
Structural Organization of Enzymes of the Phenylacetate Catabolic Hybrid Pathway

Abstract
:1. Introduction
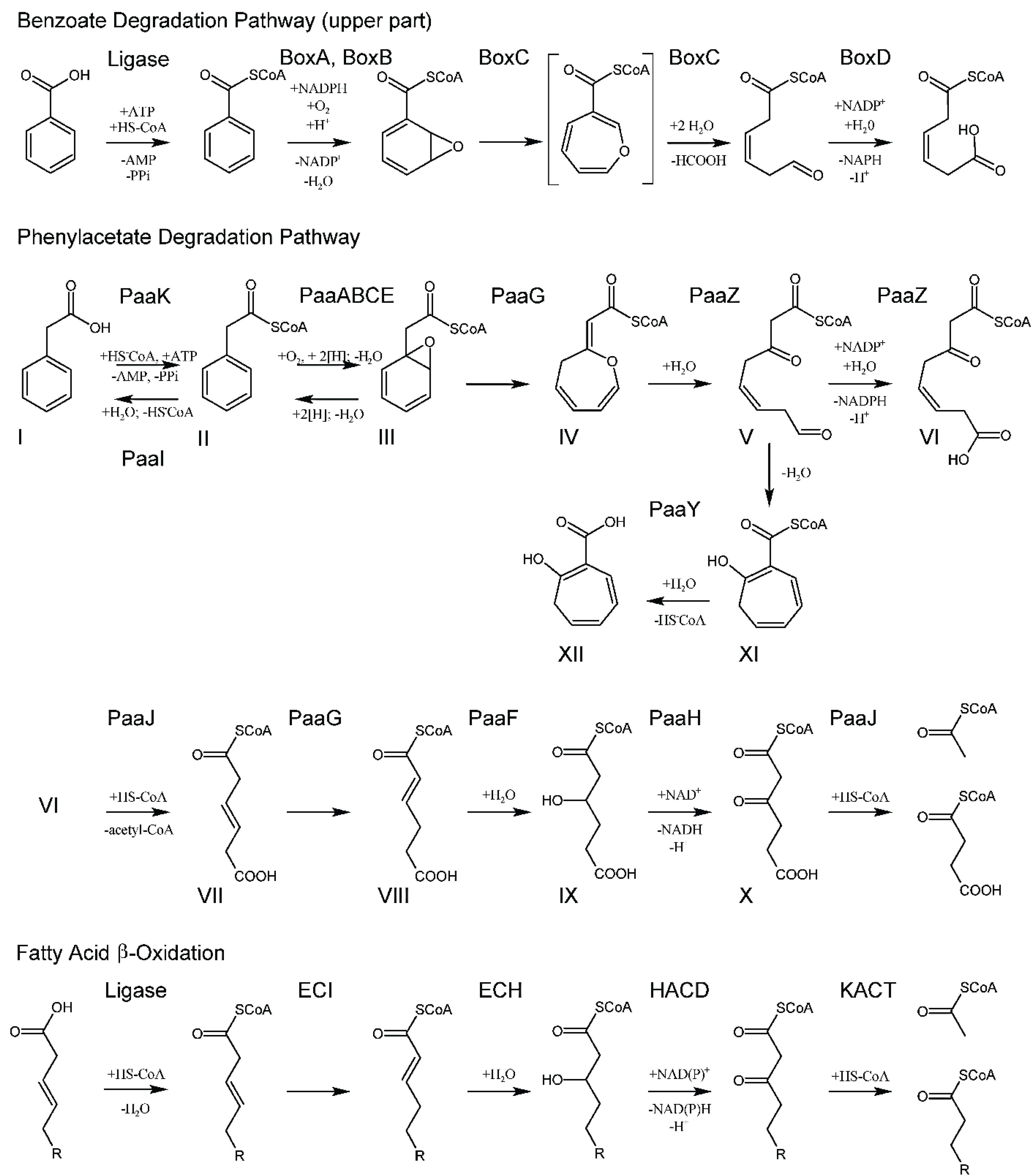
2. Phenylacetate Hybrid Degradation Pathway

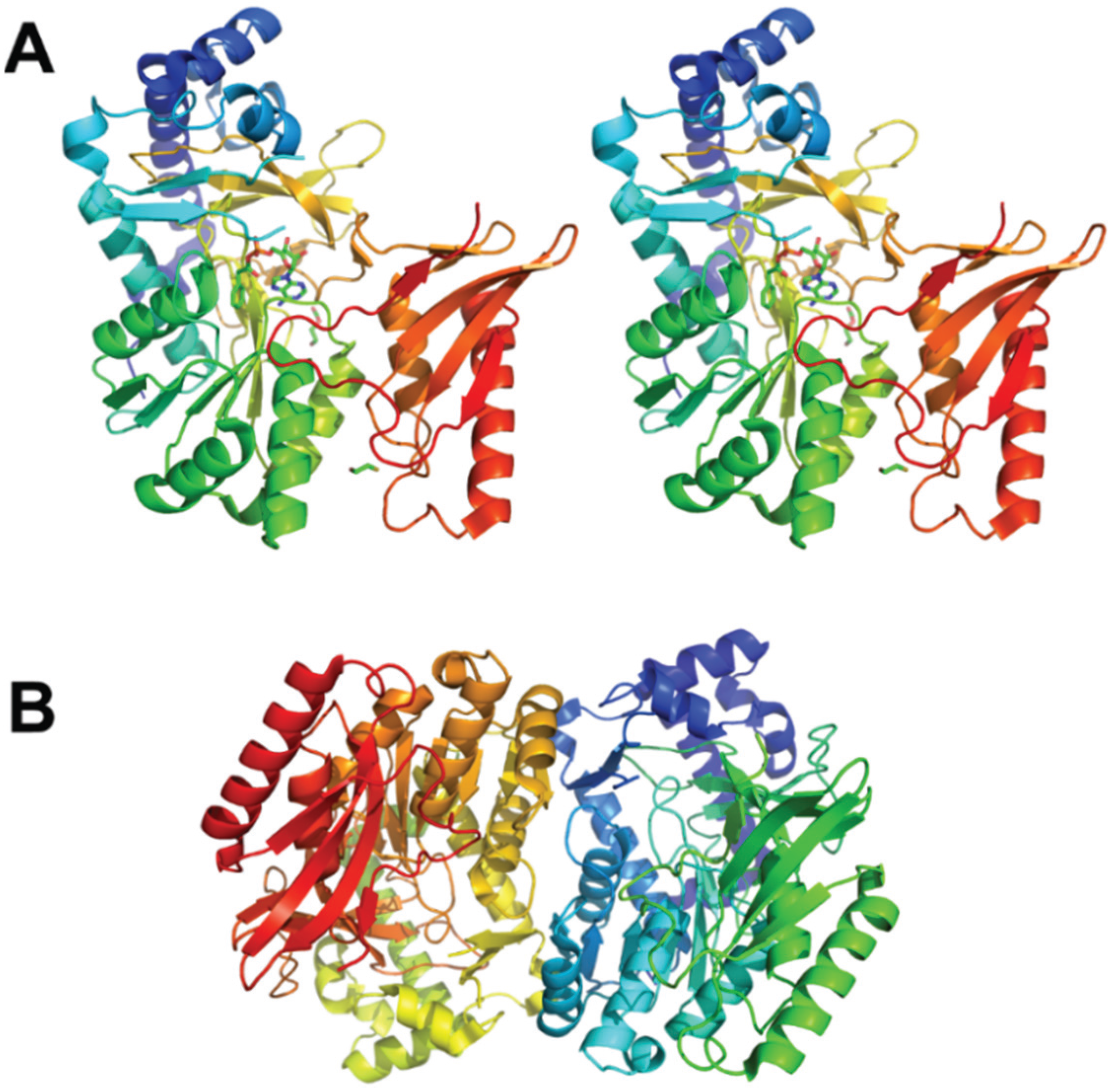{kind=link}
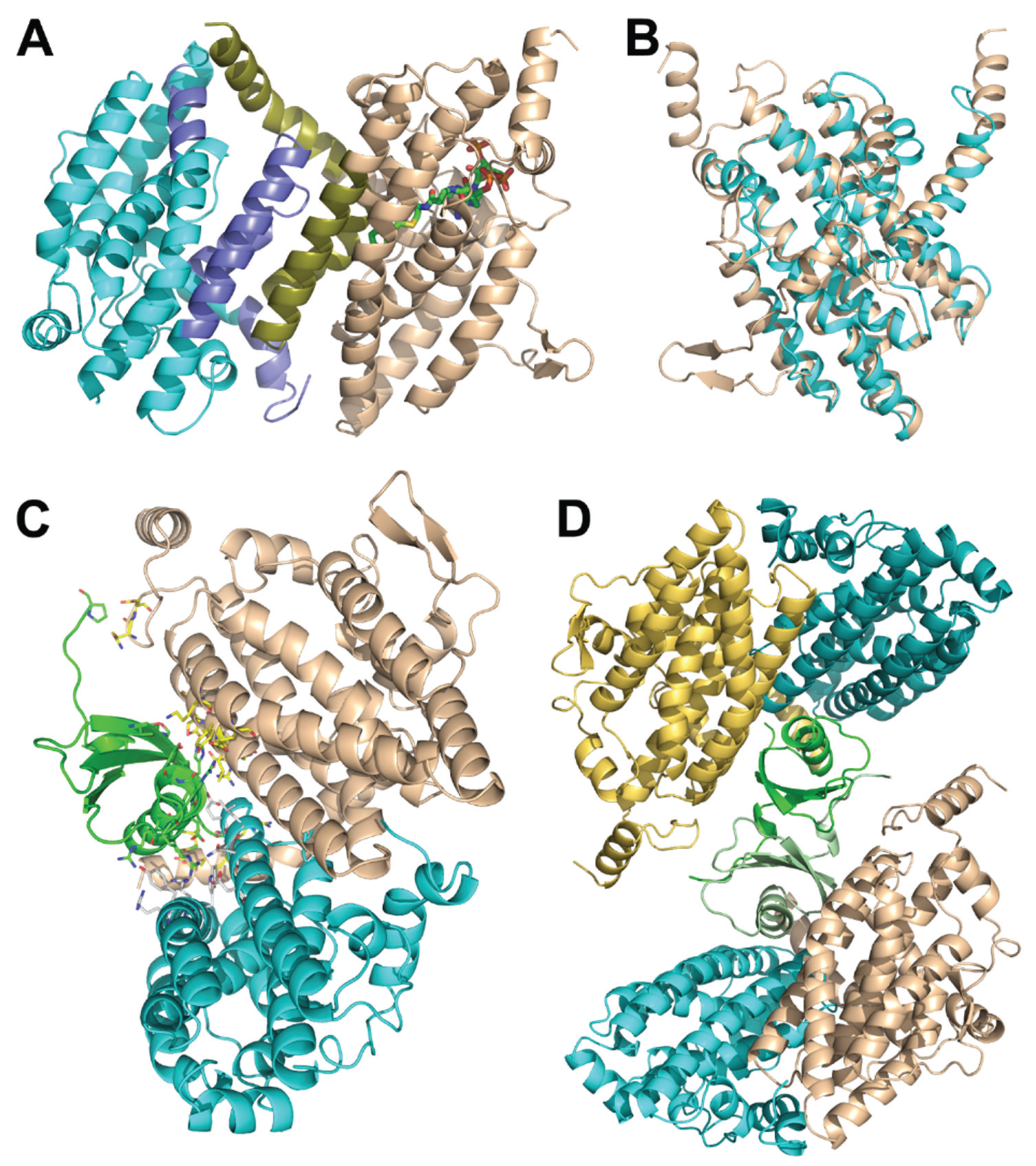{kind=link}
{kind=link}
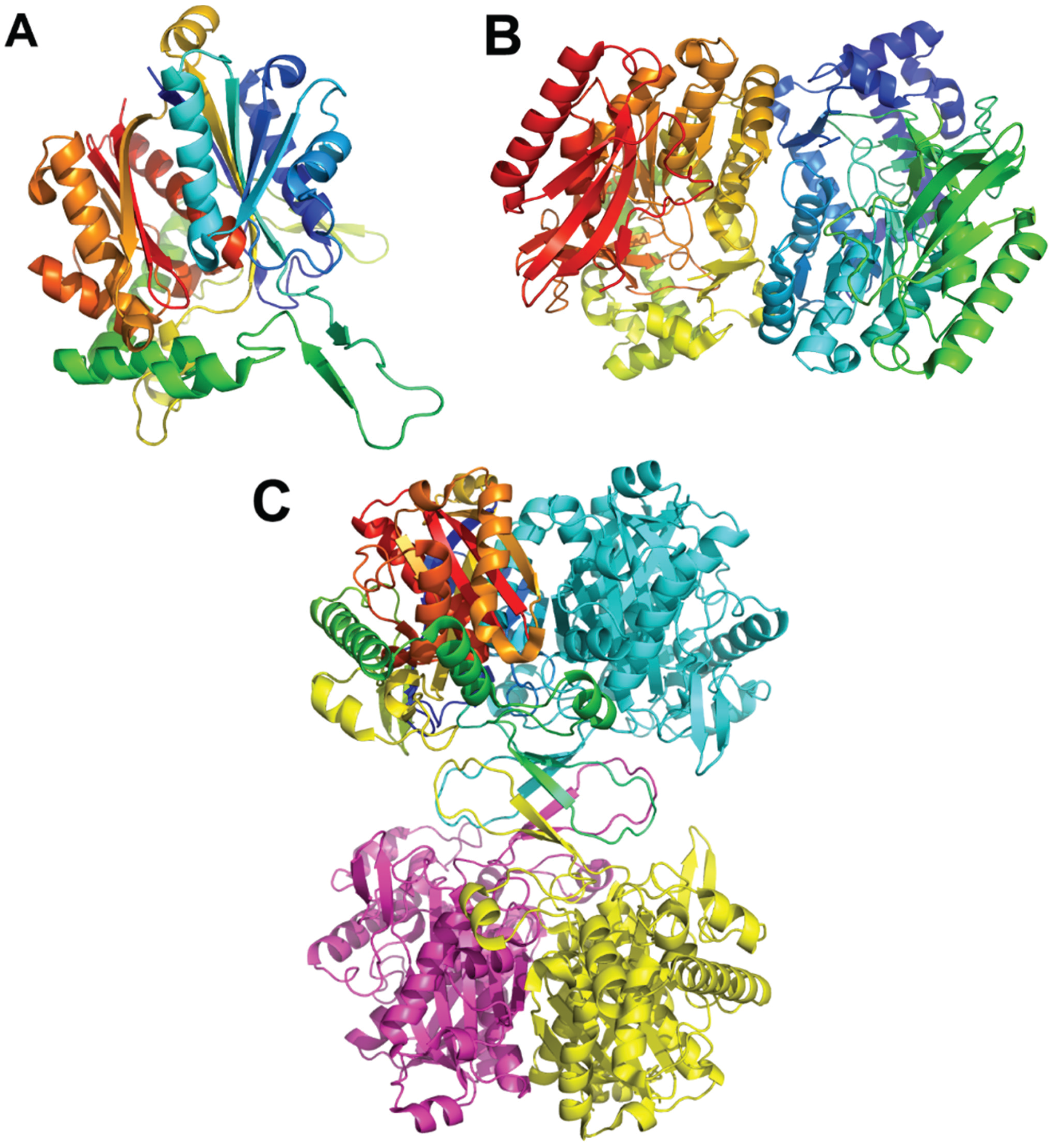{kind=link}
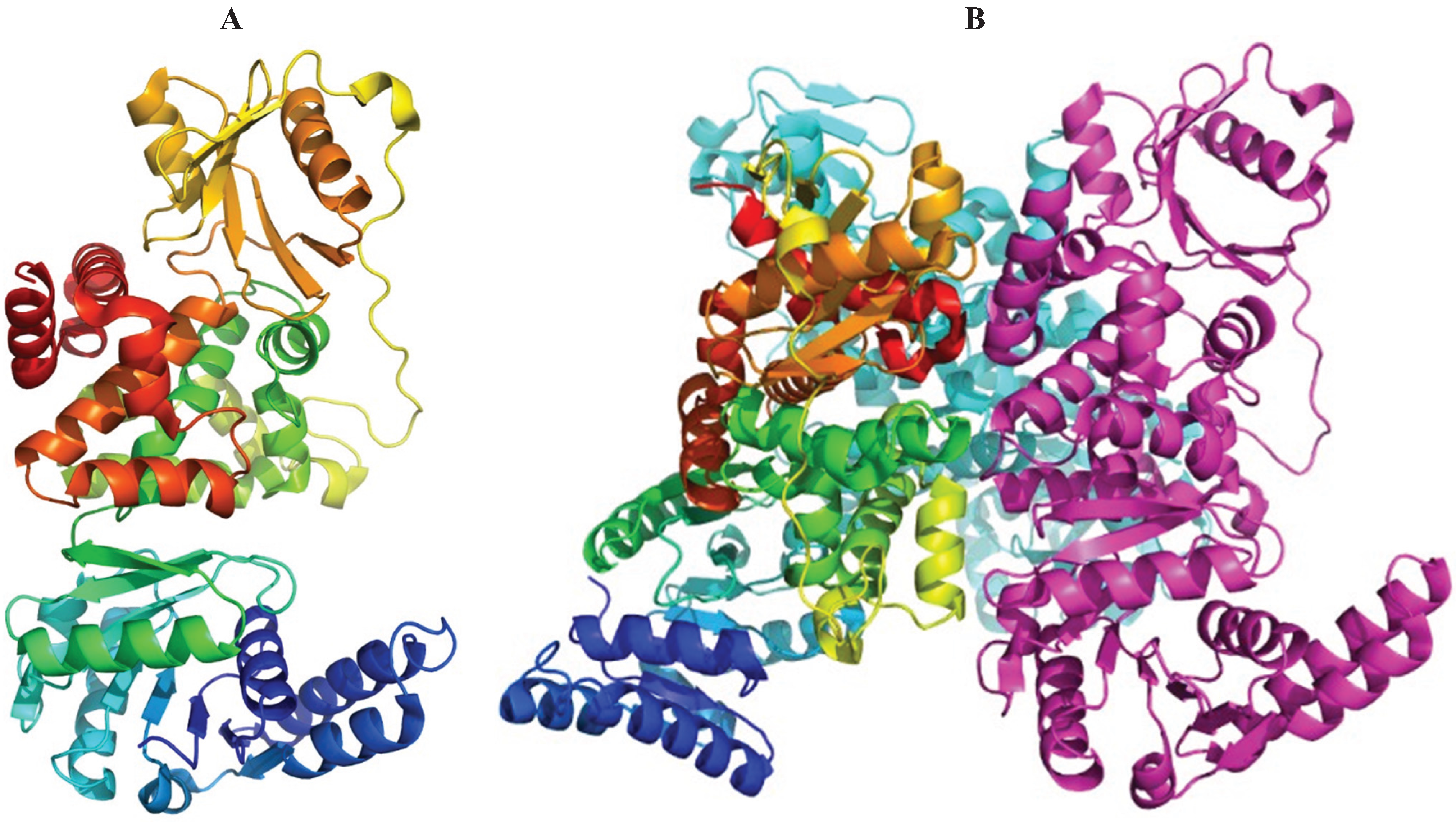{kind=link}
{kind=link}
| Protein | Function | Structure | Organism | Homologue | PDB ID | Organism | Family/fold |
|---|---|---|---|---|---|---|---|
| PaaK | Phenylacetate-CoA ligase | 2Y4O (PaaK2) 2Y27 (PaaK1) | Burkholderia cenopacia | Benzoate-CoA ligase | 2V7B | Burkholderia xenovorans | Adenylate-forming domain, class I |
| PaaI | Phenylacetyl-CoA Thioesterase | 1PSU | E. coli | PaaI-like protein | 4M20 | Staphylococcus aureus | Thioesterase group of hotdog superfamily |
| 1J1Y | Thermus thermophilus | 4-hydroxybenzoyl-CoA thioesterase | 3R37 | Arthrobacter sp. | |||
| PaaA | Catalytic subunit of 1,2-phenylacetyl-CoA epoxidase | 3PW1 | E. coli | BoxB | 3Q1G | Azoarcus evansii | Bacterial multicomponent monooxygenase |
| MMOH | 4GAM | Methylococcus capsulatus | |||||
| PaaB | Bridging subunit of 1,2-phenylacetyl-CoA epoxidase | 3EGR | Ralstonia eutropha | PaaB-like proteins | |||
| PaaC | Structural subunit of 1,2-phenylacetyl-CoA epoxidase | 3PW1 | E. coli | MMOH | 4GAM | Methylococcus capsulatus | Bacterial multicomponent monooxygenase |
| PaaE | Reductase subunit of 1,2-phenylacetyl-CoA epoxidase | Phtalate Dehydrogenase | 2PIA | Burkholderia cepacia | Class IA Reductase | ||
| PaaD | unknown | Duf59 | 3LNO | Bacillus Anthracis | Domain of the unknown function 59 | ||
| PaaG | 1,2-epoxyphenylacetyl-CoA isomerase | 4FZW | E. coli | BoxC | 2W3P | Burkholderia xenovorans | Enoyl-CoA isomerase; Crotonase superfamily |
| 3HRX | Thermus thermophilus | ||||||
| PaaZ | Oxepin-CoA hydrolase | BoxD | No structure | Azoarcus evansii | N-terminal: NAD(P)+-dependent aldehyde dehydrogenase C-terminal: (De)Hydratase group of hotdog superfamily | ||
| Aldehyde dehydrogenase | 2VRO | Burkholderia xenovorans | |||||
