
Simulation of Cellular Energy Restriction in Quiescence (ERiQ)—A Theoretical Model for Aging

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
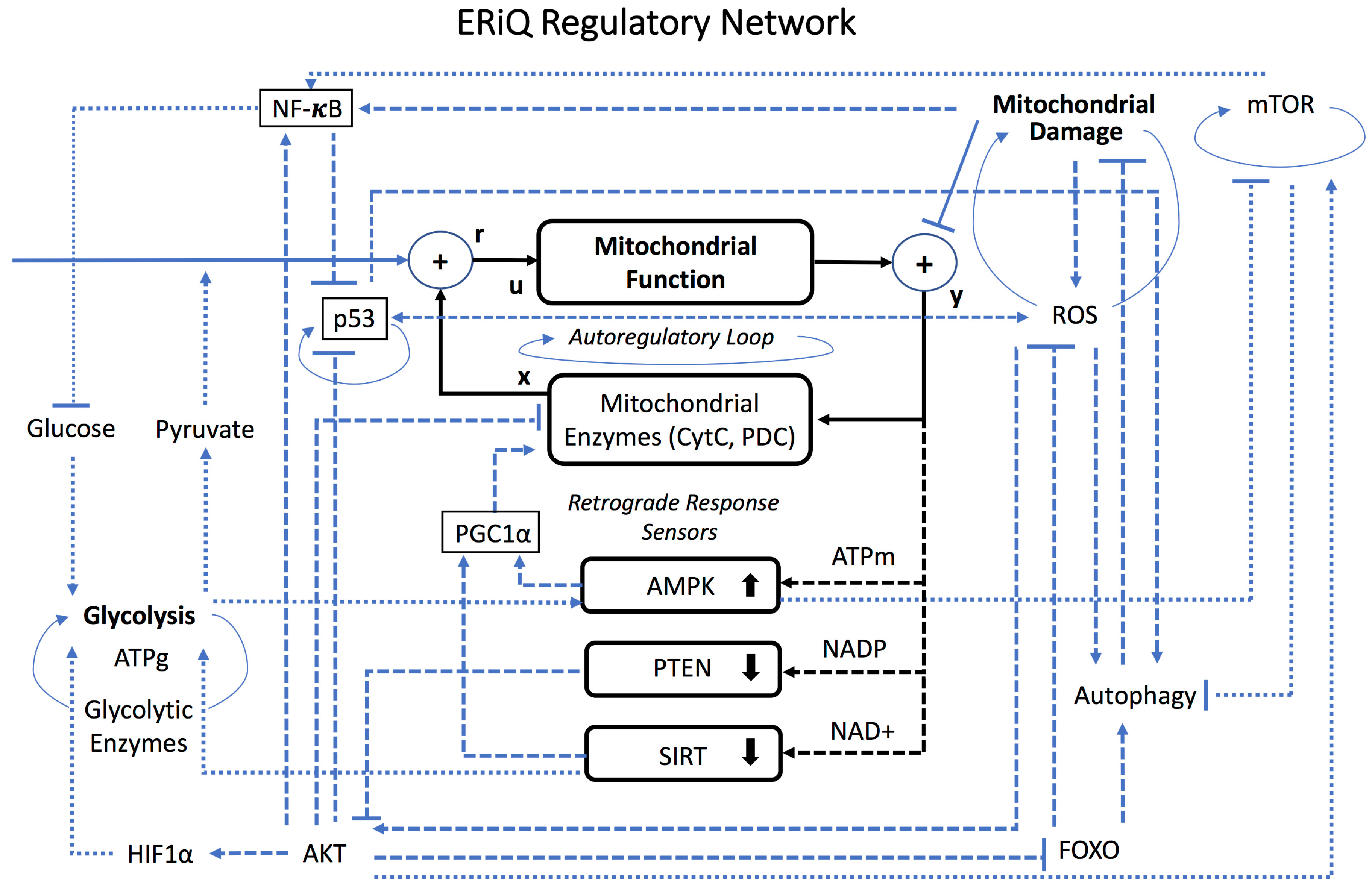
2.1. Graphical Network
2.2. Computer Implementation
2.3. Model Analysis
3. Results
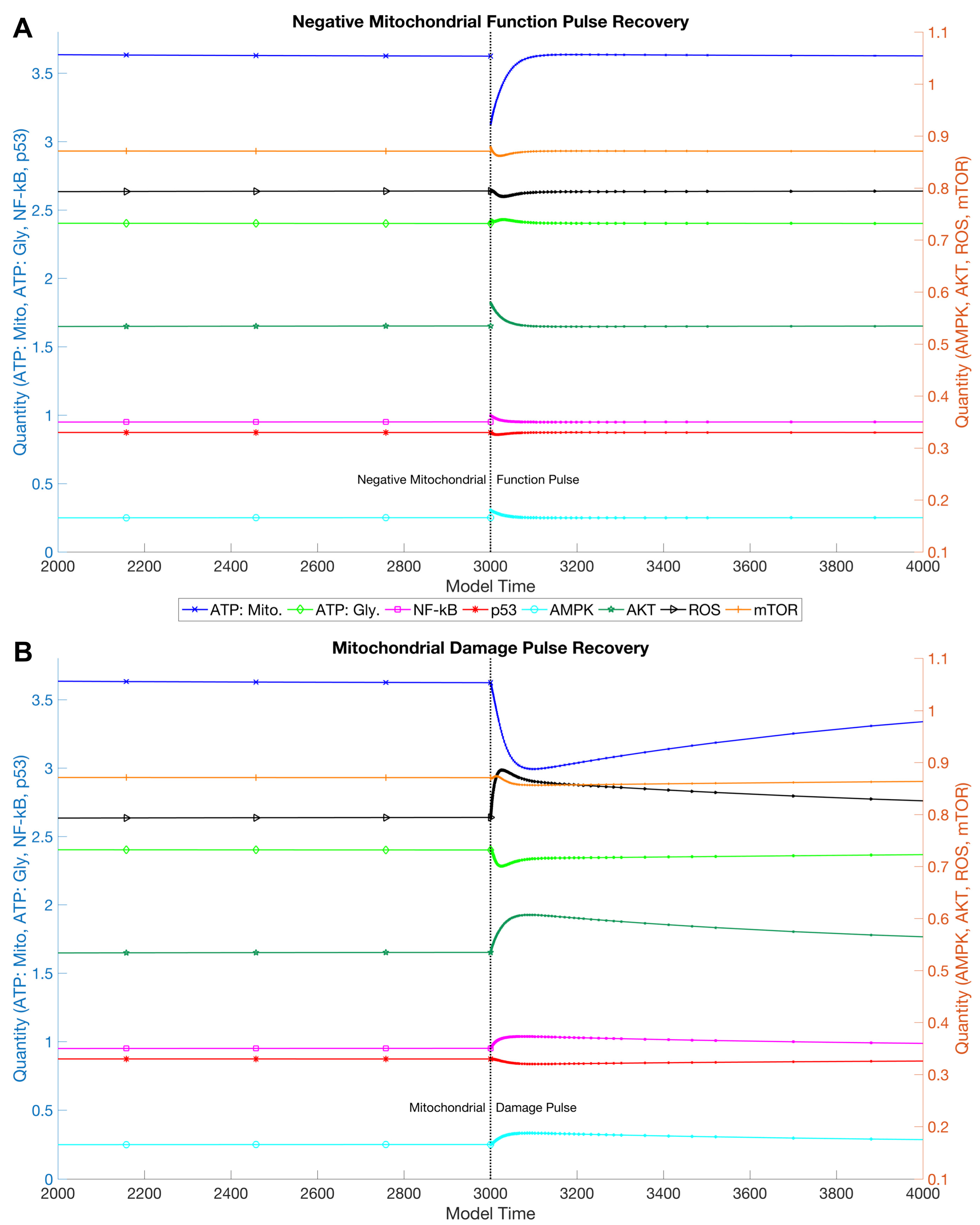
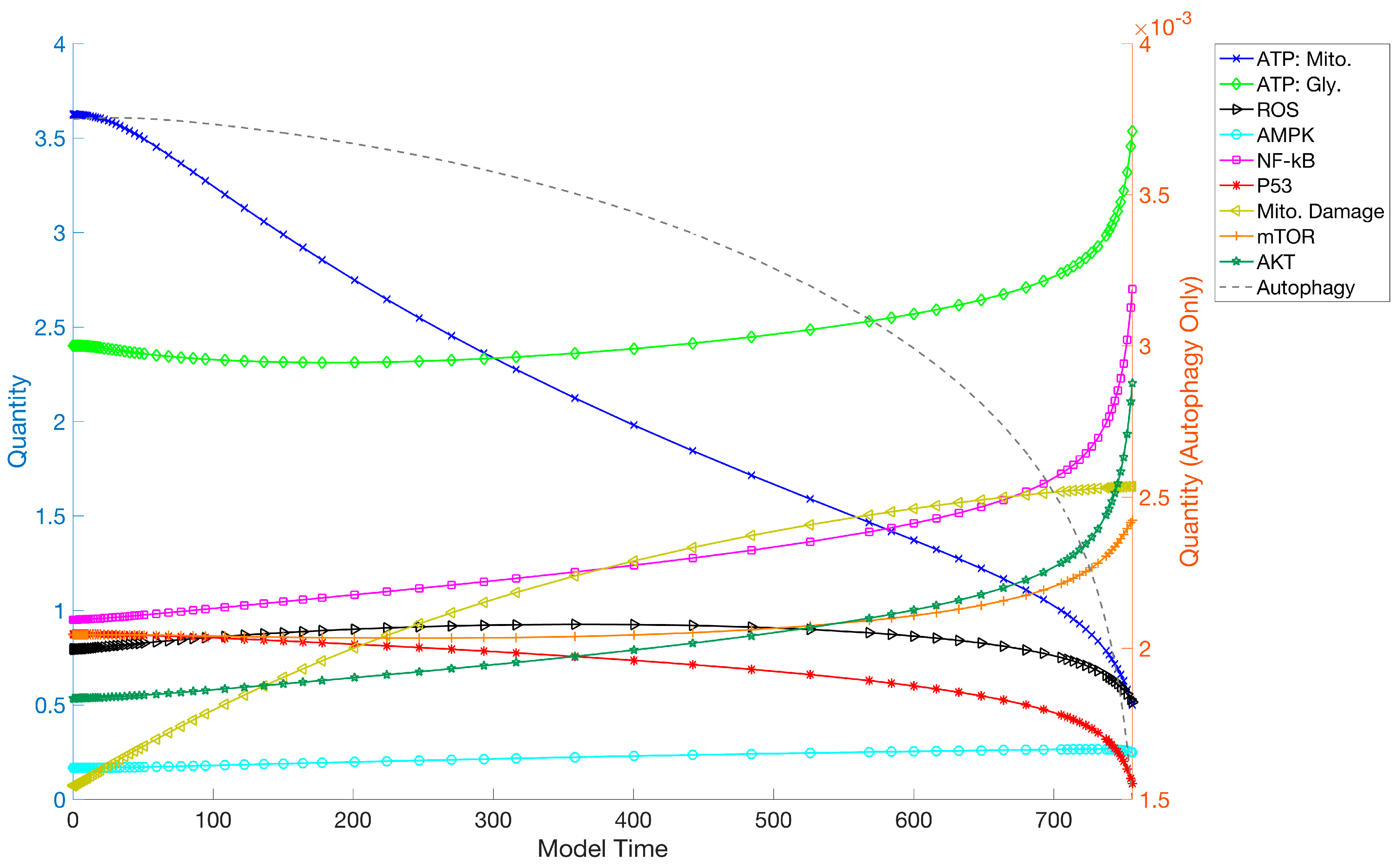
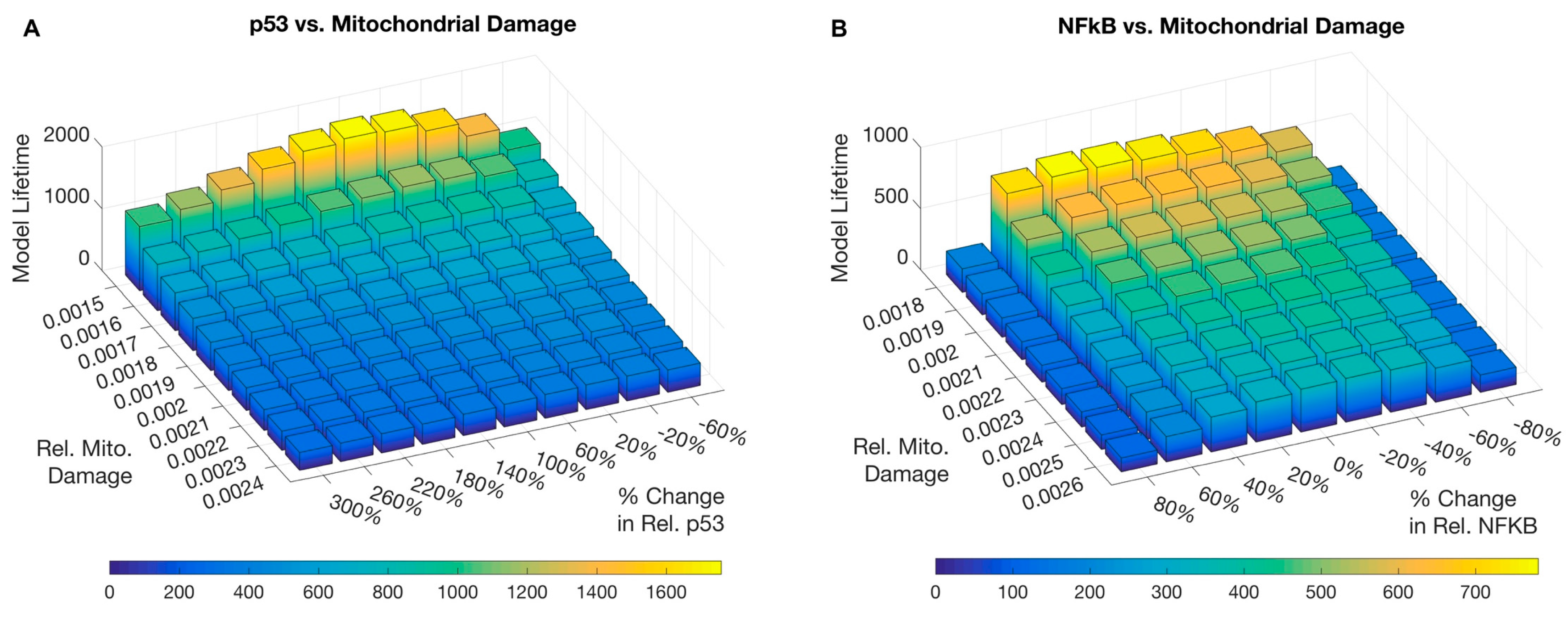
3.1. Simulations and Perturbations
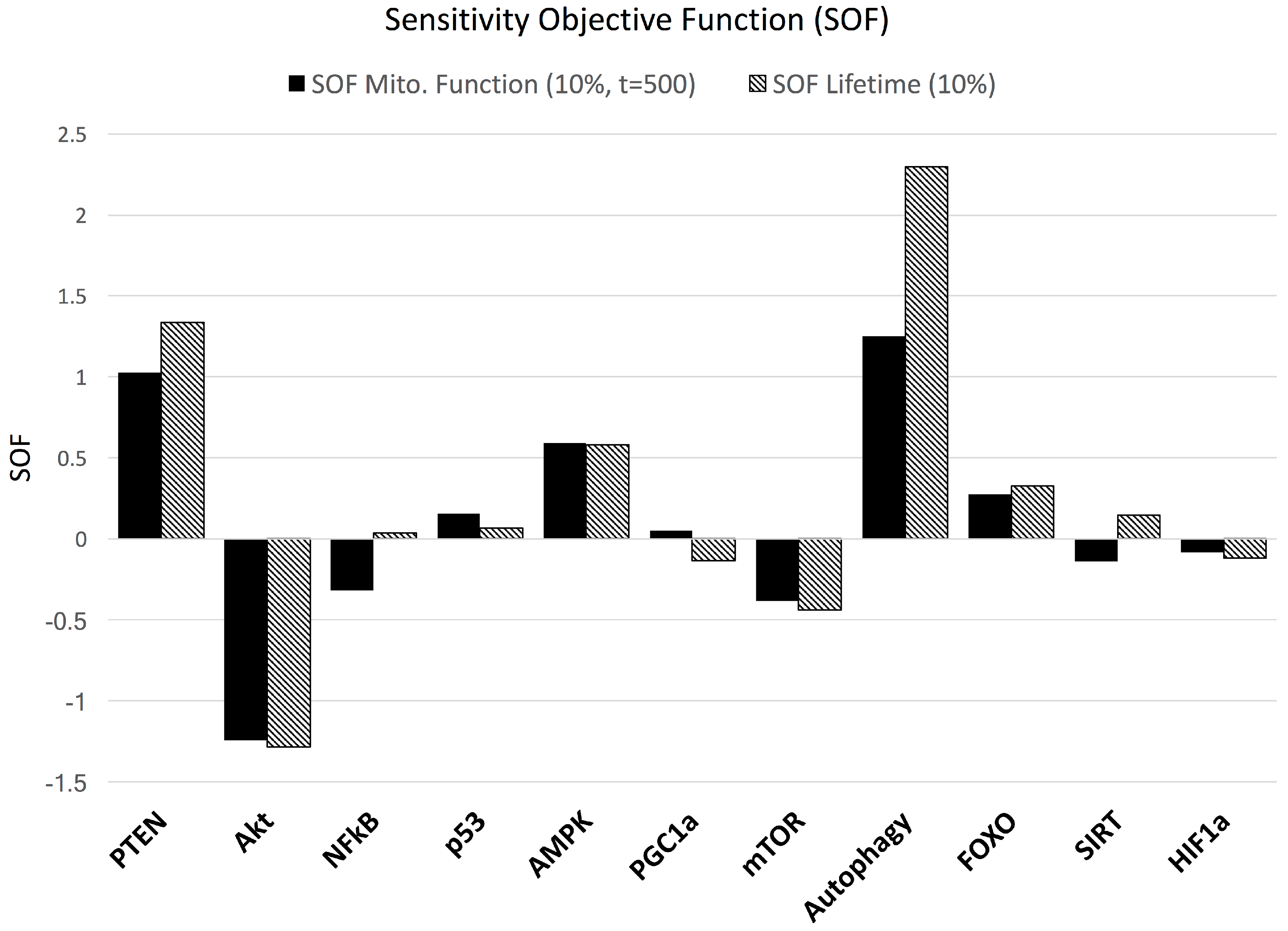
3.2. Sensitivity Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Figge, M.T.; Reichert, A.S.; Meyer-Hermann, M.; Osiewacz, H.D. Deceleration of fusion-fission cycles improves mitochondrial quality control during aging. PLoS Comput. Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Kowald, A.; Kirkwood, T.B. Evolution of the mitochondrial fusion-fission cycle and its role in aging. Proc. Natl. Acad. Sci. USA 2011, 108, 10237–10242. [Google Scholar] [CrossRef] [PubMed]
- Proctor, C.J.; Lorimer, I.A. Modelling the role of the Hsp70/Hsp90 system in the maintenance of protein homeostasis. PLoS ONE 2011, 6, e22038. [Google Scholar] [CrossRef] [PubMed]
- Aviv, A.; Levy, D.; Mangel, M. Growth, telomere dynamics and successful and unsuccessful human aging. Mech. Ageing Dev. 2003, 124, 829–837. [Google Scholar] [CrossRef]
- op den Buijs, J.; van den Bosch, P.P.; Musters, M.W.; van Riel, N.A. Mathematical modeling confirms the length-dependency of telomere shortening. Mech. Ageing Dev. 2004, 125, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Golubev, A.; Khrustalev, S.; Butov, A. An in silico investigation into the causes of telomere length heterogeneity and its implications for the Hayflick limit. J. Theor. Biol. 2003, 225, 153–170. [Google Scholar] [CrossRef]
- Xue, H.; Xian, B.; Dong, D.; Xia, K.; Zhu, S.; Zhang, Z.; Hou, L.; Zhang, Q.; Zhang, Y.; Han, J.D. A modular network model of aging. Mol. Syst. Biol. 2007, 3. [Google Scholar] [CrossRef] [PubMed]
- Soltow, Q.A.; Jones, D.P.; Promislow, D.E. A Network Perspective on Metabolism and Aging. Integr. Comp. Biol. 2010, 50, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Borklu Yucel, E.; Ulgen, K.O. A network-based approach on elucidating the multi-faceted nature of chronological aging in S. cerevisiae. PLoS ONE 2011. [Google Scholar] [CrossRef] [PubMed]
