Is There Such a Thing as a Genuine Cancer Stem Cell Marker? Perspectives from the Gut, the Brain and the Dental Pulp

 , , , ,
, , , ,  and
and
Simple Summary
Abstract
1. Introduction
2. Cell Markers and Pluripotency Core Factors
2.1. Cell Surface or Membrane Markers
2.2. Cytoplasmic Markers
2.3. Nuclear Proteins
3. Oncogenic Signaling
3.1. Wingless (Wg)-Related Integration Site (Wnt)
3.2. Transforming Growth Factor Beta (TGF-ß) Signaling
4. Telomerase Activity
5. Pathways and Obstacles to Malignant Transformation: The Surprising Case of DPSCs
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Williams, J.M.; Duckworth, C.A.; Burkitt, M.D.; Watson, A.J.M.; Campbell, B.J.; Pritchard, D.M. Epithelial cell shedding and barrier function: A matter of life and death at the small intestinal villus tip. Vet. Pathol. 2015, 52, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Umar, S. Intestinal Stem Cells. Curr. Gastroenterol. Rep. 2010, 12, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Cliffe, L.J.; Humphreys, N.E.; Lane, T.E.; Potten, C.S.; Booth, C.; Grencis, R.K. Accelerated intestinal epithelial cell turnover: A new mechanism of parasite expulsion. Science 2005, 308, 1463–1465. [Google Scholar] [CrossRef] [PubMed]
- Paredes, M.F.; Sorrells, S.F.; Cebrian-Silla, A.; Sandoval, K.; Qi, D.; Kelley, K.W.; James, D.; Mayer, S.; Chang, J.; Auguste, K.I.; et al. Does Adult Neurogenesis Persist in the Human Hippocampus? Cell Stem Cell 2018, 23, 780–781. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Jiménez, E.P.; Flor-García, M.; Terreros-Roncal, J.; Rábano, A.; Cafini, F.; Pallas-Bazarra, N.; Ávila, J.; Llorens-Martín, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019, 25, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Scoville, W.B.; Milner, B. Loss of recent memory after bilateral hippocampal lesions. J. Neurol. Neurosurg. Psychiatry 1957, 20, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Rusznák, Z.; Henskens, W.; Schofield, E.; Kim, W.S.; Fu, Y. Adult Neurogenesis and Gliogenesis: Possible Mechanisms for Neurorestoration. Exp. Neurobiol. 2016, 25, 103–112. [Google Scholar] [CrossRef]
- Arvidsson, A.; Collin, T.; Kirik, D.; Kokaia, Z.; Lindvall, O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat. Med. 2002, 8, 963–970. [Google Scholar] [CrossRef]
- Robel, S.; Berninger, B.; Götz, M. The stem cell potential of glia: Lessons from reactive gliosis. Nat. Rev. Neurosci. 2011, 12, 88–104. [Google Scholar] [CrossRef]
- Sierra, A.; Martin-Suarez, S.; Valcarcel-Martin, R.; Pascual-Brazo, J.; Aelvoet, S.A.; Abiega, O.; Deudero, J.J.; Brewster, A.L.; Bernales, I.; Anderson, A.E.; et al. Neuronal hyperactivity accelerates depletion of neural stem cells and impairs hippocampal neurogenesis. Cell Stem Cell 2015, 16, 488–503. [Google Scholar] [CrossRef]
- Brenner, H.; Kloor, M.; Pox, C.P. Colorectal cancer. Lancet 2014, 383, 1490–1502. [Google Scholar] [CrossRef]
- Seo, Y.-S.; Ko, I.O.; Park, H.; Jeong, Y.J.; Park, J.-A.; Kim, K.S.; Park, M.-J.; Lee, H.-J. Radiation-Induced Changes in Tumor Vessels and Microenvironment Contribute to Therapeutic Resistance in Glioblastoma. Front. Oncol. 2019, 9, 1259. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, C.; Wang, L.; Chen, Y. A comprehensive review in improving delivery of small-molecule chemotherapeutic agents overcoming the blood-brain/brain tumor barriers for glioblastoma treatment. Drug Deliv. 2019, 26, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Sell, S. Stem cell origin of cancer and differentiation therapy. Crit. Rev. Oncol. Hematol. 2004, 51, 1–28. [Google Scholar] [CrossRef]
- Wang, K.; Wu, X.; Wang, J.; Huang, J. Cancer stem cell theory: Therapeutic implications for nanomedicine. Int. J. Nanomed. 2013, 8, 899–908. [Google Scholar] [CrossRef]
- Kleinsmith, L.J.; Pierce, G.B. MULTIPOTENTIALITY OF SINGLE EMBRYONAL CARCINOMA CELLS. Cancer Res. 1964, 24, 1544–1551. [Google Scholar]
- Neuhaus, K.W. Teeth: Malignant neoplasms in the dental pulp? Lancet Oncol. 2007, 8, 75–78. [Google Scholar] [CrossRef]
- Neuhaus, K.W. Dental Pulp Neoplasms. In Encyclopedia of Cancer; Schwab, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 1084–1086. ISBN 978-3-642-16483-5. [Google Scholar]
- Gronthos, S.; Mankani, M.; Brahim, J.; Robey, P.G.; Shi, S. Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2000, 97, 13625–13630. [Google Scholar] [CrossRef]
- Mitsiadis, T.A.; Woloszyk, A. Odyssey of human dental pulp stem cells and their remarkable ability to survive in extremely adverse conditions. Front. Physiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Gronthos, S. Perivascular niche of postnatal mesenchymal stem cells in human bone marrow and dental pulp. J. Bone Miner. Res. 2003, 18, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Kaukua, N.; Shahidi, M.K.; Konstantinidou, C.; Dyachuk, V.; Kaucka, M.; Furlan, A.; An, Z.; Wang, L.; Hultman, I.; Ahrlund-Richter, L.; et al. Glial origin of mesenchymal stem cells in a tooth model system. Nature 2014, 513, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Wada, N.; Hasegawa, D.; Miyaji, H.; Mitarai, H.; Tomokiyo, A.; Hamano, S.; Maeda, H. Semaphorin 3A Induces Odontoblastic Phenotype in Dental Pulp Stem Cells. J. Dent. Res. 2016, 95, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, S.; Fan, M.; Li, X.; Liu, T.; Yao, Y. Effects of interleukin-1β on mineralization potential of dental pulp stem cells. Zhonghua Kou Qiang Yi Xue Za Zhi 2011, 46, 406–411. [Google Scholar] [PubMed]
- Rombouts, C.; Jeanneau, C.; Bakopoulou, A.; About, I. Dental Pulp Stem Cell Recruitment Signals within Injured Dental Pulp Tissue. Dent. J. 2016, 4, 8. [Google Scholar] [CrossRef]
- Ullah, I.; Subbarao, R.B.; Rho, G.J. Human mesenchymal stem cells—Current trends and future prospective. Biosci. Rep. 2015, 35. [Google Scholar] [CrossRef]
- Huang, G.T.-J.; Gronthos, S.; Shi, S. Mesenchymal stem cells derived from dental tissues vs. those from other sources: Their biology and role in regenerative medicine. J. Dent. Res. 2009, 88, 792–806. [Google Scholar] [CrossRef]
- Ducimetière, F.; Lurkin, A.; Ranchère-Vince, D.; Decouvelaere, A.-V.; Péoc’h, M.; Istier, L.; Chalabreysse, P.; Muller, C.; Alberti, L.; Bringuier, P.-P.; et al. Incidence of Sarcoma Histotypes and Molecular Subtypes in a Prospective Epidemiological Study with Central Pathology Review and Molecular Testing. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Burningham, Z.; Hashibe, M.; Spector, L.; Schiffman, J.D. The Epidemiology of Sarcoma. Clin. Sarcoma Res. 2012, 2, 14. [Google Scholar] [CrossRef]
- Lv, F.-J.; Tuan, R.S.; Cheung, K.M.C.; Leung, V.Y.L. Concise review: The surface markers and identity of human mesenchymal stem cells. Stem Cells 2014, 32, 1408–1419. [Google Scholar] [CrossRef] [PubMed]
- Popov, A.; Scotchford, C.; Grant, D.; Sottile, V. Impact of Serum Source on Human Mesenchymal Stem Cell Osteogenic Differentiation in Culture. Int. J. Mol. Sci. 2019, 20, 5051. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Lü, L.; Sun, H.; Zhang, J.; Ma, W.; Zhang, T. Effect of serum on the differentiation of neural stem cells. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi 2018, 32, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Kermani, S.; Megat Abdul Wahab, R.; Zarina Zainol Abidin, I.; Zainal Ariffin, Z.; Senafi, S.; Hisham Zainal Ariffin, S. Differentiation capacity of mouse dental pulp stem cells into osteoblasts and osteoclasts. Cell J. 2014, 16, 31–42. [Google Scholar]
- Hong, X.; Chedid, K.; Kalkanis, S.N. Glioblastoma cell line-derived spheres in serum-containing medium versus serum-free medium: A comparison of cancer stem cell properties. Int. J. Oncol. 2012, 41, 1693–1700. [Google Scholar] [CrossRef]