| PaaJ | 3-oxoadipyl-CoA/3-oxo-5,6-dehydrosuberyl-CoA thiolase | 1ULQ | Thermus thermophilus | Acetyl-CoA acetyltransferase | 4N44 | Clostridium acetobutylicum | 3-ketoacyl-CoA thiolase (thiolase I) |
| ThlA2 | 4E1L | Clostridium difficile | |||||
| PaaF | 2.3-dehydroadipyl-CoA hydratase | 4FZW | E. coli | BoxC | 2W3P | Burkholderia xenovorans | Enoyl-CoA hydratase; Crotonase superfamily |
| PaaH | 3-hydroxyadipyl-CoA dehydrogenase | 3MOG | E. coli | 3-hydroxyacyl-CoA dehydrogenase | |||
| PaaY | 2-hydroxycyclohepta-1,4,6-triene-1-carboxyl-CoA thioesterase | GK2848 | 3VNP | Geobacillus kaustophilus | |||
| PaaX | Transcriptional repressor | PaaX-like protein | 3LO9 | Jannaschia sp. | PaaX-like proteins containing helix-turn-helix motif |
3. The Upper Part of the Pathway
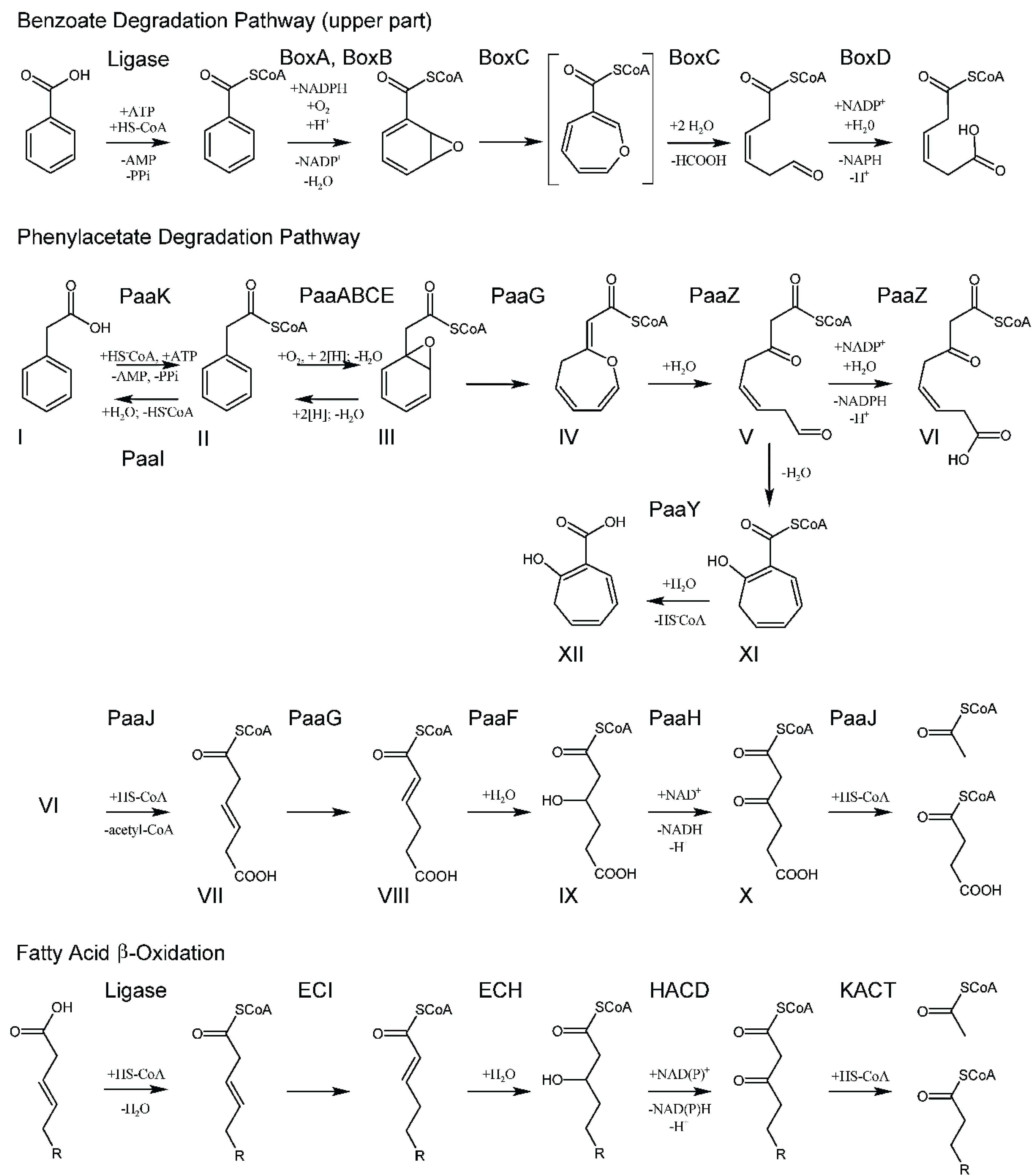
3.1. Activation of the Aromatic Compound
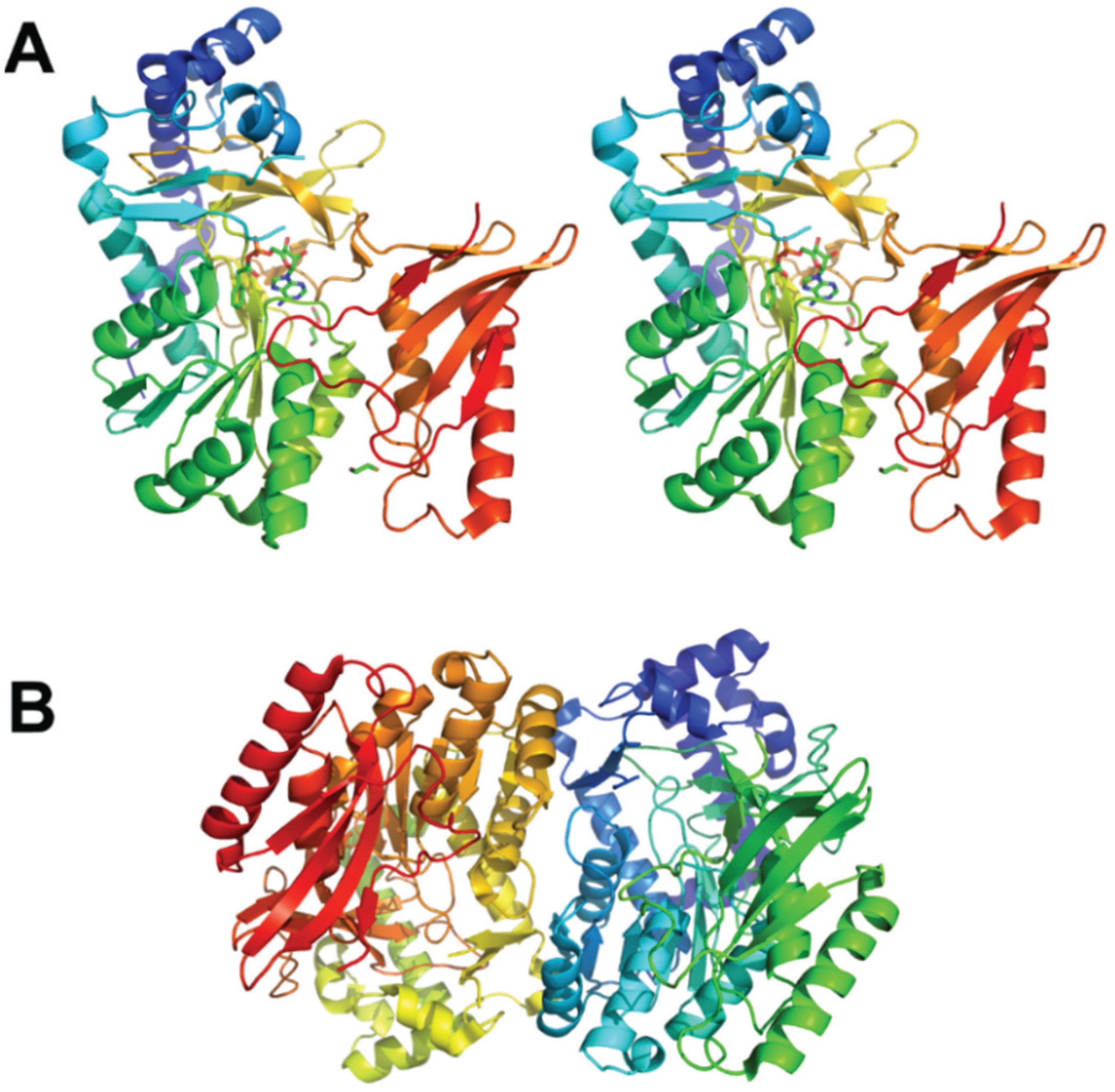
3.2. Epoxidation of the Aromatic Ring
3.3. Ring Opening
3.4. Controlling the Fate of Toxic Epoxide
The Lower Part of the Paa Pathway
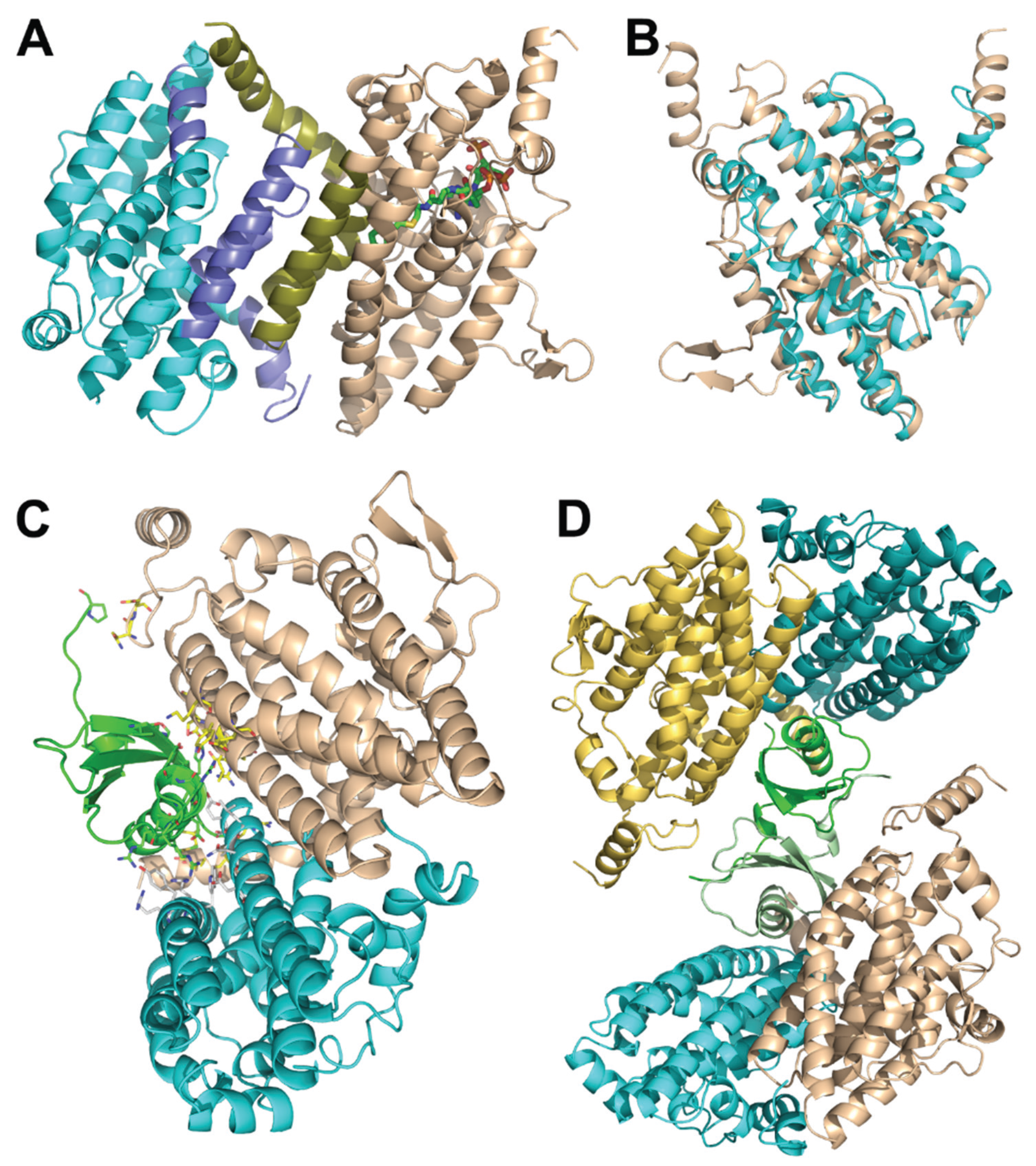
3.5. Protein-protein Interactions Among Enzymes of the Lower Part of the Paa Pathway
3.6. Structure of the PaaFG Complex
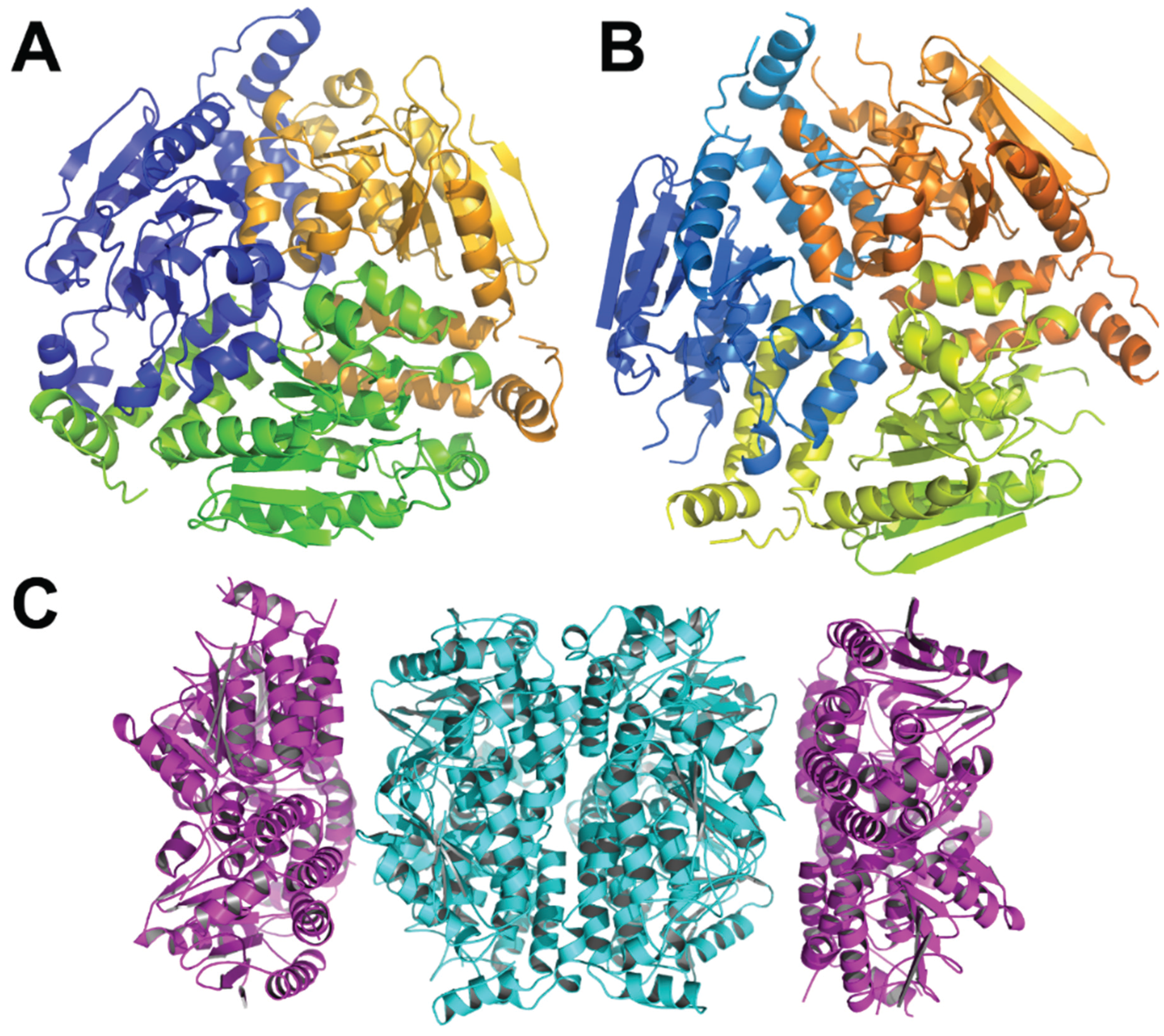
3.7. Other Enzymes Involved in the Lower Part of the Paa Pathway
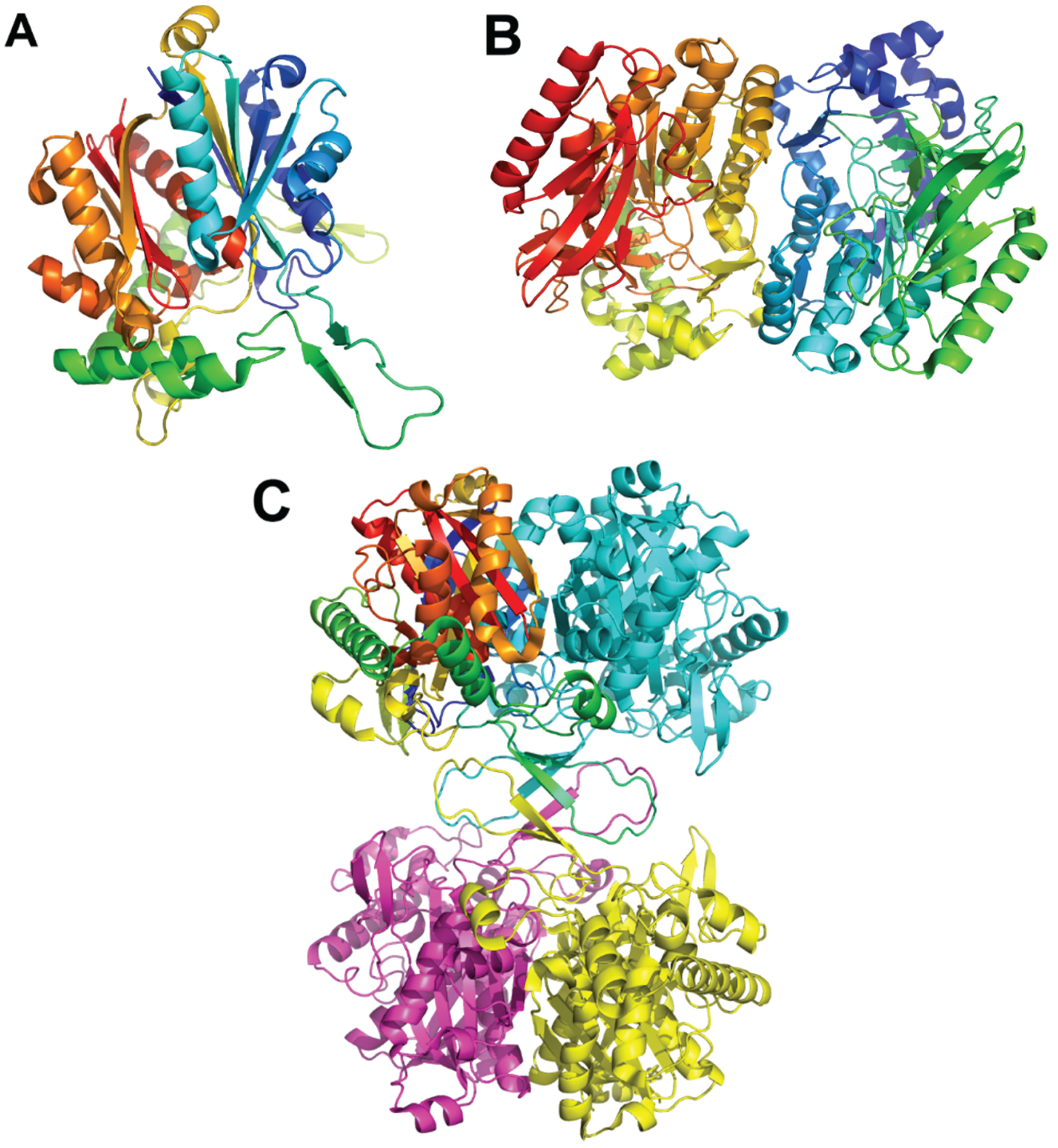
Other Proteins of the Paa Operon
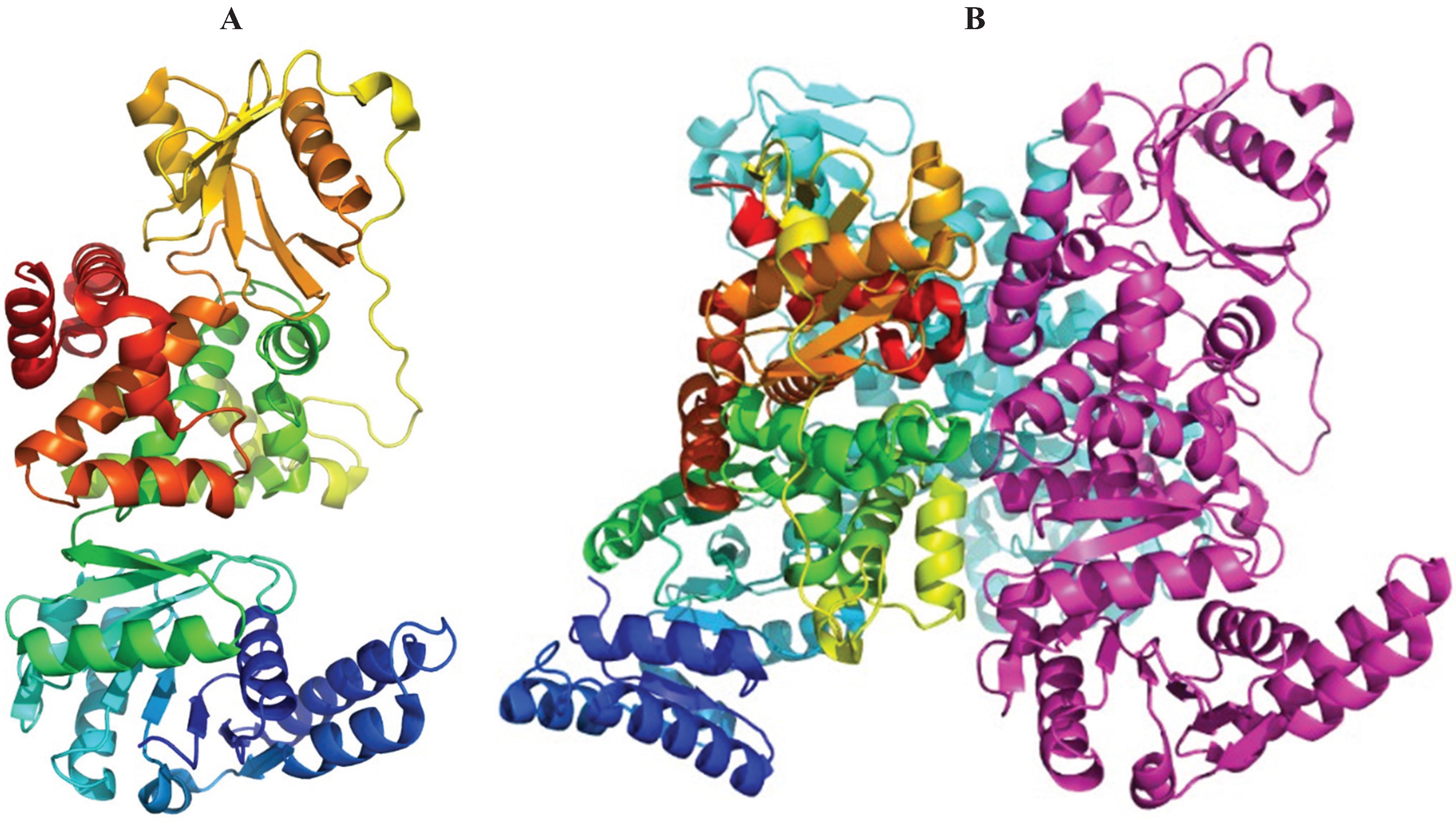
4. Similarities with Other Metabolic Pathways
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bugg, T.D.; Ahmad, M.; Hardiman, E.M.; Rahmanpour, R. Pathways for degradation of lignin in bacteria and fungi. Nat. Prod. Rep. 2011, 28, 1883–1896. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Nagarajan, K.; Loh, K.C. Biodegradation of aromatic compounds: Current status and opportunities for biomolecular approaches. Appl. Microbiol. Biotechnol. 2009, 85, 207–228. [Google Scholar] [CrossRef] [PubMed]
- Metzler, D. Biochemistry, 2nd ed.; Academic Press: San Diego, CA, USA, 2003. [Google Scholar]