- Kowald, A.; Kirkwood, T.B. A network theory of ageing: The interactions of defective mitochondria, aberrant proteins, free radicals and scavengers in the ageing process. Mutat. Res. 1996, 316, 209–236. [Google Scholar] [CrossRef]
- Kriete, A.; Bosl, W.J.; Booker, G. Rule-based cell systems model of aging using feedback loop motifs mediated by stress responses. PLoS Comput. Biol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Yao, G. Modelling mammalian cellular quiescence. Interface Focus 2014. [Google Scholar] [CrossRef] [PubMed]
- Helenius, M.; Hanninen, M.; Lehtinen, S.K.; Salminen, A. Changes associated with aging and replicative senescence in the regulation of transcription factor nuclear factor-kappa B. Biochem. J. 1996, 318, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Yalamanchili, N.; Kriete, A.; Alfego, D.; Danowski, K.M.; Kari, C.; Rodeck, U. Distinct Cell Stress Responses Induced by ATP Restriction in Quiescent Human Fibroblasts. Front. Genet. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Butow, R.A.; Avadhani, N.G. Mitochondrial signaling: The retrograde response. Mol. Cell 2004, 14, 1–15. [Google Scholar] [CrossRef]
- Liu, Z.; Butow, R.A. Mitochondrial retrograde signaling. Annu. Rev. Genet. 2006, 40, 159–185. [Google Scholar] [CrossRef] [PubMed]
- Parikh, V.S.; Morgan, M.M.; Scott, R.; Clements, L.S.; Butow, R.A. The mitochondrial genotype can influence nuclear gene expression in yeast. Science 1987, 235, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Kirchman, P.A.; Kim, S.; Lai, C.Y.; Jazwinski, S.M. Interorganelle signaling is a determinant of longevity in Saccharomyces cerevisiae. Genetics 1999, 152, 179–190. [Google Scholar] [PubMed]
- Biswas, G.; Anandatheerthavarada, H.K.; Zaidi, M.; Avadhani, N.G. Mitochondria to nucleus stress signaling: A distinctive mechanism of NFkappaB/Rel activation through calcineurin-mediated inactivation of IkappaBbeta. J. Cell Biol. 2003, 161, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Biswas, G.; Tang, W.; Sondheimer, N.; Guha, M.; Bansal, S.; Avadhani, N.G. A distinctive physiological role for IkappaBbeta in the propagation of mitochondrial respiratory stress signaling. J. Biol. Chem. 2008, 283, 12586–12594. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Roy Choudhury, A.; Guha, M.; Huang, L.; Van Winkle, T.; Rustgi, A.K.; Avadhani, N.G. Silencing of IkBbeta mRNA Causes Disruption of Mitochondrial Retrograde Signaling and Suppression of Tumor Growth In Vivo. Carcinogenesis 2012, 33. [Google Scholar] [CrossRef] [PubMed]
- Jazwinski, S.M.; Kriete, A. The yeast retrograde response as a model of intracellular signaling of mitochondrial dysfunction. Front. Physiol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, V.; Kriete, A.; Sacan, A.; Jazwinski, S.M. Comparing the yeast retrograde response and NF-kappaB stress responses: Implications for aging. Aging Cell 2010, 9, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K. NF-kappaB signaling in the aging process. J. Clin. Immunol. 2009, 29, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Tilstra, J.S.; Clauson, C.L.; Niedernhofer, L.J.; Robbins, P.D. NF-kappaB in Aging and Disease. Aging Dis. 2011, 2, 449–465. [Google Scholar] [PubMed]
- Fontanesi, F. Mechanisms of mitochondrial translational regulation. IUBMB Life 2013, 65, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Yoshida, H. Organelle autoregulation-stress responses in the ER, Golgi, mitochondria and lysosome. J. Biochem. 2015, 157, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Donahue, R.J.; Razmara, M.; Hoek, J.B.; Knudsen, T.B. Direct influence of the p53 tumor suppressor on mitochondrial biogenesis and function. FASEB J. 2001, 15, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Lombard, D.B.; Tishkoff, D.X.; Bao, J. Mitochondrial sirtuins in the regulation of mitochondrial activity and metabolic adaptation. Handb. Exp. Pharmacol. 2011, 206, 163–188. [Google Scholar] [PubMed]
- Brenmoehl, J.; Hoeflich, A. Dual control of mitochondrial biogenesis by sirtuin 1 and sirtuin 3. Mitochondrion 2013, 13, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Gong, B.; Almasan, A. Distinct stages of cytochrome c release from mitochondria: Evidence for a feedback amplification loop linking caspase activation to mitochondrial dysfunction in genotoxic stress induced apoptosis. Cell Death Differ. 2000, 7, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef] [PubMed]