- Pisciotta, A.; Riccio, M.; Carnevale, G.; Beretti, F.; Gibellini, L.; Maraldi, T.; Cavallini, G.M.; Ferrari, A.; Bruzzesi, G.; De Pol, A. Human serum promotes osteogenic differentiation of human dental pulp stem cells in vitro and in vivo. PLoS ONE 2012, 7, e50542. [Google Scholar] [CrossRef]
- Pisciotta, A.; Bertoni, L.; Riccio, M.; Mapelli, J.; Bigiani, A.; La Noce, M.; Orciani, M.; de Pol, A.; Carnevale, G. Use of a 3D Floating Sphere Culture System to Maintain the Neural Crest-Related Properties of Human Dental Pulp Stem Cells. Front. Physiol 2018, 9. [Google Scholar] [CrossRef]
- Snippert, H.J.; van Es, J.H.; van den Born, M.; Begthel, H.; Stange, D.E.; Barker, N.; Clevers, H. Prominin-1/CD133 marks stem cells and early progenitors in mouse small intestine. Gastroenterology 2009, 136, 2187–2194.e1. [Google Scholar] [CrossRef]
- Todaro, M.; Perez Alea, M.; Scopelliti, A.; Medema, J.P.; Stassi, G. IL-4-mediated drug resistance in colon cancer stem cells. Cell Cycle 2008, 7, 309–313. [Google Scholar] [CrossRef]
- Choi, D.; Lee, H.-W.; Hur, K.-Y.; Kim, J.-J.; Park, G.-S.; Jang, S.-H.; Song, Y.-S.; Jang, K.-S.; Paik, S.-S. Cancer stem cell markers CD133 and CD24 correlate with invasiveness and differentiation in colorectal adenocarcinoma. World J. Gastroenterol. 2009, 15, 2258–2264. [Google Scholar] [CrossRef]
- Florek, M.; Haase, M.; Marzesco, A.-M.; Freund, D.; Ehninger, G.; Huttner, W.B.; Corbeil, D. Prominin-1/CD133, a neural and hematopoietic stem cell marker, is expressed in adult human differentiated cells and certain types of kidney cancer. Cell Tissue Res. 2005, 319, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Pfenninger, C.V.; Roschupkina, T.; Hertwig, F.; Kottwitz, D.; Englund, E.; Bengzon, J.; Jacobsen, S.E.; Nuber, U.A. CD133 is not present on neurogenic astrocytes in the adult subventricular zone, but on embryonic neural stem cells, ependymal cells, and glioblastoma cells. Cancer Res. 2007, 67, 5727–5736. [Google Scholar] [CrossRef] [PubMed]
- Virant-Klun, I.; Skerl, P.; Novakovic, S.; Vrtacnik-Bokal, E.; Smrkolj, S. Similar Population of CD133+ and DDX4+ VSEL-Like Stem Cells Sorted from Human Embryonic Stem Cell, Ovarian, and Ovarian Cancer Ascites Cell Cultures: The Real Embryonic Stem Cells? Cells 2019, 8, 706. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Viña-Almunia, J.; Mas-Bargues, C.; Borras, C.; Gambini, J.; El Alami, M.; Sanz-Ros, J.; Peñarrocha, M.; Vina, J. Influence of Partial O₂ Pressure on the Adhesion, Proliferation, and Osteogenic Differentiation of Human Dental Pulp Stem Cells on β-Tricalcium Phosphate Scaffold. Int. J. Oral. Maxillofac. Implants 2017, 32, 1251–1256. [Google Scholar] [CrossRef] [PubMed]
- Bonnamain, V.; Thinard, R.; Sergent-Tanguy, S.; Huet, P.; Bienvenu, G.; Naveilhan, P.; Farges, J.-C.; Alliot-Licht, B. Human dental pulp stem cells cultured in serum-free supplemented medium. Front. Physiol 2013, 4. [Google Scholar] [CrossRef]
- Jang, T.J.; Park, J.B.; Lee, J.I. The Expression of CD10 and CD15 Is Progressively Increased during Colorectal Cancer Development. Korean J. Pathol. 2013, 47, 340–347. [Google Scholar] [CrossRef]
- Daynac, M.; Tirou, L.; Faure, H.; Mouthon, M.-A.; Gauthier, L.R.; Hahn, H.; Boussin, F.D.; Ruat, M. Hedgehog Controls Quiescence and Activation of Neural Stem Cells in the Adult Ventricular-Subventricular Zone. Stem Cell Rep. 2016, 7, 735–748. [Google Scholar] [CrossRef]
- Daynac, M.; Chicheportiche, A.; Pineda, J.R.; Gauthier, L.R.; Boussin, F.D.; Mouthon, M.A. Quiescent neural stem cells exit dormancy upon alteration of GABAAR signaling following radiation damage. Stem Cell Res. 2013, 11, 516–528. [Google Scholar] [CrossRef]
- Son, M.J.; Woolard, K.; Nam, D.-H.; Lee, J.; Fine, H.A. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell 2009, 4, 440–452. [Google Scholar] [CrossRef]
- Silvestre, D.C.; Pineda, J.R.; Hoffschir, F.; Studler, J.M.; Mouthon, M.A.; Pflumio, F.; Junier, M.P.; Chneiweiss, H.; Boussin, F.D. Alternative lengthening of telomeres in human glioma stem cells. Stem Cells 2011, 29, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.-G.; Zhang, X.; Xue, X.-Y.; Guo, G.; Wang, P.; Zhang, W.; Fei, Z.; Zhen, H.-N.; You, S.-W.; Yang, H. Brain Tumor Stem-Like Cells Identified by Neural Stem Cell Marker CD15. Transl. Oncol. 2009, 2, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Uribe-Etxebarria, V.; Luzuriaga, J.; Garcia-Gallastegui, P.; Agliano, A.; Unda, F.; Ibarretxe, G. Notch/Wnt cross-signalling regulates stemness of dental pulp stem cells through expression of neural crest and core pluripotency factors. Eur. Cell Mater. 2017, 34, 249–270. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Clevers, H. Tracking down the stem cells of the intestine: Strategies to identify adult stem cells. Gastroenterology 2007, 133, 1755–1760. [Google Scholar] [CrossRef]
- de Sousa e Melo, F.; Kurtova, A.V.; Harnoss, J.M.; Kljavin, N.; Hoeck, J.D.; Hung, J.; Anderson, J.E.; Storm, E.E.; Modrusan, Z.; Koeppen, H.; et al. A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature 2017, 543, 676–680. [Google Scholar] [CrossRef]
- Satoh, J.; Obayashi, S.; Tabunoki, H.; Wakana, T.; Kim, S.U. Stable expression of neurogenin 1 induces LGR5, a novel stem cell marker, in an immortalized human neural stem cell line HB1.F3. Cell. Mol. Neurobiol. 2010, 30, 415–426. [Google Scholar] [CrossRef]
- Yu, Y.; Moberly, A.H.; Bhattarai, J.P.; Duan, C.; Zheng, Q.; Li, F.; Huang, H.; Olson, W.; Luo, W.; Wen, T.; et al. The Stem Cell Marker Lgr5 Defines a Subset of Postmitotic Neurons in the Olfactory Bulb. J. Neurosci. 2017, 37, 9403–9414. [Google Scholar] [CrossRef]
- Hiraoka, K.; Hayashi, T.; Kaneko, R.; Nasu-Nishimura, Y.; Koyama-Nasu, R.; Kawasaki, Y.; Akiyama, T. SOX9-mediated upregulation of LGR5 is important for glioblastoma tumorigenicity. Biochem. Biophys. Res. Commun. 2015, 460, 216–221. [Google Scholar] [CrossRef]
- Wang, F.; Scoville, D.; He, X.C.; Mahe, M.M.; Box, A.; Perry, J.M.; Smith, N.R.; Lei, N.Y.; Davies, P.S.; Fuller, M.K.; et al. Isolation and characterization of intestinal stem cells based on surface marker combinations and colony-formation assay. Gastroenterology 2013, 145, 383–395.e1–21. [Google Scholar] [CrossRef]
- Levin, T.G.; Powell, A.E.; Davies, P.S.; Silk, A.D.; Dismuke, A.D.; Anderson, E.C.; Swain, J.R.; Wong, M.H. Characterization of the intestinal cancer stem cell marker CD166 in the human and mouse gastrointestinal tract. Gastroenterology 2010, 139, 2072–2082.e5. [Google Scholar] [CrossRef]
- Fu, L.; Zhu, L.; Huang, Y.; Lee, T.D.; Forman, S.J.; Shih, C.-C. Derivation of Neural Stem Cells from Mesenchymal Stem Cells: Evidence for a Bipotential Stem Cell Population. Stem Cells Dev. 2008, 17, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Kijima, N.; Hosen, N.; Kagawa, N.; Hashimoto, N.; Nakano, A.; Fujimoto, Y.; Kinoshita, M.; Sugiyama, H.; Yoshimine, T. CD166/activated leukocyte cell adhesion molecule is expressed on glioblastoma progenitor cells and involved in the regulation of tumor cell invasion. Neuro-Oncology 2012, 14, 1254–1264. [Google Scholar] [CrossRef] [PubMed]
- Hadaegh, Y.; Niknam, M.; Attar, A.; Maharlooei, M.K.; Tavangar, M.S.; Aarabi, A.M.; Monabati, A. Characterization of stem cells from the pulp of unerupted third molar tooth. Indian J. Dent. Res. 2014, 25, 14–21. [Google Scholar] [CrossRef] [PubMed]
- D’ Alimonte, I.; Nargi, E.; Mastrangelo, F.; Falco, G.; Lanuti, P.; Marchisio, M.; Miscia, S.; Robuffo, I.; Capogreco, M.; Buccella, S.; et al. Vascular endothelial growth factor enhances in vitro proliferation and osteogenic differentiation of human dental pulp stem cells. J. Biol. Regul. Homeost. Agents 2011, 25, 57–69. [Google Scholar]