- Fuchs, G.; Boll, M.; Heider, J. Microbial degradation of aromatic compounds—From one strategy to four. Nat. Rev. Microbiol. 2011, 9, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Carmona, M.; Zamarro, M.T.; Blazquez, B.; Durante-Rodriguez, G.; Juarez, J.F.; Valderrama, J.A.; Barragan, M.J.; Garcia, J.L.; Diaz, E. Anaerobic catabolism of aromatic compounds: A genetic and genomic view. Microbiol. Mol. Biol. Rev. 2009, 73, 71–133. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, G. Anaerobic metabolism of aromatic compounds. Ann. N.Y. Acad. Sci. 2008, 1125, 82–99. [Google Scholar] [CrossRef] [PubMed]
- Ferrandez, A.; Minambres, B.; Garcia, B.; Olivera, E.R.; Luengo, J.M.; Garcia, J.L.; Diaz, E. Catabolism of phenylacetic acid in Escherichia coli. Characterization of a new aerobic hybrid pathway. J. Biol. Chem. 1998, 273, 25974–25986. [Google Scholar] [CrossRef] [PubMed]
- Ismail, W.; El-Said Mohamed, M.; Wanner, B.L.; Datsenko, K.A.; Eisenreich, W.; Rohdich, F.; Bacher, A.; Fuchs, G. Functional genomics by NMR spectroscopy. Phenylacetate catabolism in Escherichia coli. Eur. J. Biochem. 2003, 270, 3047–3054. [Google Scholar] [CrossRef] [PubMed]
- Teufel, R.; Mascaraque, V.; Ismail, W.; Voss, M.; Perera, J.; Eisenreich, W.; Haehnel, W.; Fuchs, G. Bacterial phenylalanine and phenylacetate catabolic pathway revealed. Proc. Natl. Acad. Sci. USA 2010, 107, 14390–14395. [Google Scholar] [CrossRef] [PubMed]
- Rather, L.J.; Knapp, B.; Haehnel, W.; Fuchs, G. Coenzyme A-dependent aerobic metabolism of benzoate via epoxide formation. J. Biol. Chem. 2010, 285, 20615–20624. [Google Scholar] [CrossRef] [PubMed]
- Boll, M.; Fuchs, G. Benzoyl-coenzyme A reductase (dearomatizing), a key enzyme of anaerobic aromatic metabolism. ATP dependence of the reaction, purification and some properties of the enzyme from Thauera aromatica strain K172. Eur. J. Biochem. 1995, 234, 921–933. [Google Scholar] [CrossRef] [PubMed]
- Kung, J.W.; Loffler, C.; Dorner, K.; Heintz, D.; Gallien, S.; van Dorsselaer, A.; Friedrich, T.; Boll, M. Identification and characterization of the tungsten-containing class of benzoyl-coenzyme A reductases. Proc. Natl. Acad. Sci. USA 2009, 106, 17687–17692. [Google Scholar] [CrossRef] [PubMed]