- Terada, S.; Goto, M.; Kato, M.; Kawanaka, K.; Shimokawa, T.; Tabata, I. Effects of low-intensity prolonged exercise on PGC-1 mRNA expression in rat epitrochlearis muscle. Biochem. Biophys. Res. Commun. 2002, 296, 350–354. [Google Scholar] [CrossRef]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Polak, P.; Hall, M.N. mTOR and the control of whole body metabolism. Curr. Opin. Cell Biol. 2009, 21, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M. FOXOphagy path to inducing stress resistance and cell survival. Nat. Cell Biol. 2012, 14, 786–788. [Google Scholar] [CrossRef] [PubMed]
- Chalhoub, N.; Baker, S.J. PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. 2009, 4, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Pelicano, H.; Xu, R.H.; Du, M.; Feng, L.; Sasaki, R.; Carew, J.S.; Hu, Y.; Ramdas, L.; Hu, L.; Keating, M.J.; et al. Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. J. Cell Biol. 2006, 175, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Guha, M.; Fang, J.K.; Monks, R.; Birnbaum, M.J.; Avadhani, N.G. Activation of Akt is essential for the propagation of mitochondrial respiratory stress signaling and activation of the transcriptional coactivator heterogeneous ribonucleoprotein A2. Mol. Biol. Cell 2010, 21, 3578–3589. [Google Scholar] [CrossRef] [PubMed]
- Dan, H.C.; Cooper, M.J.; Cogswell, P.C.; Duncan, J.A.; Ting, J.P.; Baldwin, A.S. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008, 22, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Gorlach, A.; Bonello, S. The cross-talk between NF-kappaB and HIF-1: Further evidence for a significant liaison. Biochem. J. 2008, 412, e17–e19. [Google Scholar] [CrossRef] [PubMed]
- Yeung, S.J.; Pan, J.; Lee, M.H. Roles of p53, MYC and HIF-1 in regulating glycolysis—The seventh hallmark of cancer. Cell. Mol. Life Sci. 2008, 65, 3981–3999. [Google Scholar] [CrossRef] [PubMed]