- Jing, F.; Kim, H.J.; Kim, C.H.; Kim, Y.J.; Lee, J.H.; Kim, H.R. Colon cancer stem cell markers CD44 and CD133 in patients with colorectal cancer and synchronous hepatic metastases. Int. J. Oncol. 2015, 46, 1582–1588. [Google Scholar] [CrossRef]
- Su, W.; Foster, S.C.; Xing, R.; Feistel, K.; Olsen, R.H.J.; Acevedo, S.F.; Raber, J.; Sherman, L.S. CD44 Transmembrane Receptor and Hyaluronan Regulate Adult Hippocampal Neural Stem Cell Quiescence and Differentiation. J. Biol. Chem. 2017, 292, 4434–4445. [Google Scholar] [CrossRef]
- Nishikawa, M.; Inoue, A.; Ohnishi, T.; Kohno, S.; Ohue, S.; Matsumoto, S.; Suehiro, S.; Yamashita, D.; Ozaki, S.; Watanabe, H.; et al. Significance of Glioma Stem-Like Cells in the Tumor Periphery That Express High Levels of CD44 in Tumor Invasion, Early Progression, and Poor Prognosis in Glioblastoma. Stem Cells Int. 2018, 2018, 5387041. [Google Scholar] [CrossRef]
- Rimkus, T.K.; Carpenter, R.L.; Sirkisoon, S.; Zhu, D.; Pasche, B.C.; Chan, M.D.; Lesser, G.J.; Tatter, S.B.; Watabe, K.; Debinski, W.; et al. Truncated Glioma-Associated Oncogene Homolog 1 (tGLI1) Mediates Mesenchymal Glioblastoma via Transcriptional Activation of CD44. Cancer Res. 2018, 78, 2589–2600. [Google Scholar] [CrossRef]
- Wang, H.-H.; Liao, C.-C.; Chow, N.-H.; Huang, L.L.-H.; Chuang, J.-I.; Wei, K.-C.; Shin, J.-W. Whether CD44 is an applicable marker for glioma stem cells. Am. J. Transl Res. 2017, 9, 4785–4806. [Google Scholar]
- Macrin, D.; Alghadeer, A.; Zhao, Y.T.; Miklas, J.W.; Hussein, A.M.; Detraux, D.; Robitaille, A.M.; Madan, A.; Moon, R.T.; Wang, Y.; et al. Metabolism as an early predictor of DPSCs aging. Sci. Rep. 2019, 9, 2195. [Google Scholar] [CrossRef]
- Karpus, O.N.; Westendorp, B.F.; Vermeulen, J.L.M.; Meisner, S.; Koster, J.; Muncan, V.; Wildenberg, M.E.; van den Brink, G.R. Colonic CD90+ Crypt Fibroblasts Secrete Semaphorins to Support Epithelial Growth. Cell Rep. 2019, 26, 3698–3708.e5. [Google Scholar] [CrossRef] [PubMed]
- Hirashima, K.; Yue, F.; Kobayashi, M.; Uchida, Y.; Nakamura, S.; Tomotsune, D.; Matsumoto, K.; Takizawa-Shirasawa, S.; Yokoyama, T.; Kanno, H.; et al. Cell biological profiling of reprogrammed cancer stem cell-like colon cancer cells maintained in culture. Cell Tissue Res. 2019, 375, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yi, D.-Y.; Xue, B.-Z.; Wen, W.-W.; Lu, Y.-P.; Abdelmaksou, A.; Sun, M.-X.; Yuan, D.-T.; Zhao, H.-Y.; Xiong, N.-X.; et al. CD90 determined two subpopulations of glioma-associated mesenchymal stem cells with different roles in tumour progression. Cell Death Dis. 2018, 9, 1101. [Google Scholar] [CrossRef] [PubMed]
- Luzuriaga, J.; Pineda, J.R.; Irastorza, I.; Uribe-Etxebarria, V.; García-Gallastegui, P.; Encinas, J.M.; Chamero, P.; Unda, F.; Ibarretxe, G. BDNF and NT3 Reprogram Human Ectomesenchymal Dental Pulp Stem Cells to Neurogenic and Gliogenic Neural Crest Progenitors Cultured in Serum-Free Medium. Cell. Physiol. Biochem. 2019, 52, 1361–1380. [Google Scholar] [CrossRef] [PubMed]
- Hilkens, P.; Gervois, P.; Fanton, Y.; Vanormelingen, J.; Martens, W.; Struys, T.; Politis, C.; Lambrichts, I.; Bronckaers, A. Effect of isolation methodology on stem cell properties and multilineage differentiation potential of human dental pulp stem cells. Cell Tissue Res. 2013, 353, 65–78. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, C.-Q.; Fan, L.-F. Correlation of Musashi-1, Lgr5, and pEGFR expressions in human small intestinal adenocarcinomas. Tumour Biol. 2015, 36, 6075–6082. [Google Scholar] [CrossRef]
- Zhang, S.; Han, Z.; Jing, Y.; Tao, S.; Li, T.; Wang, H.; Wang, Y.; Li, R.; Yang, Y.; Zhao, X.; et al. CD133(+)CXCR4(+) colon cancer cells exhibit metastatic potential and predict poor prognosis of patients. BMC Med. 2012, 10, 85. [Google Scholar] [CrossRef]
- Ho, S.-Y.; Ling, T.-Y.; Lin, H.-Y.; Liou, J.T.-J.; Liu, F.-C.; Chen, I.-C.; Lee, S.-W.; Hsu, Y.; Lai, D.-M.; Liou, H.-H. SDF-1/CXCR4 Signaling Maintains Stemness Signature in Mouse Neural Stem/Progenitor Cells. Stem Cells Int. 2017, 2017, 2493752. [Google Scholar] [CrossRef]
- Yi, L.; Zhou, X.; Li, T.; Liu, P.; Hai, L.; Tong, L.; Ma, H.; Tao, Z.; Xie, Y.; Zhang, C.; et al. Notch1 signaling pathway promotes invasion, self-renewal and growth of glioma initiating cells via modulating chemokine system CXCL12/CXCR4. J. Exp. Clin. Cancer Res. 2019, 38, 339. [Google Scholar] [CrossRef]
- Li, M.; Sun, X.; Ma, L.; Jin, L.; Zhang, W.; Xiao, M.; Yu, Q. SDF-1/CXCR4 axis induces human dental pulp stem cell migration through FAK/PI3K/Akt and GSK3β/β-catenin pathways. Sci. Rep. 2017, 7, 40161. [Google Scholar] [CrossRef]
- Li, Y.; Wang, L.; Pappan, L.; Galliher-Beckley, A.; Shi, J. IL-1β promotes stemness and invasiveness of colon cancer cells through Zeb1 activation. Mol. Cancer 2012, 11, 87. [Google Scholar] [CrossRef] [PubMed]
- Ehrmann, J.; Kolár, Z.; Mokry, J. Nestin as a diagnostic and prognostic marker: Immunohistochemical analysis of its expression in different tumours. J. Clin. Pathol. 2005, 58, 222–223. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; DiMaio, T.A.; Scheef, E.A.; Sorenson, C.M.; Sheibani, N. PECAM-1 regulates proangiogenic properties of endothelial cells through modulation of cell-cell and cell-matrix interactions. Am. J. Physiol. Cell Physiol. 2010, 299, C1468–C1484. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Potten, C.S.; Booth, C.; Tudor, G.L.; Booth, D.; Brady, G.; Hurley, P.; Ashton, G.; Clarke, R.; Sakakibara, S.; Okano, H. Identification of a putative intestinal stem cell and early lineage marker; musashi-1. Differentiation 2003, 71, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Chiou, G.-Y.; Yang, T.-W.; Huang, C.-C.; Tang, C.-Y.; Yen, J.-Y.; Tsai, M.-C.; Chen, H.-Y.; Fadhilah, N.; Lin, C.-C.; Jong, Y.-J. Musashi-1 promotes a cancer stem cell lineage and chemoresistance in colorectal cancer cells. Sci. Rep. 2017, 7, 2172. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, S.; Imai, T.; Hamaguchi, K.; Okabe, M.; Aruga, J.; Nakajima, K.; Yasutomi, D.; Nagata, T.; Kurihara, Y.; Uesugi, S.; et al. Mouse-Musashi-1, a neural RNA-binding protein highly enriched in the mammalian CNS stem cell. Dev. Biol. 1996, 176, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-Y.; Lin, L.-T.; Wang, M.-L.; Lee, S.-H.; Tsai, M.-L.; Tsai, C.-C.; Liu, W.-H.; Chen, T.-C.; Yang, Y.-P.; Lee, Y.-Y.; et al. Musashi-1 regulates AKT-derived IL-6 autocrinal/paracrinal malignancy and chemoresistance in glioblastoma. Oncotarget 2016, 7, 42485–42501. [Google Scholar] [CrossRef] [PubMed]
- Padial-Molina, M.; de Buitrago, J.G.; Sainz-Urruela, R.; Abril-Garcia, D.; Anderson, P.; O’Valle, F.; Galindo-Moreno, P. Expression of Musashi-1 During Osteogenic Differentiation of Oral MSC: An In Vitro Study. Int. J. Mol. Sci. 2019, 20, 2171. [Google Scholar] [CrossRef]
- Lundberg, I.V.; Edin, S.; Eklöf, V.; Öberg, Å.; Palmqvist, R.; Wikberg, M.L. SOX2 expression is associated with a cancer stem cell state and down-regulation of CDX2 in colorectal cancer. BMC Cancer 2016, 16, 471. [Google Scholar] [CrossRef]
- Zhang, S.; Cui, W. Sox2, a key factor in the regulation of pluripotency and neural differentiation. World J. Stem Cells 2014, 6, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Hattermann, K.; Flüh, C.; Engel, D.; Mehdorn, H.M.; Synowitz, M.; Mentlein, R.; Held-Feindt, J. Stem cell markers in glioma progression and recurrence. Int. J. Oncol. 2016, 49, 1899–1910. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Xu, H.; Huang, M.; Ma, W.; Saxena, D.; Lustig, R.A.; Alonso-Basanta, M.; Zhang, Z.; O’Rourke, D.M.; Zhang, L.; et al. Circulating Glioma Cells Exhibit Stem Cell-like Properties. Cancer Res. 2018, 78, 6632–6642. [Google Scholar] [CrossRef] [PubMed]