- Teufel, R.; Gantert, C.; Voss, M.; Eisenreich, W.; Haehnel, W.; Fuchs, G. Studies on the mechanism of ring hydrolysis in phenylacetate degradation: A metabolic branching point. J. Biol. Chem. 2011, 286, 11021–11034. [Google Scholar] [CrossRef] [PubMed]
- Teufel, R.; Friedrich, T.; Fuchs, G. An oxygenase that forms and deoxygenates toxic epoxide. Nature 2012, 483, 359–362. [Google Scholar] [CrossRef] [PubMed]
- El-Said Mohamed, M. Biochemical and molecular characterization of phenylacetate-coenzyme A ligase, an enzyme catalyzing the first step in aerobic metabolism of phenylacetic acid in Azoarcus evansii. J. Bacteriol. 2000, 182, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Grishin, A.M.; Ajamian, E.; Tao, L.; Zhang, L.; Menard, R.; Cygler, M. Structural and functional studies of the Escherichia coli phenylacetyl-CoA monooxygenase complex. J. Biol. Chem. 2011, 286, 10735–10743. [Google Scholar] [CrossRef] [PubMed]
- Nogales, J.; Macchi, R.; Franchi, F.; Barzaghi, D.; Fernandez, C.; Garcia, J.L.; Bertoni, G.; Diaz, E. Characterization of the last step of the aerobic phenylacetic acid degradation pathway. Microbiology 2007, 153, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Gulick, A.M. Conformational dynamics in the Acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase. ACS chemical biology 2009, 4, 811–827. [Google Scholar] [CrossRef] [PubMed]
- Erb, T.J.; Ismail, W.; Fuchs, G. Phenylacetate metabolism in thermophiles: Characterization of phenylacetate-CoA ligase, the initial enzyme of the hybrid pathway in Thermus thermophilus. Curr. Microbiol. 2008, 57, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Law, A.; Boulanger, M.J. Defining a structural and kinetic rationale for paralogous copies of phenylacetate-CoA ligases from the cystic fibrosis pathogen Burkholderia cenocepacia J2315. J. Biol. Chem. 2011, 286, 15577–15585. [Google Scholar] [CrossRef] [PubMed]