- Nakae, J.; Oki, M.; Cao, Y. The FoxO transcription factors and metabolic regulation. FEBS Lett. 2008, 582, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Compton, S.; Kim, C.; Griner, N.B.; Potluri, P.; Scheffler, I.E.; Sen, S.; Jerry, D.J.; Schneider, S.; Yadava, N. Mitochondrial dysfunction impairs tumor suppressor p53 expression/function. J. Biol. Chem. 2011, 286, 20297–20312. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, T.M.; Leal, J.F.; Seger, R.; Taya, Y.; Oren, M. Cross-talk between Akt, p53 and Mdm2: Possible implications for the regulation of apoptosis. Oncogene 2002, 21, 1299–1303. [Google Scholar] [CrossRef] [PubMed]
- Webster, G.A.; Perkins, N.D. Transcriptional cross talk between NF-kappaB and p53. Mol. Cell. Biol. 1999, 19, 3485–3495. [Google Scholar] [CrossRef] [PubMed]
- Ak, P.; Levine, A.J. p53 and NF-kappaB: Different strategies for responding to stress lead to a functional antagonism. FASEB J. 2010, 24, 3643–3652. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Li, X.M.; Meinkoth, J.; Pittman, R.N. Akt regulates cell survival and apoptosis at a postmitochondrial level. J. Cell Biol. 2000, 151, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Auwerx, J. Targeting sirtuin 1 to improve metabolism: All you need is NAD(+)? Pharmacol. Rev. 2012, 64, 166–187. [Google Scholar] [CrossRef] [PubMed]
- Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxid. Redox Signal. 2008, 10, 179–206. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef] [PubMed]
- De Luca, C.; Olefsky, J.M. Inflammation and insulin resistance. FEBS Lett. 2008, 582, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Galluzzi, L.; Morselli, E.; Kepp, O.; Malik, S.A.; Kroemer, G. Autophagy regulation by p53. Curr. Opin. Cell Biol. 2010, 22, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Van Houten, B.; Hunter, S.E.; Meyer, J.N. Mitochondrial DNA damage induced autophagy, cell death, and disease. Front. Biosci. 2016, 21, 42–54. [Google Scholar] [CrossRef]
- Vaseva, A.V.; Moll, U.M. The mitochondrial p53 pathway. Biochim. Biophys. Acta 2009, 1787, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Koh, H. Role of FOXO transcription factors in crosstalk between mitochondria and the nucleus. J. Bioenerg. Biomembr. 2017. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Chen, Y.; St Clair, D.K. ROS and p53: A versatile partnership. Free Radic. Biol. Med. 2008, 44, 1529–1535. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol. Cancer 2017, 16. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Epstein, T.; Xu, L.; Gillies, R.J.; Gatenby, R.A. Separation of metabolic supply and demand: Aerobic glycolysis as a normal physiological response to fluctuating energetic demands in the membrane. Cancer Metab. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Gunawan, R.; Cao, Y.; Petzold, L.; Doyle, F.J., III. Sensitivity analysis of discrete stochastic systems. Biophys. J. 2005, 88, 2530–2540. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, B.B.; Burke, J.M.; Lauffenburger, D.A.; Sorger, P.K. Physicochemical modelling of cell signalling pathways. Nat. Cell Biol. 2006, 8, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Szigeti, B.; Roth, Y.D.; Sekar, J.A.P.; Goldberg, A.P.; Pochiraju, S.C.; Karr, J.R. A blueprint for human whole-cell modeling. Curr. Opin. Syst. Biol. 2018, 7, 8–15. [Google Scholar] [CrossRef]
- Preston, C.C.; Oberlin, A.S.; Holmuhamedov, E.L.; Gupta, A.; Sagar, S.; Syed, R.H.; Siddiqui, S.A.; Raghavakaimal, S.; Terzic, A.; Jahangir, A. Aging-induced alterations in gene transcripts and functional activity of mitochondrial oxidative phosphorylation complexes in the heart. Mech. Ageing Dev. 2008, 129, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Zahn, J.M.; Sonu, R.; Vogel, H.; Crane, E.; Mazan-Mamczarz, K.; Rabkin, R.; Davis, R.W.; Becker, K.G.; Owen, A.B.; Kim, S.K. Transcriptional profiling of aging in human muscle reveals a common aging signature. PLoS Genet. 2006, 2, e115. [Google Scholar] [CrossRef]
- Nojima, A.; Yamashita, M.; Yoshida, Y.; Shimizu, I.; Ichimiya, H.; Kamimura, N.; Kobayashi, Y.; Ohta, S.; Ishii, N.; Minamino, T. Haploinsufficiency of akt1 prolongs the lifespan of mice. PLoS ONE 2013, 8, e69178. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, H.; Minamino, T.; Tateno, K.; Kunieda, T.; Toko, H.; Komuro, I. Akt negatively regulates the in vitro lifespan of human endothelial cells via a p53/p21-dependent pathway. EMBO J. 2004, 23, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.P.; Price, N.L.; Ling, A.J.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [PubMed]