- Uribe-Etxebarria, V.; Agliano, A.; Unda, F.; Ibarretxe, G. Wnt signaling reprograms metabolism in dental pulp stem cells. J. Cell. Physiol. 2019, 234, 13068–13082. [Google Scholar] [CrossRef] [PubMed]
- Uribe-Etxebarria, V.; García-Gallastegui, P.; Pérez-Garrastachu, M.; Casado-Andrés, M.; Irastorza, I.; Unda, F.; Ibarretxe, G.; Subirán, N. Wnt-3a Induces Epigenetic Remodeling in Human Dental Pulp Stem Cells. Cells 2020, 9, 652. [Google Scholar] [CrossRef] [PubMed]
- Wen, K.; Fu, Z.; Wu, X.; Feng, J.; Chen, W.; Qian, J. Oct-4 is required for an antiapoptotic behavior of chemoresistant colorectal cancer cells enriched for cancer stem cells: Effects associated with STAT3/Survivin. Cancer Lett. 2013, 333, 56–65. [Google Scholar] [CrossRef]
- Vincent, P.H.; Benedikz, E.; Uhlén, P.; Hovatta, O.; Sundström, E. Expression of Pluripotency Markers in Nonpluripotent Human Neural Stem and Progenitor Cells. Stem Cells Dev. 2017, 26, 876–887. [Google Scholar] [CrossRef]
- Asadi, M.H.; Khalifeh, K.; Mowla, S.J. OCT4 spliced variants are highly expressed in brain cancer tissues and inhibition of OCT4B1 causes G2/M arrest in brain cancer cells. J. Neurooncol. 2016, 130, 455–463. [Google Scholar] [CrossRef]
- Luzuriaga, J.; Pastor-Alonso, O.; Encinas, J.M.; Unda, F.; Ibarretxe, G.; Pineda, J.R. Human Dental Pulp Stem Cells Grown in Neurogenic Media Differentiate Into Endothelial Cells and Promote Neovasculogenesis in the Mouse Brain. Front. Physiol. 2019, 10, 347. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, C.; Wang, H.-Z.; Peng, Y.-N.; Li, H.-O.; Zhou, Y.-J.; Liu, S.; Wang, F.; Liu, L.; Chang, Y.; et al. Soft fibrin matrix downregulates DAB2IP to promote Nanog-dependent growth of colon tumor-repopulating cells. Cell Death Dis. 2019, 10, 151. [Google Scholar] [CrossRef]
- Gregorian, C.; Nakashima, J.; Le Belle, J.; Ohab, J.; Kim, R.; Liu, A.; Smith, K.B.; Groszer, M.; Garcia, A.D.; Sofroniew, M.V.; et al. Pten deletion in adult neural stem/progenitor cells enhances constitutive neurogenesis. J. Neurosci. 2009, 29, 1874–1886. [Google Scholar] [CrossRef] [PubMed]
- Groszer, M.; Erickson, R.; Scripture-Adams, D.D.; Lesche, R.; Trumpp, A.; Zack, J.A.; Kornblum, H.I.; Liu, X.; Wu, H. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science 2001, 294, 2186–2189. [Google Scholar] [CrossRef] [PubMed]
- He, X.C.; Yin, T.; Grindley, J.C.; Tian, Q.; Sato, T.; Tao, W.A.; Dirisina, R.; Porter-Westpfahl, K.S.; Hembree, M.; Johnson, T.; et al. PTEN-deficient intestinal stem cells initiate intestinal polyposis. Nat. Genet. 2007, 39, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Yu, Y.; Padhye, S.B.; Sarkar, F.H.; Majumdar, A.P.N. Difluorinated-curcumin (CDF) restores PTEN expression in colon cancer cells by down-regulating miR-21. PLoS ONE 2013, 8, e68543. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Yuan, G.; Liu, X.; Ren, R.; Li, J.; Zhang, W.; Wu, J.; Xu, X.; Fu, L.; Li, Y.; et al. PTEN deficiency reprogrammes human neural stem cells towards a glioblastoma stem cell-like phenotype. Nat. Commun. 2015, 6, 10068. [Google Scholar] [CrossRef]
- Shen, W.-C.; Lai, Y.-C.; Li, L.-H.; Liao, K.; Lai, H.-C.; Kao, S.-Y.; Wang, J.; Chuong, C.-M.; Hung, S.-C. Methylation and PTEN activation in dental pulp mesenchymal stem cells promotes osteogenesis and reduces oncogenesis. Nat. Commun. 2019, 10, 2226. [Google Scholar] [CrossRef]
- Shmelkov, S.V.; Butler, J.M.; Hooper, A.T.; Hormigo, A.; Kushner, J.; Milde, T.; St Clair, R.; Baljevic, M.; White, I.; Jin, D.K.; et al. CD133 expression is not restricted to stem cells, and both CD133+ and CD133- metastatic colon cancer cells initiate tumors. J. Clin. Investig. 2008, 118, 2111–2120. [Google Scholar] [CrossRef]
- Paschall, A.V.; Yang, D.; Lu, C.; Redd, P.S.; Choi, J.-H.; Heaton, C.M.; Lee, J.R.; Nayak-Kapoor, A.; Liu, K. CD133+CD24lo defines a 5-Fluorouracil-resistant colon cancer stem cell-like phenotype. Oncotarget 2016, 7, 78698–78712. [Google Scholar] [CrossRef]
- Glumac, P.M.; LeBeau, A.M. The role of CD133 in cancer: A concise review. Clin. Transl. Med. 2018, 7, 18. [Google Scholar] [CrossRef]
- Uchida, N.; Buck, D.W.; He, D.; Reitsma, M.J.; Masek, M.; Phan, T.V.; Tsukamoto, A.S.; Gage, F.H.; Weissman, I.L. Direct isolation of human central nervous system stem cells. Proc. Natl. Acad. Sci. USA 2000, 97, 14720–14725. [Google Scholar] [CrossRef]
- Schwartz, P.H.; Bryant, P.J.; Fuja, T.J.; Su, H.; O’Dowd, D.K.; Klassen, H. Isolation and characterization of neural progenitor cells from post-mortem human cortex. J. Neurosci. Res. 2003, 74, 838–851. [Google Scholar] [CrossRef] [PubMed]
- Beckervordersandforth, R.; Tripathi, P.; Ninkovic, J.; Bayam, E.; Lepier, A.; Stempfhuber, B.; Kirchhoff, F.; Hirrlinger, J.; Haslinger, A.; Lie, D.C.; et al. In vivo fate mapping and expression analysis reveals molecular hallmarks of prospectively isolated adult neural stem cells. Cell Stem Cell 2010, 7, 744–758. [Google Scholar] [CrossRef] [PubMed]
- Lottaz, C.; Beier, D.; Meyer, K.; Kumar, P.; Hermann, A.; Schwarz, J.; Junker, M.; Oefner, P.J.; Bogdahn, U.; Wischhusen, J.; et al. Transcriptional profiles of CD133+ and CD133- glioblastoma-derived cancer stem cell lines suggest different cells of origin. Cancer Res. 2010, 70, 2030–2040. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-C.; Lee, T.-H.; Huang, Y.-H.; Chang, N.-K.; Lin, Y.-J.; Chien, P.-W.C.; Yang, W.-H.; Lin, M.H.-C. Comparison of surface markers between human and rabbit mesenchymal stem cells. PLoS ONE 2014, 9, e111390. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.-H.; Liu, A.-G.; Gu, W.-G.; Deng, L.; Cheng, X.-G.; Tong, T.-J.; Zhang, H.-Z. CD133+ subpopulation of the HT1080 human fibrosarcoma cell line exhibits cancer stem-like characteristics. Oncol. Rep. 2013, 30, 815–823. [Google Scholar] [CrossRef]
- Irollo, E.; Pirozzi, G. CD133: To be or not to be, is this the real question? Am. J. Transl Res. 2013, 5, 563–581. [Google Scholar]
- Sokol, S.Y. Maintaining embryonic stem cell pluripotency with Wnt signaling. Development 2011, 138, 4341–4350. [Google Scholar] [CrossRef]
- Barraud, P.; Stott, S.; Møllgård, K.; Parmar, M.; Björklund, A. In vitro characterization of a human neural progenitor cell coexpressing SSEA4 and CD133. J. Neurosci. Res. 2007, 85, 250–259. [Google Scholar] [CrossRef]
- Patru, C.; Romao, L.; Varlet, P.; Coulombel, L.; Raponi, E.; Cadusseau, J.; Renault-Mihara, F.; Thirant, C.; Leonard, N.; Berhneim, A.; et al. CD133, CD15/SSEA-1, CD34 or side populations do not resume tumor-initiating properties of long-term cultured cancer stem cells from human malignant glio-neuronal tumors. BMC Cancer 2010, 10, 66. [Google Scholar] [CrossRef]
- Kahlert, U.D.; Bender, N.O.; Maciaczyk, D.; Bogiel, T.; Bar, E.E.; Eberhart, C.G.; Nikkhah, G.; Maciaczyk, J. CD133/CD15 defines distinct cell subpopulations with differential in vitro clonogenic activity and stem cell-related gene expression profile in in vitro propagated glioblastoma multiforme-derived cell line with a PNET-like component. Folia Neuropathol. 2012, 50, 357–368. [Google Scholar] [CrossRef]