- Imolorhe, I.A.; Cardona, S.T. 3-Hydroxyphenylacetic acid induces the Burkholderia cenocepacia phenylacetic acid degradation pathway—Toward understanding the contribution of aromatic catabolism to pathogenesis. Front. Cell. Infect. Microbio. 2011. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, C.; Ferrandez, A.; Minambres, B.; Diaz, E.; Garcia, J.L. Genetic characterization of the phenylacetyl-coenzyme A oxygenase from the aerobic phenylacetic acid degradation pathway of Escherichia coli. Appl. Environ. Microbiol. 2006, 72, 7422–7426. [Google Scholar] [CrossRef] [PubMed]
- Grishin, A.M.; Ajamian, E.; Zhang, L.; Cygler, M. Crystallization and preliminary X-ray analysis of PaaAC, the main component of the hydroxylase of the Escherichia coli phenylacetyl-coenzyme A oxygenase complex. Acta Crystallogr. Sect. F Struct. Biolo. Cryst. Commun. 2010, 66, 1045–1049. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, A.C.; Frederick, C.A.; Lippard, S.J.; Nordlund, P. Crystal structure of a bacterial non-haem iron hydroxylase that catalyses the biological oxidation of methane. Nature 1993, 366, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Grishin, A.M.; Ajamian, E.; Tao, L.; Bostina, M.; Zhang, L.; Trempe, J.F.; Menard, R.; Rouiller, I.; Cygler, M. Family of phenylacetyl-CoA monooxygenases differs in subunit organization from other monooxygenases. J. Struct. Biol. 2013, 184, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Correll, C.C.; Batie, C.J.; Ballou, D.P.; Ludwig, M.L. Phthalate dioxygenase reductase: A modular structure for electron transfer from pyridine nucleotides to [2Fe-2S]. Science 1992, 258, 1604–1610. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.H.; Farmer, P.B. Evidence for DNA and protein binding by styrene and styrene oxide. Crit. Rev. Toxicol. 1994, 24, S35–S46. [Google Scholar] [CrossRef] [PubMed]
- Park, J.B.; Buhler, B.; Habicher, T.; Hauer, B.; Panke, S.; Witholt, B.; Schmid, A. The efficiency of recombinant Escherichia coli as biocatalyst for stereospecific epoxidation. Biotechnol. Bioeng. 2006, 95, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Law, R.J.; Hamlin, J.N.; Sivro, A.; McCorrister, S.J.; Cardama, G.A.; Cardona, S.T. A functional phenylacetic acid catabolic pathway is required for full pathogenicity of Burkholderia cenocepacia in the Caenorhabditis elegans host model. J. Bacteriol. 