; Wallace, J.A.; Hu, R.; Round, J.L.; Rutter, J.; Drummond, M.J.; et al. T Cell–Expressed microRNA-155 Reduces Lifespan in a Mouse Model of Age-Related Chronic Inflammation. J. Immunol. 2020, 204, 2064–2075. [Google Scholar] [CrossRef]
- Xia, Y.; Chen, S.; Zeng, S.; Zhao, Y.; Zhu, C.; Deng, B.; Zhu, G.; Yin, J.; Wang, W.; Hardeland, R.; et al. Melatonin in macrophage biology: Current understanding and future perspectives. J. Pineal Res. 2019, 66, e12547. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Xu, Z.; Ding, T.; Kuang, D.M.; Zheng, L. MicroRNA-155 regulates inflammatory cytokine production in tumor-associated macrophages via targeting C/EBPbeta. Cell. Mol. Immunol. 2009, 6, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Ge, J.; Huang, Z.; Liu, H.; Chen, J.; Xie, Z.; Chen, Z.; Peng, J.; Sun, J.; Hou, J.; Zhang, X. Lower Expression of MicroRNA-155 Contributes to Dysfunction of Natural Killer Cells in Patients with Chronic Hepatitis B. Front. Immunol. 2017, 8, 1173. [Google Scholar] [CrossRef] [Green Version]
- Alter, G.; Suscovich, T.J.; Kleyman, M.; Teigen, N.; Streeck, H.; Zaman, M.T.; Meier, A.; Altfeld, M. Low perforin and elevated SHIP-1 expression is associated with functional anergy of natural killer cells in chronic HIV-1 infection. AIDS 2006, 20, 1549–1551. [Google Scholar] [CrossRef]
- Stelekati, E.; Chen, Z.; Manne, S.; Kurachi, M.; Ali, M.-A.; Lewy, K.; Cai, Z.; Nzingha, K.; McLane, L.M.; Hope, J.L.; et al. Long-Term Persistence of Exhausted CD8 T Cells in Chronic Infection Is Regulated by MicroRNA-155. Cell Rep. 2018, 23, 2142–2156. [Google Scholar] [CrossRef] [PubMed]
- Hsin, J.-P.; Lu, Y.; Loeb, G.B.; Leslie, C.S.; Rudensky, A.Y. The effect of cellular context on miR-155-mediated gene regulation in four major immune cell types. Nat. Immunol. 2018, 19, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.-P.; Zhang, Y.; Do, D.C.; Ke, X.; Zhang, S.; Lambert, K.; Kumar, S.; Hu, C.; Zhou, Y.-F.; Ishmael, F.T.; et al. miR-155 Modulates Cockroach Allergen– and Oxidative Stress–Induced Cyclooxygenase-2 in Asthma. J. Immunol. 2018, 201, 916–929. [Google Scholar] [CrossRef] [PubMed]
- Comer, B.S. Does miRNA-155 Promote Cyclooxygenase-2 Expression in Cancer? Drug Dev. Res. 2015, 76, 354–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, P.R.; Marques, R.M.; Gomez, E.A.; Colas, R.A.; De Matteis, R.; Zak, A.; Patel, M.; Collier, D.J.; Dalli, J. Enriched Marine Oil Supplements Increase Peripheral Blood Specialized Pro-Resolving Mediators Concentrations and Reprogram Host Immune Responses. Circ. Res. 2020, 126, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Lannan, K.L.; Spinelli, S.L.; Blumberg, N.; Phipps, R.P. Maresin 1 induces a novel pro-resolving phenotype in human platelets. J. Thromb. Haemost. 2017, 15, 802–813. [Google Scholar] [CrossRef]