- Friedman, G.K.; Moore, B.P.; Nan, L.; Kelly, V.M.; Etminan, T.; Langford, C.P.; Xu, H.; Han, X.; Markert, J.M.; Beierle, E.A.; et al. Pediatric medulloblastoma xenografts including molecular subgroup 3 and CD133+ and CD15+ cells are sensitive to killing by oncolytic herpes simplex viruses. Neuro-Oncology 2016, 18, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Ariza, A.; López, D.; Castellà, E.M.; Muñoz, C.; Zújar, M.J.; Mate, J.L. Expression of CD15 in normal and metaplastic Paneth cells of the digestive tract. J. Clin. Pathol. 1996, 49, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Bakopoulou, A.; Apatzidou, D.; Aggelidou, E.; Gousopoulou, E.; Leyhausen, G.; Volk, J.; Kritis, A.; Koidis, P.; Geurtsen, W. Isolation and prolonged expansion of oral mesenchymal stem cells under clinical-grade, GMP-compliant conditions differentially affects “stemness” properties. Stem Cell Res. Ther. 2017, 8, 247. [Google Scholar] [CrossRef] [PubMed]
- Yudoh, K.; Matsui, H.; Tsuji, H. Nitric oxide induced by tumor cells activates tumor cell adhesion to endothelial cells and permeability of the endothelium in vitro. Clin. Exp. Metastasis 1997, 15, 557–567. [Google Scholar] [CrossRef]
- Carmon, K.S.; Lin, Q.; Gong, X.; Thomas, A.; Liu, Q. LGR5 interacts and cointernalizes with Wnt receptors to modulate Wnt/β-catenin signaling. Mol. Cell. Biol. 2012, 32, 2054–2064. [Google Scholar] [CrossRef]
- Xu, L.; Lin, W.; Wen, L.; Li, G. Lgr5 in cancer biology: Functional identification of Lgr5 in cancer progression and potential opportunities for novel therapy. Stem Cell Res. Ther. 2019, 10, 219. [Google Scholar] [CrossRef]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef]
- Snyder, J.C.; Rochelle, L.K.; Marion, S.; Lyerly, H.K.; Barak, L.S.; Caron, M.G. Lgr4 and Lgr5 drive the formation of long actin-rich cytoneme-like membrane protrusions. J. Cell Sci. 2015, 128, 1230–1240. [Google Scholar] [CrossRef]
- Schoumacher, M.; Goldman, R.D.; Louvard, D.; Vignjevic, D.M. Actin, microtubules, and vimentin intermediate filaments cooperate for elongation of invadopodia. J. Cell Biol. 2010, 189, 541–556. [Google Scholar] [CrossRef]
- Lin, W.; Xu, L.; Lin, S.; Shi, L.; Wang, B.; Pan, Q.; Lee, W.Y.W.; Li, G. Characterisation of multipotent stem cells from human peripheral blood using an improved protocol. J. Orthop. Translat. 2019, 19, 18–28. [Google Scholar] [CrossRef]
- Kim, J.-H.; Jeon, M.; Song, J.-S.; Lee, J.-H.; Choi, B.-J.; Jung, H.-S.; Moon, S.J.; DenBesten, P.K.; Kim, S.-O. Distinctive genetic activity pattern of the human dental pulp between deciduous and permanent teeth. PLoS ONE 2014, 9, e102893. [Google Scholar] [CrossRef] [PubMed]
- Rot, S.; Taubert, H.; Bache, M.; Greither, T.; Würl, P.; Eckert, A.W.; Schubert, J.; Vordermark, D.; Kappler, M. A novel splice variant of the stem cell marker LGR5/GPR49 is correlated with the risk of tumor-related death in soft-tissue sarcoma patients. BMC Cancer 2011, 11, 429. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.N.; Zamler, D.; Baker, G.J.; Kadiyala, P.; Erdreich-Epstein, A.; DeCarvalho, A.C.; Mikkelsen, T.; Castro, M.G.; Lowenstein, P.R. CXCR4 increases in-vivo glioma perivascular invasion, and reduces radiation induced apoptosis: A genetic knockdown study. Oncotarget 2016, 7, 83701–83719. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Cao, J.; Ji, Z.; Wang, J.; Jiang, T.; Ding, H. Co-expression of Lgr5 and CXCR4 characterizes cancer stem-like cells of colorectal cancer. Oncotarget 2016, 7, 81144–81155. [Google Scholar] [CrossRef]
- Ganju, R.K.; Brubaker, S.A.; Meyer, J.; Dutt, P.; Yang, Y.; Qin, S.; Newman, W.; Groopman, J.E. The alpha-chemokine, stromal cell-derived factor-1alpha, binds to the transmembrane G-protein-coupled CXCR-4 receptor and activates multiple signal transduction pathways. J. Biol. Chem. 1998, 273, 23169–23175. [Google Scholar] [CrossRef]
- Xiu, G.; Li, X.; Yin, Y.; Li, J.; Li, B.; Chen, X.; Liu, P.; Sun, J.; Ling, B. SDF-1/CXCR4 Augments the Therapeutic Effect of Bone Marrow Mesenchymal Stem Cells in the Treatment of Lipopolysaccharide-Induced Liver Injury by Promoting Their Migration Through PI3K/Akt Signaling Pathway. Cell Transplant. 2020, 29, 963689720929992. [Google Scholar] [CrossRef]
- Hermann, A.; Gastl, R.; Liebau, S.; Popa, M.O.; Fiedler, J.; Boehm, B.O.; Maisel, M.; Lerche, H.; Schwarz, J.; Brenner, R.; et al. Efficient generation of neural stem cell-like cells from adult human bone marrow stromal cells. J. Cell. Sci. 2004, 117, 4411–4422. [Google Scholar] [CrossRef]
- Shafaei, S.; Sharbatdaran, M.; Kamrani, G.; Khafri, S. The association between CD166 detection rate and clinicopathologic parameters of patients with colorectal cancer. Caspian J. Intern. Med. 2013, 4, 768–772. [Google Scholar]
- Kumar, A.; Bhanja, A.; Bhattacharyya, J.; Jaganathan, B.G. Multiple roles of CD90 in cancer. Tumour Biol. 2016, 37, 11611–11622. [Google Scholar] [CrossRef]
- Parry, P.V.; Engh, J.A. CD90 is identified as a marker for cancer stem cells in high-grade gliomas using tissue microarrays. Neurosurgery 2012, 70, N23–N24. [Google Scholar] [CrossRef][Green Version]
- Park, D.; Xiang, A.P.; Mao, F.F.; Zhang, L.; Di, C.-G.; Liu, X.-M.; Shao, Y.; Ma, B.-F.; Lee, J.-H.; Ha, K.-S.; et al. Nestin is required for the proper self-renewal of neural stem cells. Stem Cells 2010, 28, 2162–2171. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.H.; Wu, Q.L.; Yu, X.B.; Xu, C.X.; Ma, B.F.; Zhang, X.M.; Li, S.N.; Lahn, B.T.; Xiang, A.P. Nestin expression in different tumours and its relevance to malignant grade. J. Clin. Pathol. 2008, 61, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Lv, D.; Lu, L.; Hu, Z.; Fei, Z.; Liu, M.; Wei, L.; Xu, J. Nestin Expression Is Associated with Poor Clinicopathological Features and Prognosis in Glioma Patients: An Association Study and Meta-analysis. Mol. Neurobiol. 2017, 54, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef]
- Li, J.; Wang, R.; Yang, L.; Wu, Q.; Wang, Q.; Nie, Z.; Yu, Y.; Ma, J.; Pan, Q. Knockdown of Nestin inhibits proliferation and migration of colorectal cancer cells. Int. J. Clin. Exp. Pathol. 2015, 8, 6377–6386. [Google Scholar]
- Amoh, Y.; Yang, M.; Li, L.; Reynoso, J.; Bouvet, M.; Moossa, A.R.; Katsuoka, K.; Hoffman, R.M. Nestin-linked green fluorescent protein transgenic nude mouse for imaging human tumor angiogenesis. Cancer Res. 2005, 65, 5352–5357. [Google Scholar] [CrossRef]
- Gervois, P.; Struys, T.; Hilkens, P.; Bronckaers, A.; Ratajczak, J.; Politis, C.; Brone, B.; Lambrichts, I.; Martens, W. Neurogenic maturation of human dental pulp stem cells following neurosphere generation induces morphological and electrophysiological characteristics of functional neurons. Stem Cells Dev. 2015, 24, 296–311. [Google Scholar] [CrossRef]
- Martens, W.; Sanen, K.; Georgiou, M.; Struys, T.; Bronckaers, A.; Ameloot, M.; Phillips, J.; Lambrichts, I. Human dental pulp stem cells can differentiate into Schwann cells and promote and guide neurite outgrowth in an aligned tissue-engineered collagen construct in vitro. FASEB J. 2014, 28, 1634–1643. [Google Scholar] [CrossRef]
- Ibarretxe, G.; Crende, O.; Aurrekoetxea, M.; García-Murga, V.; Etxaniz, J.; Unda, F. Neural crest stem cells from dental tissues: A new hope for dental and neural regeneration. Stem Cells Int. 2012, 2012, 103503. [Google Scholar] [CrossRef]
- Aurrekoetxea, M.; Garcia-Gallastegui, P.; Irastorza, I.; Luzuriaga, J.; Uribe-Etxebarria, V.; Unda, F.; Ibarretxe, G. Dental pulp stem cells as a multifaceted tool for bioengineering and the regeneration of craniomaxillofacial tissues. Front. Physiol. 2015, 6. [Google Scholar] [CrossRef]