2008, 190, 7209–7218. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Zhuang, Z.; Finci, L.; Dunaway-Mariano, D.; Kniewel, R.; Buglino, J.A.; Solorzano, V.; Wu, J.; Lima, C.D. Structure, function, and mechanism of the phenylacetate pathway hot dog-fold thioesterase PaaI. J. Biol. Chem. 2006, 281, 11028–11038. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, C.; Diaz, E.; Garcia, J.L. Insights on the regulation of the phenylacetate degradation pathway from Escherichia coli. Environ. Microbiol. Rep. 2014, 6, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Kasaragod, P.; Venkatesan, R.; Kiema, T.R.; Hiltunen, J.K.; Wierenga, R.K. Crystal structure of liganded rat peroxisomal multifunctional enzyme type 1: A flexible molecule with two interconnected active sites. J. Biol. Chem. 2010, 285, 24089–24098. [Google Scholar] [CrossRef] [PubMed]
- Grishin, A.M.; Ajamian, E.; Zhang, L.; Rouiller, I.; Bostina, M.; Cygler, M. Protein-protein interactions in the beta-oxidation part of the phenylacetate utilization pathway: Crystal structure of the PaaF–PaaG hydratase-isomerase complex. J. Biol. Chem. 2012, 287, 37986–37996. [Google Scholar] [CrossRef] [PubMed]
- Hamed, R.B.; Batchelar, E.T.; Clifton, I.J.; Schofield, C.J. Mechanisms and structures of crotonase superfamily enzymes—How nature controls enolate and oxyanion reactivity. Cell. Mol. Life Sci. 2008, 65, 2507–2527. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Ferrandez, A.; Garcia, J.L.; Diaz, E. Transcriptional regulation of the divergent paa catabolic operons for phenylacetic acid degradation in Escherichia coli. J. Biol. Chem. 2000, 275, 12214–12222. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Altuve, A.; Carrasco-Lopez, C.; Hernandez-Rocamora, V.M.; Sanz, J.M.; Hermoso, J.A. Crystallization and preliminary X-ray diffraction studies of the transcriptional repressor PaaX, the main regulator of the phenylacetic acid degradation pathway in Escherichia coli W. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011, 67, 1278–1280. [Google Scholar] [CrossRef] [PubMed]