- Barnig, C.; Cernadas, M.; Dutile, S.; Liu, X.; Perrella, M.A.; Kazani, S.; Wechsler, M.E.; Israel, E.; Levy, B.D. Lipoxin A4 Regulates Natural Killer Cell and Type 2 Innate Lymphoid Cell Activation in Asthma. Sci. Transl. Med. 2013, 5, 174ra26. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Deng, M.; Lu, A.; Chen, Y.; Chen, Y.; Wu, C.; Tan, Z.; Boini, K.M.; Yang, T.; Zhu, Q.; et al. Sodium butyrate attenuates angiotensin II-induced cardiac hypertrophy by inhibiting COX2/PGE2 pathway via a HDAC5/HDAC6-dependent mechanism. J. Cell. Mol. Med. 2019, 23, 8139–8150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torun, A.; Enayat, S.; Sheraj, I.; Tunçer, S.; Ülgen, D.H.; Banerjee, S. Butyrate mediated regulation of RNA binding proteins in the post-transcriptional regulation of inflammatory gene expression. Cell. Signal. 2019, 64, 109410. [Google Scholar] [CrossRef] [PubMed]
- Menzies, K.J.; Zhang, H.; Katsyuba, E.; Auwerx, J. Protein acetylation in metabolism—Metabolites and cofactors. Nat. Rev. Endocrinol. 2015, 12, 43–60. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Liu, X.-X.; Hong, M.; Huang, X.-Z.; Chen, H.; Xu, J.-H.; Wang, C.; Zhang, Y.-X.; Zhong, J.; Nie, H.; et al. Sodium butyrate alleviates LPS-induced acute lung injury in mice via inhibiting HMGB1 release. Int. Immunopharmacol. 2018, 56, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Fei, X.; Li, S.; Xu, C.; Tu, C.; Jiang, L.; Wo, M. Predicting significance of COX-2 expression of peripheral blood monocyte in patients with coronary artery disease. Ann. Transl. Med. 2019, 7, 483. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, Y.; Yang, M.; Zhang, M.; Xiao, M.; Li, X. Butyrate mitigates TNF-α-induced attachment of monocytes to endothelial cells. J. Bioenerg. Biomembr. 2020, 52, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Saban, K.L.; Mathews, H.L.; Bryant, F.B.; Tell, D.; Joyce, C.; Devon, H.A.; Witek-Janusek, L. Perceived discrimination is associated with the inflammatory response to acute laboratory stress in women at risk for cardiovascular disease. Brain Behav. Immun. 2018, 73, 625–632. [Google Scholar] [CrossRef]
- Brody, G.H.; Yu, T.; Miller, G.E.; Chen, E. Discrimination, Racial Identity, and Cytokine Levels among African-American Adolescents. J. Adolesc. Health 2015, 56, 496–501. [Google Scholar] [CrossRef] [Green Version]
- Slusher, A.L.; Zúñiga, T.M.; Acevedo, E.O. Inflamm-Aging Is Associated with Lower Plasma PTX3 Concentrations and an Impaired Capacity of PBMCs to Express hTERT following LPS Stimulation. Mediat. Inflamm. 2019, 2019, 2324193. [Google Scholar] [CrossRef] [Green Version]
- Simons, R.L.; Lei, M.-K.; Beach, S.R.H.; Barr, A.B.; Cutrona, C.E.; Gibbons, F.X.; Philibert, R.A. An index of the ratio of inflammatory to antiviral cell types mediates the effects of social adversity and age on chronic illness. Soc. Sci. Med. 2017, 185, 158–165. [Google Scholar] [CrossRef]
- Hung, Y.-Y.; Wu, M.-K.; Tsai, M.-C.; Huang, Y.-L.; Kang, H.-Y. Aberrant Expression of Intracellular let-7e, miR-146a, and miR-155 Correlates with Severity of Depression in Patients with Major Depressive Disorder and Is Ameliorated after Antidepressant Treatment. Cells 2019, 8, 647. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.H.; Zhu, H.; Harris, R.A.; Dong, Y.; Su, S.; Tingen, M.S.; Kapuku, G.K.; Pollock, J.S.; Pollock, D.M.; Harshfield, G.A.; et al. Ethnic Differences in Nighttime Melatonin and Nighttime Blood Pressure: A Study in European Americans and African Americans. Am. J. Hypertens. 