- Pagella, P.; Miran, S.; Neto, E.; Martin, I.; Lamghari, M.; Mitsiadis, T.A. Human dental pulp stem cells exhibit enhanced properties in comparison to human bone marrow stem cells on neurites outgrowth. FASEB J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.-F.; Dong, W.-G.; Jiang, C.-Q.; Xia, D.; Liao, F.; Yu, Q.-F. Expression of putative stem cell genes Musashi-1 and beta1-integrin in human colorectal adenomas and adenocarcinomas. Int. J. Colorectal Dis. 2010, 25, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Sakakibara, S.; Imai, T.; Suzuki, A.; Nakamura, Y.; Sawamoto, K.; Ogawa, Y.; Toyama, Y.; Miyata, T.; Okano, H. Musashi1: An evolutionally conserved marker for CNS progenitor cells including neural stem cells. Dev. Neurosci. 2000, 22, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-Y.; Lin, L.-T.; Wang, M.-L.; Laurent, B.; Hsu, C.-H.; Pan, C.-M.; Jiang, W.-R.; Chen, P.-Y.; Ma, H.-I.; Chen, Y.-W.; et al. Musashi-1 Enhances Glioblastoma Cell Migration and Cytoskeletal Dynamics through Translational Inhibition of Tensin3. Sci. Rep. 2017, 7, 8710. [Google Scholar] [CrossRef]
- Lin, J.-C.; Tsai, J.-T.; Chao, T.-Y.; Ma, H.-I.; Liu, W.-H. Musashi-1 Enhances Glioblastoma Migration by Promoting ICAM1 Translation. Neoplasia 2019, 21, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.; Wu, H. PTEN, Stem Cells, and Cancer Stem Cells. J. Biol. Chem. 2009, 284, 11755–11759. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, F.; Ross, A.H. PTEN regulation of neural development and CNS stem cells. J. Cell Biochem. 2003, 88, 24–28. [Google Scholar] [CrossRef]
- Amiri, A.; Cho, W.; Zhou, J.; Birnbaum, S.G.; Sinton, C.M.; McKay, R.M.; Parada, L.F. Pten Deletion in Adult Hippocampal Neural Stem/Progenitor Cells Causes Cellular Abnormalities and Alters Neurogenesis. J. Neurosci. 2012, 32, 5880–5890. [Google Scholar] [CrossRef]
- Potdar, P.D.; Jethmalani, Y.D. Human dental pulp stem cells: Applications in future regenerative medicine. World J. Stem Cells 2015, 7, 839–851. [Google Scholar] [CrossRef]
- Schubert, M.; Holland, L.Z. The Wnt Gene Family and the Evolutionary Conservation of Wnt Expression; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- González-Sancho, J.M.; Aguilera, O.; García, J.M.; Pendás-Franco, N.; Peña, C.; Cal, S.; García de Herreros, A.; Bonilla, F.; Muñoz, A. The Wnt antagonist DICKKOPF-1 gene is a downstream target of beta-catenin/TCF and is downregulated in human colon cancer. Oncogene 2005, 24, 1098–1103. [Google Scholar] [CrossRef]
- Jung, Y.-S.; Park, J.-I. Wnt signaling in cancer: Therapeutic targeting of Wnt signaling beyond β-catenin and the destruction complex. Exp. Mol. Med. 2020, 52, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Duchartre, Y.; Kim, Y.-M.; Kahn, M. The Wnt signaling pathway in cancer. Crit. Rev. Oncol. Hematol. 2016, 99, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Shay, J.W. Multiple Roles of APC and its Therapeutic Implications in Colorectal Cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- de Roo, J.J.D.; Breukel, C.; Chhatta, A.R.; Linssen, M.M.; Vloemans, S.A.; Salvatori, D.; Mikkers, H.M.M.; Verbeek, S.J.; Staal, F.J.T. Axin2-mTurquoise2: A novel reporter mouse model for the detection of canonical Wnt signalling. Genesis 2017, 55. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-M.; Mao, J.; Taketo, M.M.; Shivdasani, R.A. Phases of canonical Wnt signaling during the development of mouse intestinal epithelium. Gastroenterology 2007, 133, 529–538. [Google Scholar] [CrossRef]
- Haegebarth, A.; Clevers, H. Wnt signaling, lgr5, and stem cells in the intestine and skin. Am. J. Pathol. 2009, 174, 715–721. [Google Scholar] [CrossRef]
- Kahlert, U.D.; Mooney, S.M.; Natsumeda, M.; Steiger, H.-J.; Maciaczyk, J. Targeting cancer stem-like cells in glioblastoma and colorectal cancer through metabolic pathways. Int. J. Cancer 2017, 140, 10–22. [Google Scholar] [CrossRef]
- Lie, D.-C.; Colamarino, S.A.; Song, H.-J.; Désiré, L.; Mira, H.; Consiglio, A.; Lein, E.S.; Jessberger, S.; Lansford, H.; Dearie, A.R.; et al. Wnt signalling regulates adult hippocampal neurogenesis. Nature 2005, 437, 1370–1375. [Google Scholar] [CrossRef]
- Zhou, F.; Cao, W.; Xu, R.; Zhang, J.; Yu, T.; Xu, X.; Zhi, T.; Yin, J.; Cao, S.; Liu, N.; et al. MicroRNA-206 attenuates glioma cell proliferation, migration, and invasion by blocking the WNT/β-catenin pathway via direct targeting of Frizzled 7 mRNA. Am. J. Transl Res. 2019, 11, 4584–4601. [Google Scholar]
- Augustin, I.; Goidts, V.; Bongers, A.; Kerr, G.; Vollert, G.; Radlwimmer, B.; Hartmann, C.; Herold-Mende, C.; Reifenberger, G.; von Deimling, A.; et al. The Wnt secretion protein Evi/Gpr177 promotes glioma tumourigenesis. EMBO Mol. Med. 2012, 4, 38–51. [Google Scholar] [CrossRef]
- Yu, J.M.; Jun, E.S.; Jung, J.S.; Suh, S.Y.; Han, J.Y.; Kim, J.Y.; Kim, K.W.; Jung, J.S. Role of Wnt5a in the proliferation of human glioblastoma cells. Cancer Lett. 2007, 257, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Zuccarini, M.; Giuliani, P.; Ziberi, S.; Carluccio, M.; Iorio, P.D.; Caciagli, F.; Ciccarelli, R. The Role of Wnt Signal in Glioblastoma Development and Progression: A Possible New Pharmacological Target for the Therapy of This Tumor. Genes 2018, 9, 105. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, V.; Arnotskaya, N.; Korneyko, M.; Zaytsev, S.; Khotimchenko, Y.; Sharma, H.; Bryukhovetskiy, I. Proteins of the Wnt signaling pathway as targets for the regulation of CD133+ cancer stem cells in glioblastoma. Oncol. Rep. 2019, 41, 3080–3088. [Google Scholar] [CrossRef]
- Gonçalves, C.S.; Vieira de Castro, J.; Pojo, M.; Martins, E.P.; Queirós, S.; Chautard, E.; Taipa, R.; Pires, M.M.; Pinto, A.A.; Pardal, F.; et al. WNT6 is a novel oncogenic prognostic biomarker in human glioblastoma. Theranostics 2018, 8, 4805–4823. [Google Scholar] [CrossRef] [PubMed]
- Portela, M.; Venkataramani, V.; Fahey-Lozano, N.; Seco, E.; Losada-Perez, M.; Winkler, F.; Casas-Tintó, S. Glioblastoma cells vampirize WNT from neurons and trigger a JNK/MMP signaling loop that enhances glioblastoma progression and neurodegeneration. PLoS Biol. 2019, 17, e3000545. [Google Scholar] [CrossRef]
- Rajakulendran, N.; Rowland, K.J.; Selvadurai, H.J.; Ahmadi, M.; Park, N.I.; Naumenko, S.; Dolma, S.; Ward, R.J.; So, M.; Lee, L.; et al. Wnt and Notch signaling govern self-renewal and differentiation in a subset of human glioblastoma stem cells. Genes Dev. 2019, 33, 498–510. [Google Scholar] [CrossRef]
- Oskarsson, T.; Batlle, E.; Massagué, J. Metastatic stem cells: Sources, niches, and vital pathways. Cell Stem Cell 2014, 14, 306–321. [Google Scholar] [CrossRef]
- Valdor, R.; García-Bernal, D.; Riquelme, D.; Martinez, C.M.; Moraleda, J.M.; Cuervo, A.M.; Macian, F.; Martinez, S. Glioblastoma ablates pericytes antitumor immune function through aberrant up-regulation of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2019, 116, 20655–20665. [Google Scholar] [CrossRef]
- Sakaki-Yumoto, M.; Katsuno, Y.; Derynck, R. TGF-β family signaling in stem cells. Biochim. Biophys. Acta 2013, 1830, 2280–2296. [Google Scholar] [CrossRef]
- Weiss, A.; Attisano, L. The TGFbeta superfamily signaling pathway. Wiley Interdiscip Rev. Dev. Biol. 2013, 2, 47–63. [Google Scholar] [CrossRef]
- Daynac, M.; Pineda, J.R.; Chicheportiche, A.; Gauthier, L.R.; Morizur, L.; Boussin, F.D.; Mouthon, M.-A. TGFβ lengthens the G1 phase of stem cells in aged mouse brain. Stem Cells 2014, 32, 3257–3265. [Google Scholar] [CrossRef] [PubMed]
- Pineda, J.R.; Daynac, M.; Chicheportiche, A.; Cebrian-Silla, A.; Sii Felice, K.; Garcia-Verdugo, J.M.; Boussin, F.D.; Mouthon, M.-A. Vascular-derived TGF-β increases in the stem cell niche and perturbs neurogenesis during aging and following irradiation in the adult mouse brain. EMBO Mol. Med. 2013, 5, 548–562. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Flanders, K.C.; Ren, R.F.; Lippa, C.F. Transforming growth factor-betas in neurodegenerative disease. Prog. Neurobiol. 1998, 54, 71–85. [Google Scholar] [CrossRef]