- Rather, L.J.; Weinert, T.; Demmer, U.; Bill, E.; Ismail, W.; Fuchs, G.; Ermler, U. Structure and mechanism of the diiron benzoyl-coenzyme A epoxidase BoxB. J. Biol. Chem. 2011, 286, 29241–29248. [Google Scholar] [CrossRef] [PubMed]
- Zaar, A.; Gescher, J.; Eisenreich, W.; Bacher, A.; Fuchs, G. New enzymes involved in aerobic benzoate metabolism in Azoarcus evansii. Mol. Microbiol. 2004, 54, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Bains, J.; Leon, R.; Boulanger, M.J. Structural and biophysical characterization of BoxC from Burkholderia xenovorans LB400: A novel ring-cleaving enzyme in the crotonase superfamily. J. Biol. Chem. 2009, 284, 16377–16385. [Google Scholar] [CrossRef] [PubMed]
- Bains, J.; Boulanger, M.J. Structural and biochemical characterization of a novel aldehyde dehydrogenase encoded by the benzoate oxidation pathway in Burkholderia xenovorans LB400. J. Mol. Biol. 2008, 379, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Mursula, A.M.; Hiltunen, J.K.; Wierenga, R.K. Structural studies on delta3-delta2-enoyl-CoA isomerase: The variable mode of assembly of the trimeric disks of the crotonase superfamily. FEBS Lett. 2004, 557, 81–87. [Google Scholar] [CrossRef]
- Partanen, S.T.; Novikov, D.K.; Popov, A.N.; Mursula, A.M.; Hiltunen, J.K.; Wierenga, R.K. The 1.3 Å crystal structure of human mitochondrial Delta3-Delta2-enoyl-CoA isomerase shows a novel mode of binding for the fatty acyl group. J. Mol. Biol. 2004, 342, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Mursula, A.M.; van Aalten, D.M.; Hiltunen, J.K.; Wierenga, R.K. The crystal structure of delta3-delta2-enoyl-CoA isomerase. J. Mol. Biol. 2001, 309, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Modis, Y.; Filppula, S.A.; Novikov, D.K.; Norledge, B.; Hiltunen, J.K.; Wierenga, R.K. The crystal structure of dienoyl-CoA isomerase at 1.5 Å resolution reveals the importance of aspartate and glutamate sidechains for catalysis. Structure 1998, 6, 957–970. [Google Scholar] [CrossRef]