2019, 32, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Kueht, M.L.; McFarlin, B.K.; Lee, R.E. Severely obese have greater LPS-stimulated TNF-alpha production than normal weight African-American women. Obesity 2008, 17, 447–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millett, G.A.; Jones, A.T.; Benkeser, D.; Baral, S.; Mercer, L.; Beyrer, C.; Honermann, B.; Lankiewicz, E.; Mena, L.; Crowley, J.S.; et al. Assessing Differential Impacts of COVID-19 on Black Communities. Ann. Epidemiol. 2020, 47, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Sanford, Z.; Taylor, H.; Fiorentino, A.; Broda, A.; Zaidi, A.; Turcotte, J.; Patton, C. Racial Disparities in Surgical Outcomes After Spine Surgery: An ACS-NSQIP Analysis. Glob. Spine J. 2018, 9, 583–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folsom, A.R.; Basu, S.; Hong, C.-P.; Heckbert, S.R.; Lutsey, P.L.; Rosamond, W.D.; Cushman, M. Atherosclerosis Risk in Communities (ARIC) Study Reasons for Differences in the Incidence of Venous Thromboembolism in Black Versus White Americans. Am. J. Med. 2019, 132, 970–976. [Google Scholar] [CrossRef]
- Sanaiha, Y.; Bailey, K.L.; Aguayo, E.; Seo, Y.-J.; Dobaria, V.; Lin, A.Y.; Benharash, P. Racial Disparities in the Incidence of Pulmonary Embolism after Colectomy. Am. Surg. 2018, 84, 1560–1564. [Google Scholar] [CrossRef]
- Pellom, S.T.; Arnold, T.; Williams, M.; Brown, V.L.; Samuels, A.D. Examining breast cancer disparities in African Americans with suggestions for policy. Cancer Causes Control 2020, 31, 795–800. [Google Scholar] [CrossRef]
- Troy, C.; Brunson, A.; Goldsmith, A.; Noblet, S.; Steck, S.E.; Hebert, J.R.; Payne, J.; McCormick, D.; Friedman, D.B. Implementing Community-Based Prostate Cancer Education in Rural South Carolina: A Collaborative Approach Through a Statewide Cancer Alliance. J. Cancer Educ. 2020. [Google Scholar] [CrossRef]
- Schurman, S.H.; O’Hanlon, T.P.; McGrath, J.; Gruzdev, A.; Bektas, A.; Xu, H.; Garantziotis, S.; Zeldin, D.C.; Miller, F. Transethnic associations among immune-mediated diseases and single-nucleotide polymorphisms of the aryl hydrocarbon response gene ARNT and the PTPN22 immune regulatory gene. J. Autoimmun. 2019, 107, 102363. [Google Scholar] [CrossRef]
- Iwane, M.K.; Chaves, S.S.; Szilagyi, P.G.; Edwards, K.M.; Hall, C.B.; Staat, M.A.; Brown, C.J.; Griffin, M.R.; Weinberg, G.A.; Poehling, K.A.; et al. Disparities Between Black and White Children in Hospitalizations Associated With Acute Respiratory Illness and Laboratory-confirmed Influenza and Respiratory Syncytial Virus in 3 US Counties--2002–2009. Am. J. Epidemiol. 2013, 177, 656–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turski, W.A.; Wnorowski, A.; Turski, G.N.; Turski, C.A.; Turski, L. AhR and IDO1 in pathogenesis of Covid-19 and the “Systemic AhR Activation Syndrome” Translational review and therapeutic perspectives. Restor. Neurol. Neurosci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Venkataraman, A.; Jain, P.C.; Wiesler, E.P.; DeBlasio, M.; Klein, J.; Tu, S.S.; Lee, S.; Medzhitov, R.; Iwasaki, A. Vitamin B12 and folic acid alleviate symptoms of nutritional deficiency by antagonizing aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 15837–15845. [Google Scholar] [CrossRef] [PubMed]
- Kandeel, M.; Al-Nazawi, M. Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci. 2020, 251, 117627. [Google Scholar] [CrossRef] [PubMed]