- Derynck, R.; Goeddel, D.V.; Ullrich, A.; Gutterman, J.U.; Williams, R.D.; Bringman, T.S.; Berger, W.H. Synthesis of messenger RNAs for transforming growth factors alpha and beta and the epidermal growth factor receptor by human tumors. Cancer Res. 1987, 47, 707–712. [Google Scholar]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef]
- Platten, M.; Wick, W.; Weller, M. Malignant glioma biology: Role for TGF-beta in growth, motility, angiogenesis, and immune escape. Microsc. Res. Tech. 2001, 52, 401–410. [Google Scholar] [CrossRef]
- TGFβ Promotes Immune Evasion to Limit the Efficacy of Anti-PD-1/PD-L1. Cancer Discov 2018, 8, OF10. [CrossRef]
- Bellomo, C.; Caja, L.; Moustakas, A. Transforming growth factor β as regulator of cancer stemness and metastasis. Br. J. Cancer 2016, 115, 761–769. [Google Scholar] [CrossRef]
- Vallier, L.; Mendjan, S.; Brown, S.; Chng, Z.; Teo, A.; Smithers, L.E.; Trotter, M.W.B.; Cho, C.H.-H.; Martinez, A.; Rugg-Gunn, P.; et al. Activin/Nodal signalling maintains pluripotency by controlling Nanog expression. Development 2009, 136, 1339–1349. [Google Scholar] [CrossRef]
- Liu, J.; Jin, T.; Chang, S.; Ritchie, H.H.; Smith, A.J.; Clarkson, B.H. Matrix and TGF-beta-related gene expression during human dental pulp stem cell (DPSC) mineralization. In Vitro Cell. Dev. Biol. Anim. 2007, 43, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Li, Y.; Zhao, B.; Xu, C.; Liu, Y.; Li, H.; Zhang, B.; Wang, X.; Yang, X.; Xie, W.; et al. BMP restricts stemness of intestinal Lgr5+ stem cells by directly suppressing their signature genes. Nat. Commun. 2017, 8, 13824. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, A.; Najafi, M.; Farhood, B.; Mortezaee, K. Transforming growth factor-β signaling: Tumorigenesis and targeting for cancer therapy. J. Cell. Physiol. 2019, 234, 12173–12187. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Geng, L.; Wang, D.; Yi, H.; Talmon, G.; Wang, J. R-Spondin1/LGR5 Activates TGFβ Signaling and Suppresses Colon Cancer Metastasis. Cancer Res. 2017, 77, 6589–6602. [Google Scholar] [CrossRef]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.F.; Iglesias, M.; Céspedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.-F.; et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef]
- Ulaner, G.A.; Hu, J.F.; Vu, T.H.; Giudice, L.C.; Hoffman, A.R. Telomerase activity in human development is regulated by human telomerase reverse transcriptase (hTERT) transcription and by alternate splicing of hTERT transcripts. Cancer Res. 1998, 58, 4168–4172. [Google Scholar] [PubMed]
- Chen, K.; Chen, L.; Li, L.; Qu, S.; Yu, B.; Sun, Y.; Wan, F.; Chen, X.; Liang, R.; Zhu, X. A positive feedback loop between Wnt/β-catenin signaling and hTERT regulates the cancer stem cell-like traits in radioresistant nasopharyngeal carcinoma cells. J. Cell. Biochem. 2020. [Google Scholar] [CrossRef]
- Park, J.-I.; Venteicher, A.S.; Hong, J.Y.; Choi, J.; Jun, S.; Shkreli, M.; Chang, W.; Meng, Z.; Cheung, P.; Ji, H.; et al. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 2009, 460, 66–72. [Google Scholar] [CrossRef]
- Saha, A.; Shree Padhi, S.; Roy, S.; Banerjee, B. HCT116 colonospheres shows elevated expression of hTERT and β-catenin protein—A short report. J. Stem Cells 2014, 9, 243–251. [Google Scholar]
- Kerem Terali, K. On the Far Side of Telomeres: The Many Roles of Telomerase in the Acquisition and Retention of Cancer Stemness. In Telomere—A Complex End of a Chromosome; Intechopen: London, UK, 2016; pp. 1–28. ISBN 978-953-51-2753-6. [Google Scholar]
- Rhyu, M.S. Telomeres, telomerase, and immortality. J. Natl. Cancer Inst. 1995, 87, 884–894. [Google Scholar] [CrossRef]
- Beck, S.; Jin, X.; Sohn, Y.-W.; Kim, J.-K.; Kim, S.-H.; Yin, J.; Pian, X.; Kim, S.-C.; Nam, D.-H.; Choi, Y.-J.; et al. Telomerase Activity-Independent Function of TERT Allows Glioma Cells to Attain Cancer Stem Cell Characteristics by Inducing EGFR Expression. Mol. Cells 2011, 31, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Gunes, C.; Avila, A.I.; Rudolph, K.L. Telomeres in cancer. Differentiation 2018, 99, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Hirota, T. Chromosomal instability: A common feature and a therapeutic target of cancer. Biochim. Biophys. Acta 2016, 1866, 64–75. [Google Scholar] [CrossRef] [PubMed]
- McClelland, S.E. Role of chromosomal instability in cancer progression. Endocr. Relat. Cancer 2017, 24, T23–T31. [Google Scholar] [CrossRef]
- Ishaq, A.; Hanson, P.S.; Morris, C.M.; Saretzki, G. Telomerase Activity is Downregulated Early During Human Brain Development. Genes 2016, 7, 27. [Google Scholar] [CrossRef]
- Hiyama, E.; Hiyama, K. Telomere and telomerase in stem cells. Br. J. Cancer 2007, 96, 1020–1024. [Google Scholar] [CrossRef]
- Schepers, A.G.; Vries, R.; van den Born, M.; van de Wetering, M.; Clevers, H. Lgr5 intestinal stem cells have high telomerase activity and randomly segregate their chromosomes. EMBO J. 2011, 30, 1104–1109. [Google Scholar] [CrossRef]
- Ninagawa, N.; Murakami, R.; Isobe, E.; Tanaka, Y.; Nakagawa, H.; Torihashi, S. Mesenchymal stem cells originating from ES cells show high telomerase activity and therapeutic benefits. Differentiation 2011, 82, 153–164. [Google Scholar] [CrossRef][Green Version]
- Horibe, H.; Murakami, M.; Iohara, K.; Hayashi, Y.; Takeuchi, N.; Takei, Y.; Kurita, K.; Nakashima, M. Isolation of a stable subpopulation of mobilized dental pulp stem cells (MDPSCs) with high proliferation, migration, and regeneration potential is independent of age. PLoS ONE 2014, 9, e98553. [Google Scholar] [CrossRef]
- Jeon, B.-G.; Kang, E.-J.; Kumar, B.M.; Maeng, G.-H.; Ock, S.-A.; Kwack, D.-O.; Park, B.-W.; Rho, G.-J. Comparative analysis of telomere length, telomerase and reverse transcriptase activity in human dental stem cells. Cell Transplant. 2011, 20, 1693–1705. [Google Scholar] [CrossRef]
- Caporaso, G.L.; Lim, D.A.; Alvarez-Buylla, A.; Chao, M.V. Telomerase activity in the subventricular zone of adult mice. Mol. Cell Neurosci. 2003, 23, 693–702. [Google Scholar] [CrossRef]
- Ferrón, S.; Mira, H.; Franco, S.; Cano-Jaimez, M.; Bellmunt, E.; Ramírez, C.; Fariñas, I.; Blasco, M.A. Telomere shortening and chromosomal instability abrogates proliferation of adult but not embryonic neural stem cells. Development 2004, 131, 4059–4070. [Google Scholar] [CrossRef] [PubMed]
- Ferrón, S.R.; Marqués-Torrejón, M.A.; Mira, H.; Flores, I.; Taylor, K.; Blasco, M.A.; Fariñas, I. Telomere shortening in neural stem cells disrupts neuronal differentiation and neuritogenesis. J. Neurosci. 2009, 29, 14394–14407. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.-Y.; Nemes, A.; Zhou, Q.-G. The Emerging Roles for Telomerase in the Central Nervous System. Front. Mol. Neurosci. 2018, 11, 160. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Katakura, Y.; Yamamoto, K.; Uehara, N.; Tsuchiya, T.; Kim, E.H.; Shirahata, S. Neural stem cells lose telomerase activity upon differentiating into astrocytes. Cytotechnology 2001, 36, 137–144. [Google Scholar] [CrossRef]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Wang, F.; Liu, L. Alternative Lengthening of Telomeres (ALT) in Tumors and Pluripotent Stem Cells. Genes 2019, 10, 1030. [Google Scholar] [CrossRef]
- Arnoult, N.; Karlseder, J. ALT telomeres borrow from meiosis to get moving. Cell 2014, 159, 11–12. [Google Scholar] [CrossRef]
- Farooqi, A.; Yang, J.; Sharin, V.; Ezhilarasan, R.; Danussi, C.; Alvarez, C.; Dharmaiah, S.; Irvin, D.; Huse, J.; Sulman, E.P. Identification of patient-derived glioblastoma stem cell (GSC) lines with the alternative lengthening of telomeres phenotype. Acta Neuropathol. Commun 2019, 7, 76. [Google Scholar] [CrossRef]