- Engel, C.K.; Kiema, T.R.; Hiltunen, J.K.; Wierenga, R.K. The crystal structure of enoyl-CoA hydratase complexed with octanoyl-CoA reveals the structural adaptations required for binding of a long chain fatty acid-CoA molecule. J. Mol. Biol. 1998, 275, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Engel, C.K.; Mathieu, M.; Zeelen, J.P.; Hiltunen, J.K.; Wierenga, R.K. Crystal structure of enoyl-coenzyme A (CoA) hydratase at 2.5 angstroms resolution: A spiral fold defines the CoA-binding pocket. EMBO J. 1996, 15, 5135–5145. [Google Scholar] [PubMed]
- Ishikawa, M.; Tsuchiya, D.; Oyama, T.; Tsunaka, Y.; Morikawa, K. Structural basis for channelling mechanism of a fatty acid beta-oxidation multienzyme complex. EMBO J. 2004, 23, 2745–2754. [Google Scholar] [CrossRef] [PubMed]
- Barycki, J.J.; O’Brien, L.K.; Bratt, J.M.; Zhang, R.; Sanishvili, R.; Strauss, A.W.; Banaszak, L.J. Biochemical characterization and crystal structure determination of human heart short chain L-3-hydroxyacyl-CoA dehydrogenase provide insights into catalytic mechanism. Biochemistry 1999, 38, 5786–5798. [Google Scholar] [CrossRef] [PubMed]
- Haapalainen, A.M.; Merilainen, G.; Pirila, P.L.; Kondo, N.; Fukao, T.; Wierenga, R.K. Crystallographic and kinetic studies of human mitochondrial acetoacetyl-CoA thiolase: The importance of potassium and chloride ions for its structure and function. Biochemistry 2007, 46, 4305–4321. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grishin, A.M.; Cygler, M. Structural Organization of Enzymes of the Phenylacetate Catabolic Hybrid Pathway. Biology 2015, 4, 424-442. https://doi.org/10.3390/biology4020424
Grishin AM, Cygler M. Structural Organization of Enzymes of the Phenylacetate Catabolic Hybrid Pathway. Biology. 2015; 4(2):424-442. https://doi.org/10.3390/biology4020424
Chicago/Turabian StyleGrishin, Andrey M., and Miroslaw Cygler. 2015. "Structural Organization of Enzymes of the Phenylacetate Catabolic Hybrid Pathway" Biology 4, no. 2: 424-442. https://doi.org/10.3390/biology4020424
APA StyleGrishin, A. M., & Cygler, M. (2015). Structural Organization of Enzymes of the Phenylacetate Catabolic Hybrid Pathway. Biology, 4(2), 424-442. https://doi.org/10.3390/biology4020424