- Partearroyo, T.; Úbeda, N.; Montero, A.; Achón, M.; Varela-Moreiras, G. Vitamin B12 and Folic Acid Imbalance Modifies NK Cytotoxicity, Lymphocytes B and Lymphoprolipheration in Aged Rats. Nutrients 2013, 5, 4836–4848. [Google Scholar] [CrossRef] [Green Version]
- Rojas, I.Y.; Moyer, B.J.; Ringelberg, C.S.; Tomlinson, C.R. Reversal of obesity and liver steatosis in mice via inhibition of aryl hydrocarbon receptor and altered gene expression of CYP1B1, PPARα, SCD1, and osteopontin. Int. J. Obes. 2020, 44, 948–963. [Google Scholar] [CrossRef]
- Palermo, C.M.; Hernando, J.I.M.; Dertinger, S.D.; Kende, A.S.; Gasiewicz, T.A. Identification of Potential Aryl Hydrocarbon Receptor Antagonists in Green Tea. Chem. Res. Toxicol. 2003, 16, 865–872. [Google Scholar] [CrossRef]
- Ghosh, R.; Chakraborty, A.; Biswas, A.; Chowdhuri, S. Evaluation of green tea polyphenols as novel corona virus (SARS CoV-2) main protease (Mpro) inhibitors—An in silico docking and molecular dynamics simulation study. J. Biomol. Struct. Dyn. 2020, 22, 1–13. [Google Scholar] [CrossRef]
- Bhardwaj, V.K.; Singh, R.; Sharma, J.; Rajendran, V.; Purohit, R.; Kumar, S. Identification of bioactive molecules from tea plant as SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2020, 20, 1–10. [Google Scholar] [CrossRef]
- Lee, Y.H.; Jang, Y.H.; Kim, Y.-S.; Kim, J.; Seong, B.L. Evaluation of green tea extract as a safe personal hygiene against viral infections. J. Boil. Eng. 2018, 12, 1. [Google Scholar] [CrossRef] [Green Version]
- Bordoni, V.; Sacchi, A.; Cimini, E.; Notari, S.; Grassi, G.; Tartaglia, E.; Casetti, R.; Giancola, L.; Bevilacqua, N.; Maeurer, M.; et al. An inflammatory profile correlates with decreased frequency of cytotoxic cells in COVID-19. Clin. Infect. Dis. 2020, ciaa577. [Google Scholar] [CrossRef]
- Agrati, C.; Sacchi, A.; Bordoni, V.; Cimini, E.; Notari, S.; Grassi, G.; Casetti, R.; Tartaglia, E.; Lalle, E.; D’Abramo, A.; et al. Expansion of myeloid-derived suppressor cells in patients with severe coronavirus disease (COVID-19). Cell Death Differ. 2020. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Yan, F.; Zhao, Y.; Chen, X.; Sun, S.; Wang, Y.; Ying, L. Green Tea Polyphenol EGCG Attenuates MDSCs-mediated Immunosuppression through Canonical and Non-Canonical Pathways in a 4T1 Murine Breast Cancer Model. Nutrients 2020, 12, 1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, T.H.; Lee, J.H.; Kil Song, C.; Han, H.D.; Shin, B.C.; Pai, S.I.; Hung, C.-F.; Trimble, C.; Lim, J.-S.; Kim, T.W.; et al. Epigallocatechin-3-gallate enhances CD8+ T cell-mediated antitumor immunity induced by DNA vaccination. Cancer Res. 2007, 67, 802–811. [Google Scholar] [CrossRef] [Green Version]
- Ademosun, A.O.; Oboh, G. Comparison of the Inhibition of Monoamine Oxidase and Butyrylcholinesterase Activities by Infusions from Green Tea and Some Citrus Peels. Int. J. Alzheimer’s Dis. 2014, 2014, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, O.; Amit, T.; Youdim, M.B.H. A novel approach of proteomics and transcriptomics to study the mechanism of action of the antioxidant-iron chelator green tea polyphenol (-)-epigallocatechin-3-gallate. Free. Radic. Boil. Med. 2007, 43, 546–556. [Google Scholar] [CrossRef]