- Lafferty-Whyte, K.; Cairney, C.J.; Will, M.B.; Serakinci, N.; Daidone, M.-G.; Zaffaroni, N.; Bilsland, A.; Keith, W.N. A gene expression signature classifying telomerase and ALT immortalization reveals an hTERT regulatory network and suggests a mesenchymal stem cell origin for ALT. Oncogene 2009, 28, 3765–3774. [Google Scholar] [CrossRef]
- Pompili, L.; Maresca, C.; Dello Stritto, A.; Biroccio, A.; Salvati, E. BRCA2 Deletion Induces Alternative Lengthening of Telomeres in Telomerase Positive Colon Cancer Cells. Genes 2019, 10, 697. [Google Scholar] [CrossRef] [PubMed]
- Heaphy, C.M.; Subhawong, A.P.; Hong, S.-M.; Goggins, M.G.; Montgomery, E.A.; Gabrielson, E.; Netto, G.J.; Epstein, J.I.; Lotan, T.L.; Westra, W.H.; et al. Prevalence of the Alternative Lengthening of Telomeres Telomere Maintenance Mechanism in Human Cancer Subtypes. Am. J. Pathol. 2011, 179, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, R.T.; Veronese, N.; Pea, A.; Nottegar, A.; Smith, L.; Pilati, C.; Demurtas, J.; Fassan, M.; Cheng, L.; Luchini, C. Alternative lengthening of telomeres (ALT) influences survival in soft tissue sarcomas: A systematic review with meta-analysis. BMC Cancer 2019, 19, 232. [Google Scholar] [CrossRef] [PubMed]
- Venturini, L.; Motta, R.; Gronchi, A.; Daidone, M.; Zaffaroni, N. Prognostic relevance of ALT-associated markers in liposarcoma: A comparative analysis. BMC Cancer 2010, 10, 254. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer, World Health Organization. Cancer Today. Available online: https://gco.iarc.fr/today/home (accessed on 10 October 2020).
- Szaryńska, M.; Olejniczak, A.; Kobiela, J.; Spychalski, P.; Kmieć, Z. Therapeutic strategies against cancer stem cells in human colorectal cancer. Oncol. Lett. 2017, 14, 7653–7668. [Google Scholar] [CrossRef]
- Alcantara Llaguno, S.; Sun, D.; Pedraza, A.M.; Vera, E.; Wang, Z.; Burns, D.K.; Parada, L.F. Cell-of-origin susceptibility to glioblastoma formation declines with neural lineage restriction. Nat. Neurosci. 2019, 22, 545–555. [Google Scholar] [CrossRef]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef]
- Chow, L.M.L.; Endersby, R.; Zhu, X.; Rankin, S.; Qu, C.; Zhang, J.; Broniscer, A.; Ellison, D.W.; Baker, S.J. Cooperativity within and among Pten, p53, and Rb pathways induces high-grade astrocytoma in adult brain. Cancer Cell 2011, 19, 305–316. [Google Scholar] [CrossRef]
- Bachoo, R.M.; Maher, E.A.; Ligon, K.L.; Sharpless, N.E.; Chan, S.S.; You, M.J.; Tang, Y.; DeFrances, J.; Stover, E.; Weissleder, R.; et al. Epidermal growth factor receptor and Ink4a/Arf: Convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell 2002, 1, 269–277. [Google Scholar] [CrossRef]
- Afify, S.M.; Seno, M. Conversion of Stem Cells to Cancer Stem Cells: Undercurrent of Cancer Initiation. Cancers 2019, 11, 345. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Virchow, R. Die krankhaften Geschwülste. Dreissig Vorlesungen, gehalten wahrend des Wintersemesters 1862–1863 an Der Universität Zu Berlin; Springer: Berlin/Heidelberg, Germany, 1867; Volume 3. [Google Scholar]
- Zanini, M.; Meyer, E.; Simon, S. Pulp Inflammation Diagnosis from Clinical to Inflammatory Mediators: A Systematic Review. J. Endod. 2017, 43, 1033–1051. [Google Scholar] [CrossRef] [PubMed]
- Thuringer, JM Incipient dental tumor involving pulp and parodontium. J. Dent. Res. 1937, 16, 387–399. [CrossRef]
- Wilson, R.; Urraca, N.; Skobowiat, C.; Hope, K.A.; Miravalle, L.; Chamberlin, R.; Donaldson, M.; Seagroves, T.N.; Reiter, L.T. Assessment of the Tumorigenic Potential of Spontaneously Immortalized and hTERT-Immortalized Cultured Dental Pulp Stem Cells. Stem. Cells Transl. Med. 2015, 4, 905–912. [Google Scholar] [CrossRef]
- Orimoto, A.; Kyakumoto, S.; Eitsuka, T.; Nakagawa, K.; Kiyono, T.; Fukuda, T. Efficient immortalization of human dental pulp stem cells with expression of cell cycle regulators with the intact chromosomal condition. PLoS ONE 2020, 15, e0229996. [Google Scholar] [CrossRef]
- Inada, E.; Saitoh, I.; Kubota, N.; Iwase, Y.; Kiyokawa, Y.; Shibasaki, S.; Noguchi, H.; Yamasaki, Y.; Sato, M. piggyBac Transposon-Based Immortalization of Human Deciduous Tooth Dental Pulp Cells with Multipotency and Non-Tumorigenic Potential. Int. J. Mol. Sci. 2019, 20, 4904. [Google Scholar] [CrossRef]
- Liu, G.-X.; Ma, S.; Li, Y.; Yu, Y.; Zhou, Y.-X.; Lu, Y.-D.; Jin, L.; Wang, Z.-L.; Yu, J.-H. Hsa-let-7c controls the committed differentiation of IGF-1-treated mesenchymal stem cells derived from dental pulps by targeting IGF-1R via the MAPK pathways. Exp. Mol. Med. 2018, 50, 25. [Google Scholar] [CrossRef]
- Egbuniwe, O.; Idowu, B.D.; Funes, J.M.; Grant, A.D.; Renton, T.; Di Silvio, L. P16/p53 expression and telomerase activity in immortalized human dental pulp cells. Cell Cycle 2011, 10, 3912–3919. [Google Scholar] [CrossRef]
- Seifrtová, M.; Havelek, R.; Cmielová, J.; Jiroutová, A.; Soukup, T.; Brůčková, L.; Mokrý, J.; English, D.; Rezáčová, M. The response of human ectomesenchymal dental pulp stem cells to cisplatin treatment. Int. Endod. J. 2012, 45, 401–412. [Google Scholar] [CrossRef]
- Badiola, I.; Santaolalla, F.; Garcia-Gallastegui, P.; Ana, S.-D.R.; Unda, F.; Ibarretxe, G. Biomolecular bases of the senescence process and cancer. A new approach to oncological treatment linked to ageing. Ageing Res. Rev. 2015, 23, 125–138. [Google Scholar] [CrossRef] [PubMed]



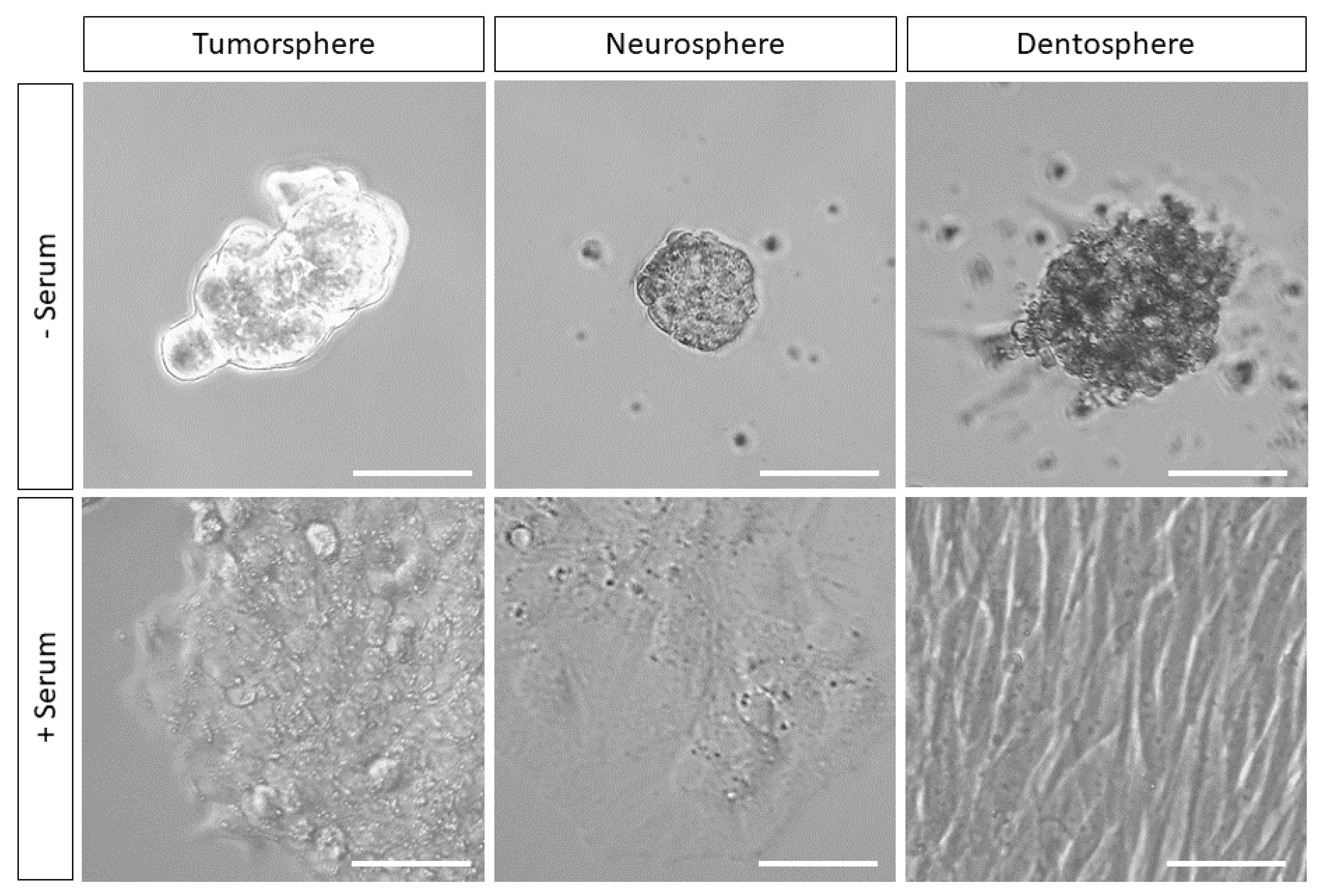{kind=link}
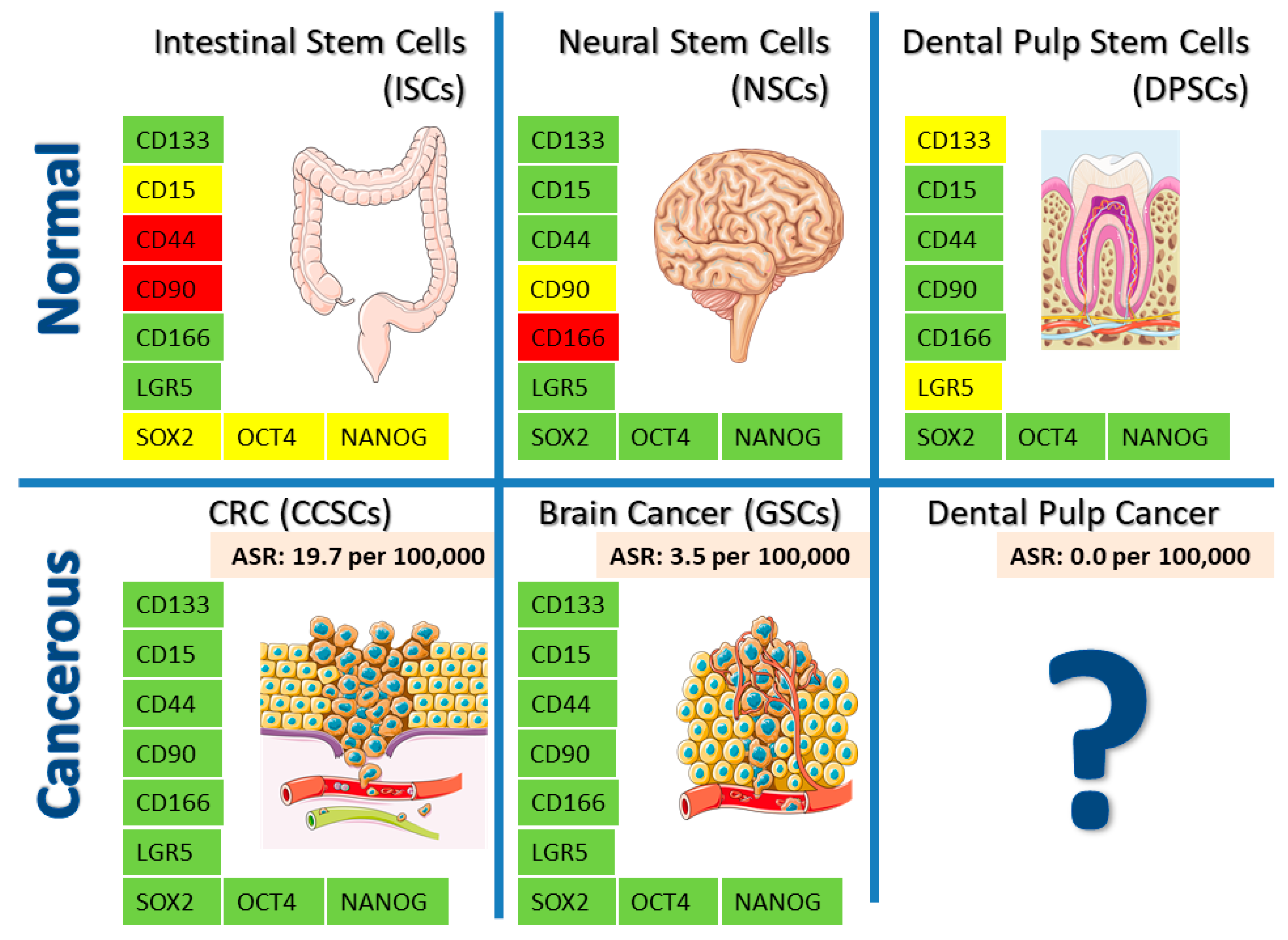{kind=link}
| Intestinal | Neural | Dental | |||
|---|---|---|---|---|---|
| ISC | CSC/CCSC | NSC | CSC/GSC | DPSC | |
| CD133 | + [39] | + ↑ [40,41] | + [42,43,44] | + ↑ [43,45] | +/- [46,47] |
| CD15 | ? | + [48] | + [49,50] | + [51,52,53] | + [54] |
| LGR5 | + [55] | + [56] | + [57,58] | + [59] | ? |
| CD166 | + [60,61] | + [61] | - [62] | + ↑ [63] | + [64,65] |
| CD44 | - [60] | + ↑ [66] | + [67] | + ↑ [68,69,70] | + [71] |
| CD90 | - [72] | + ↑ [73] | - [62] | + ↑ [74] | + [75,76] |
| CXCR4 | + [77] | + ↑ [78] | + [79] | + ↑ [80] | + [81] |
| NESTIN | + [82] | + ↑ [83] | + [84] | + ↑ [83,85] | + [54,75] |
| MUSASHI | + [86] | + ↑ [87] | + [88] | + ↑ [89] | + [90] |
| SOX2 | ? | + ↑ [91] | + [52,92] | + ↑ [52,93,94] | + [54,75,95,96] |
| OCT4 | ? | + ↑ [97] | + [98] | + ↑ [93,94,99] | + [54,95,96,100] |
| NANOG | ? | + ↑ [101] | + [98,102,103] | + ↑ [93,94] | + [54,95,96,100] |
| PTEN | + [104] | + ↓ [105] | + [106] | + ↓ [106] | + [107] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olatz, C.; Patricia, G.-G.; Jon, L.; Iker, B.; Carmen, d.l.H.; Fernando, U.; Gaskon, I.; Ramon, P.J. Is There Such a Thing as a Genuine Cancer Stem Cell Marker? Perspectives from the Gut, the Brain and the Dental Pulp. Biology 2020, 9, 426. https://doi.org/10.3390/biology9120426
Olatz C, Patricia G-G, Jon L, Iker B, Carmen dlH, Fernando U, Gaskon I, Ramon PJ. Is There Such a Thing as a Genuine Cancer Stem Cell Marker? Perspectives from the Gut, the Brain and the Dental Pulp. Biology. 2020; 9(12):426. https://doi.org/10.3390/biology9120426
Chicago/Turabian StyleOlatz, Crende, García-Gallastegui Patricia, Luzuriaga Jon, Badiola Iker, de la Hoz Carmen, Unda Fernando, Ibarretxe Gaskon, and Pineda Jose Ramon. 2020. "Is There Such a Thing as a Genuine Cancer Stem Cell Marker? Perspectives from the Gut, the Brain and the Dental Pulp" Biology 9, no. 12: 426. https://doi.org/10.3390/biology9120426
APA StyleOlatz, C., Patricia, G.-G., Jon, L., Iker, B., Carmen, d. l. H., Fernando, U., Gaskon, I., & Ramon, P. J. (2020). Is There Such a Thing as a Genuine Cancer Stem Cell Marker? Perspectives from the Gut, the Brain and the Dental Pulp. Biology, 9(12), 426. https://doi.org/10.3390/biology9120426