- Baar, E.L.; Carbajal, K.A.; Ong, I.M.; Lamming, D.W. Sex- and tissue-specific changes in mTOR signaling with age in C57BL/6J mice. Aging Cell 2016, 15, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Kriete, A.; Mayo, K.L. Atypical pathways of NF-kappaB activation and aging. Exp. Gerontol. 2009, 44, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, W.; Ndlovu, M.N.; Hoya-Arias, R.; Dijsselbloem, N.; Gerlo, S.; Haegeman, G. Keeping up NF-kappaB appearances: Epigenetic control of immunity or inflammation-triggered epigenetics. Biochem. Pharmacol. 2006, 72, 1114–1131. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, P.; Solovieff, N.; Dewan, A.T.; Walsh, K.M.; Puca, A.; Hartley, S.W.; Melista, E.; Andersen, S.; Dworkis, D.A.; Wilk, J.B.; et al. Genetic signatures of exceptional longevity in humans. PLoS ONE 2012, 7, e29848. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.S.; Kawahara, T.L.; Segal, E.; Chang, H.Y. Reversal of aging by NFkappaB blockade. Cell Cycle 2008, 7, 556–559. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Jimi, E.; Zeiss, C.; Hayden, M.S.; Ghosh, S. Constitutively active NF-kappaB triggers systemic TNFalpha-dependent inflammation and localized TNFalpha-independent inflammatory disease. Genes Dev. 2010, 24, 1709–1717. [Google Scholar] [CrossRef] [PubMed]
- Alcamo, E.; Mizgerd, J.P.; Horwitz, B.H.; Bronson, R.; Beg, A.A.; Scott, M.; Doerschuk, C.M.; Hynes, R.O.; Baltimore, D. Targeted mutation of TNF receptor I rescues the RelA-deficient mouse and reveals a critical role for NF-kappa B in leukocyte recruitment. J. Immunol. 2001, 167, 1592–1600. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Jimi, E.; Zhong, H.; Hayden, M.S.; Ghosh, S. Repression of gene expression by unphosphorylated NF-kappaB p65 through epigenetic mechanisms. Genes Dev. 2008, 22, 1159–1173. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Maynard, S.; Bayne, A.C.; Sykora, P.; Tian, J.; de Souza-Pinto, N.C.; Croteau, D.L.; Bohr, V.A. The mitochondrial transcription factor A functions in mitochondrial base excision repair. DNA Repair 2010, 9, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Artandi, S.E.; Attardi, L.D. Pathways connecting telomeres and p53 in senescence, apoptosis, and cancer. Biochem. Biophys. Res. Commun. 