- Wei, R.; Mao, L.; Xu, P.; Zheng, X.; Hackman, R.M.; MacKenzie, G.G.; Wang, Y. Suppressing glucose metabolism with epigallocatechin-3-gallate (EGCG) reduces breast cancer cell growth in preclinical models. Food Funct. 2018, 9, 5682–5696. [Google Scholar] [CrossRef]
- Castellano-Gonzalez, G.; Pichaud, N.; Ballard, J.W.O.; Bessede, A.; Marçal, H.; Guillemin, G.J. Epigallocatechin-3-gallate induces oxidative phosphorylation by activating cytochrome c oxidase in human cultured neurons and astrocytes. Oncotarget 2016, 7, 7426–7440. [Google Scholar] [CrossRef]
- Almatroodi, S.A.; Almatroudi, A.; Alsahli, M.A.; Aljasir, M.A.; Syed, M.A.; Rahmani, A.H. Epigallocatechin-3-Gallate (EGCG), an Active Compound of Green Tea Attenuates Acute Lung Injury Regulating Macrophage Polarization and Krüpple-Like-Factor 4 (KLF4) Expression. Molecules 2020, 25, 2853. [Google Scholar] [CrossRef]
- Vassallo, A.; Wood, A.J.; Subburayalu, J.; Summers, C.; Chilvers, E.R.; Subburayalu, J. The counter-intuitive role of the neutrophil in the acute respiratory distress syndrome. Br. Med. Bull. 2019, 131, 43–55. [Google Scholar] [CrossRef]
- Yang, P.; Ding, G.-B.; Liu, W.; Fu, R.; Sajid, A.; Li, Z. Tannic acid directly targets pyruvate kinase isoenzyme M2 to attenuate colon cancer cell proliferation. Food Funct. 2018, 9, 5547–5559. [Google Scholar] [CrossRef] [PubMed]
- Gdynia, G.; Sauer, S.W.; Kopitz, J.; Fuchs, D.; Duglova, K.; Ruppert, T.; Miller, M.; Pahl, J.; Cerwenka, A.; Enders, M.; et al. The HMGB1 protein induces a metabolic type of tumour cell death by blocking aerobic respiration. Nat. Commun. 2016, 7, 10764. [Google Scholar] [CrossRef] [PubMed]
- Palsson-McDermott, E.M.; Dyck, L.; Zasłona, Z.; Menon, D.; McGettrick, A.F.; Mills, K.H.G.; O’Neill, L.A. Pyruvate Kinase M2 Is Required for the Expression of the Immune Checkpoint PD-L1 in Immune Cells and Tumors. Front. Immunol. 2017, 8, 1300. [Google Scholar] [CrossRef] [Green Version]
- Di Cosimo, S.; Malfettone, A.; Pérez-García, J.M.; Llombart-Cussac, A.; Miceli, R.; Curigliano, G.; Cortés, J. Immune checkpoint inhibitors: A physiology-driven approach to the treatment of coronavirus disease 2019. Eur. J. Cancer 2020, 135, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Hou, R.; Wei, Y.; Wang, T.; Wan, X.; Yang, C.S.; Reiter, R.J.; Zhang, J. Melatonin attenuates (-)-epigallocatehin-3-gallate-triggered hepatotoxicity without compromising its downregulation of hepatic gluconeogenic and lipogenic genes in mice. J. Pineal Res. 2015, 59, 497–507. [Google Scholar] [CrossRef]
- Anderson, G.; Reiter, R.J. Melatonin: Roles in influenza, Covid-19, and other viral infections. Rev. Med. Virol. 2020, 30, 2109. [Google Scholar] [CrossRef]
- Jehi, L.; Ji, X.; Milinovich, A.; Erzurum, S.; Rubin, B.P.; Gordon, S.; Young, J.B.; Kattan, M.W. Individualizing Risk Prediction for Positive Coronavirus Disease 2019 Testing. Chest 2020. [Google Scholar] [CrossRef]
- Zhou, Y.; Hou, Y.; Shen, J.; Kallianpur, A.; Zein, J.; Culver, D.A.; Farha, S.; Comhair, S.; Fiocchi, C.; Gack, M.U.; et al. A Network Medicine Approach to Investigation and Population-based Validation of Disease Manifestations and Drug Repurposing for COVID-19. ChemRxiv 2020. preprint. [Google Scholar] [CrossRef]
- Han, S.; Li, Z.; Han, F.; Jia, Y.; Qi, L.; Wu, G.; Cai, W.; Xu, Y.; Li, C.; Zhang, W.; et al. ROR alpha protects against LPS-induced inflammation by down-regulating SIRT1/NF-kappa B pathway. Arch. Biochem. Biophys. 2019, 668, 1–8. [Google Scholar] [CrossRef]
- Tiong, Y.L.; Ng, K.Y.; Koh, R.Y.; Ponnudurai, G.; Chye, S.M. Melatonin inhibits high glucose-induced ox-LDL/LDL expression and apoptosis in human umbilical endothelial cells. Horm. Mol. Biol. Clin. Investig. 2020. [Google Scholar] [CrossRef]
- Koizumi, N.; Okumura, N.; Ueno, M.; Kinoshita, S. New Therapeutic Modality for Corneal Endothelial Disease Using Rho-Associated Kinase Inhibitor Eye Drops. Cornea 2014, 33 (Suppl. S11), S25–S31. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.; Akhbanbetova, A.; Quantock, A.J.; Heard, C.M. Topical delivery of a Rho-kinase inhibitor to the cornea via mucoadhesive film. Eur. J. Pharm. Sci. 2016, 91, 256–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, K.; Duan, X.; Cai, H.; Liu, X.; Yang, Y.; Li, M.; Zhang, X.; Wang, J. Curcumin inhibits LPA-induced invasion by attenuating RhoA/ROCK/MMPs pathway in MCF7 breast cancer cells. Clin. Exp. Med. 2015, 16, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Karlsson, H. Antiviral Effect of IDO in Mouse Fibroblast Cells during Influenza Virus Infection. Viral Immunol. 2017, 30, 542–544. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, P.; Hartjen, P.; Kohsar, M.; Kummer, S.; Schmiedel, S.; Bockmann, J.-H.; Fathi, A.; Huber, S.; Haag, F.; Wiesch, J.S.Z. Defining the CD39/CD73 Axis in SARS-CoV-2 Infection: The CD73- Phenotype Identifies Polyfunctional Cytotoxic Lymphocytes. Cells 2020, 9, 1750. [Google Scholar] [CrossRef]
- Rodríguez-Rubio, M.; Figueira, J.C.; Acuña-Castroviejo, D.; Borobia, A.M.; Escames, G.; De La Oliva, P. A phase II, single-center, double-blind, randomized placebo-controlled trial to explore the efficacy and safety of intravenous melatonin in patients with COVID-19 admitted to the intensive care unit (MelCOVID study): A structured summary of a study protocol for a randomized controlled trial. Trials 2020, 21, 1–3. [Google Scholar] [CrossRef]
- González-Barbosa, E.; Mejía-García, A.; Bautista, E.; Gonzalez, F.J.; Segovia, J.; Elizondo, G. TCDD induces UbcH7 expression and synphilin-1 protein degradation in the mouse ventral midbrain. J. Biochem. Mol. Toxicol. 2017, 31, e21947. [Google Scholar] [CrossRef]
- Tal, Y.; Adini, A.; Eran, A.; Adini, I. Racial disparity in Covid-19 mortality rates—A plausible explanation. Clin. Immunol. 2020, 217, 108481. [Google Scholar] [CrossRef]
- Zhang, M.; Jin, F. 1α,25-Dihydroxyvitamin D3 Ameliorates Seawater Aspiration-Induced Lung Injury By Inhibiting The Translocation Of NF-κB and RhoA. Inflammation 2017, 40, 832–839. [Google Scholar] [CrossRef] [Green Version]
- Adapa, S.; Chenna, A.; Balla, M.; Merugu, G.P.; Koduri, N.M.; Daggubati, S.R.; Gayam, V.; Naramala, S.; Konala, V.M. COVID-19 Pandemic Causing Acute Kidney Injury and Impact on Patients With Chronic Kidney Disease and Renal Transplantation. J. Clin. Med. Res. 2020, 12, 352–361. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, S.; Huang, Y.; Yang, K.; Liu, Y.; Bi, X.; Liu, C.; Xiong, J.; Zhang, B.; Zhao, J.; et al. Inhibition of CYP1B1 ameliorates cardiac hypertrophy induced by uremic toxin. Mol. Med. Rep. 2019, 21, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Yoo, T.-H.; Hwang, Y.; Lee, G.H.; Kim, B.; Jang, J.; Yu, H.T.; Kim, M.C.; Cho, J.-Y.; Lee, C.J.; et al. Indoxyl sulfate (IS)-mediated immune dysfunction provokes endothelial damage in patients with end-stage renal disease (ESRD). Sci. Rep. 2017, 7, 3057. [Google Scholar] [CrossRef] [Green Version]