2005, 331, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Lawless, C.; Jurk, D.; Gillespie, C.S.; Shanley, D.; Saretzki, G.; von Zglinicki, T.; Passos, J.F. A stochastic step model of replicative senescence explains ROS production rate in ageing cell populations. PLoS ONE 2012, 7, e32117. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hu, W.; Teresky, A.K.; Hernando, E.; Cordon-Cardo, C.; Levine, A.J. Declining p53 function in the aging process: A possible mechanism for the increased tumor incidence in older populations. Proc. Natl. Acad. Sci. USA 2007, 104, 16633–16638. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A. Does p53 affect organismal aging? J. Cell. Physiol. 2002, 192, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Witten, T.M. Modeling Cellular Aging: An Introduction—Mathematical and Computational Approaches. In Cellular Ageing and Replicative Senescence; Rattan, S., Hayflick, L., Eds.; Springer: Cham, Switzerland, 2016; pp. 117–141. [Google Scholar]
- Mooney, K.M.; Morgan, A.E.; Mc Auley, M.T. Aging and computational systems biology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Kriete, A.; Lechner, M.; Clearfield, D.; Bohmann, D. Computational systems biology of aging. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011, 3, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Kriete, A. Robustness and aging—A systems-level perspective. Biosystems 2013, 112, 37–48. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfego, D.; Kriete, A. Simulation of Cellular Energy Restriction in Quiescence (ERiQ)—A Theoretical Model for Aging. Biology 2017, 6, 44. https://doi.org/10.3390/biology6040044
Alfego D, Kriete A. Simulation of Cellular Energy Restriction in Quiescence (ERiQ)—A Theoretical Model for Aging. Biology. 2017; 6(4):44. https://doi.org/10.3390/biology6040044
Chicago/Turabian StyleAlfego, David, and Andres Kriete. 2017. "Simulation of Cellular Energy Restriction in Quiescence (ERiQ)—A Theoretical Model for Aging" Biology 6, no. 4: 44. https://doi.org/10.3390/biology6040044
APA StyleAlfego, D., & Kriete, A. (2017). Simulation of Cellular Energy Restriction in Quiescence (ERiQ)—A Theoretical Model for Aging. Biology, 6(4), 44. https://doi.org/10.3390/biology6040044