- Chu, S.; Mao, X.; Guo, H.; Wang, L.; Li, Z.; Zhang, Y.; Wang, Y.; Wang, H.; Zhang, X.; Peng, W. Indoxyl sulfate potentiates endothelial dysfunction via reciprocal role for reactive oxygen species and RhoA/ROCK signaling in 5/6 nephrectomized rats. Free. Radic. Res. 2017, 51, 1–43. [Google Scholar] [CrossRef]
- Karbowska, M.; Kaminski, T.W.; Znorko, B.; Domaniewski, T.; Misztal, T.; Rusak, T.; Pryczynicz, A.; Guzinska-Ustymowicz, K.; Pawlak, K.; Pawlak, D. Indoxyl Sulfate Promotes Arterial Thrombosis in Rat Model via Increased Levels of Complex TF/VII, PAI-1, Platelet Activation as Well as Decreased Contents of SIRT1 and SIRT3. Front. Physiol. 2018, 9, 1623. [Google Scholar] [CrossRef] [Green Version]
- Kolachalama, V.B.; Shashar, M.; Alousi, F.; Shivanna, S.; Rijal, K.; Belghasem, M.E.; Walker, J.; Matsuura, S.; Chang, G.H.; Gibson, C.M.; et al. Uremic Solute-Aryl Hydrocarbon Receptor-Tissue Factor Axis Associates with Thrombosis after Vascular Injury in Humans. J. Am. Soc. Nephrol. 2018, 29, 1063–1072. [Google Scholar] [CrossRef]
- Lustgarten, M.S.; Fielding, R.A. Metabolites Associated with Circulating Interleukin-6 in Older Adults. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 2017, 72, 1277–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vyhlídalová, B.; Krasulová, K.; Pečinková, P.; Marcalíková, A.; Vrzal, R.; Zemánková, L.; Vančo, J.; Trávníček, Z.; Vondráček, J.; Karasová, M.; et al. Gut Microbial Catabolites of Tryptophan Are Ligands and Agonists of the Aryl Hydrocarbon Receptor: A Detailed Characterization. Int. J. Mol. Sci. 2020, 21, 2614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloc, M.; Ghobrial, R.M.; Kubiak, J.Z. The Role of Genetic Sex and Mitochondria in Response to COVID-19 Infection. Int. Arch. Allergy Immunol. 2020, 181, 629–634. [Google Scholar] [CrossRef]
- Kim, S.; Sieburth, D. Sphingosine Kinase Activates the Mitochondrial Unfolded Protein Response and Is Targeted to Mitochondria by Stress. Cell Rep. 2018, 24, 2932–2945. [Google Scholar] [CrossRef] [Green Version]
- Borro, M.; Di Girolamo, P.; Gentile, G.; De Luca, O.; Preissner, R.; Marcolongo, A.; Ferracuti, S.; Simmaco, M. Evidence-Based Considerations Exploring Relations between SARS-CoV-2 Pandemic and Air Pollution: Involvement of PM2.5-Mediated Up-Regulation of the Viral Receptor ACE-2. Int. J. Environ. Res. Public Health 2020, 17, 5573. [Google Scholar] [CrossRef]
- Moccia, F.; Gerbino, A.; Lionetti, V.; Miragoli, M.; Munaron, L.M.; Pagliaro, P.; Pasqua, T.; Penna, C.; Rocca, C.; Samaja, M.; et al. COVID-19-associated cardiovascular morbidity in older adults: A position paper from the Italian Society of Cardiovascular Researches. GeroScience 2020, 42, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Ramos-García, N.A.; Orozco-Ibarra, M.; Estudillo, E.; Elizondo, G.; Apo, E.G.; Macías, L.G.C.; Sosa-Ortiz, A.L.; Torres-Ramos, M.A. Aryl Hydrocarbon Receptor in Post-Mortem Hippocampus and in Serum from Young, Elder, and Alzheimer’s Patients. Int. J. Mol. Sci. 2020, 21, 1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkmann, V.; Ale-Agha, N.; Haendeler, J.; Ventura, N. The Aryl Hydrocarbon Receptor (AhR) in the Aging Process: Another Puzzling Role for This Highly Conserved Transcription Factor. Front. Physiol. 2020, 10, 1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Mode of Efficacy | References |
|---|---|---|
| Vitamin B12/Folate | AhR antagonism; could inhibit entry, cytokine storm and enhance antiviral response | [123] |
| Vitamin B12 | Mpro protease inhibitor; expect to inhibit entry | [124] |
| Optimizes NK cell cytotoxicity | [125] | |
| Resveratrol, Curcumin and Rosiglitazone | Inhibitors of the IDO-kynurenine-AhR pathway | [122] |
| Green Tea’s-Epigallocatechin gallate (EGCG) | AhR antagonist | [127] |
| - EGCG | MDSC inhibitor; increases CD8+ T cell cytotoxicity | [128] |
| - EGCG, epicatechingallate and gallocatechin-3-gallate | Mpro antagonist | [133,134] |
| - EGCG | Inhibits monoamine oxidase; increases 14-3-3 and therefore melatonergic pathway | [135,136] |
| - EGCG | Induces M2-like macrophage;dampens neutrophils | [140] |
| - tannic acid | PKM2 inhibitor; enhance NK cell cytotoxicity? | [141] |
| - caffeine | Adenosine A2A receptor inhibition increases CD8+ T cell and NK cell cytotoxicity | [155] |
| Melatonin | Inhibits SARS-CoV-2 infection risk | [147,148] |
| Increases α7nAChR levels, dampening immune activation | [27] | |
| Decreases platelet activation | [20] | |
| Seems to reset immune cell metabolism | [5] | |
| ROCK inhibitors: Y27632 | Only Y27632 clinically available but toxicity problematic; efficacy via S1P3r/RhoA/ROCK inhibition | [151] |
| Curcumin | Acts indirectly to inhibit ROCK pathway | [153] |
| IDO inhibitors | Potential, but not currently indicated | [154] |
| Nimesulide | Inhibitor of B0AT1/SLC6A19 and therefore potentially of entry via the ACE2-B0AT1 dimer | [10] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anderson, G.; Carbone, A.; Mazzoccoli, G. Aryl Hydrocarbon Receptor Role in Co-Ordinating SARS-CoV-2 Entry and Symptomatology: Linking Cytotoxicity Changes in COVID-19 and Cancers; Modulation by Racial Discrimination Stress. Biology 2020, 9, 249. https://doi.org/10.3390/biology9090249
Anderson G, Carbone A, Mazzoccoli G. Aryl Hydrocarbon Receptor Role in Co-Ordinating SARS-CoV-2 Entry and Symptomatology: Linking Cytotoxicity Changes in COVID-19 and Cancers; Modulation by Racial Discrimination Stress. Biology. 2020; 9(9):249. https://doi.org/10.3390/biology9090249
Chicago/Turabian StyleAnderson, George, Annalucia Carbone, and Gianluigi Mazzoccoli. 2020. "Aryl Hydrocarbon Receptor Role in Co-Ordinating SARS-CoV-2 Entry and Symptomatology: Linking Cytotoxicity Changes in COVID-19 and Cancers; Modulation by Racial Discrimination Stress" Biology 9, no. 9: 249. https://doi.org/10.3390/biology9090249
APA StyleAnderson, G., Carbone, A., & Mazzoccoli, G. (2020). Aryl Hydrocarbon Receptor Role in Co-Ordinating SARS-CoV-2 Entry and Symptomatology: Linking Cytotoxicity Changes in COVID-19 and Cancers; Modulation by Racial Discrimination Stress. Biology, 9(9), 249. https://doi.org/10.3390/biology9090249