Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disease characterized by the accumulation of amyloid-β (Aβ) and the loss of synapses. Aggregation of the cellular prion protein (PrPC) by Aβ oligomers induced synapse damage in cultured neurons. PrPC is attached to membranes via a glycosylphosphatidylinositol (GPI) anchor, the composition of which affects protein targeting and cell signaling. Monoacylated PrPC incorporated into neurons bound “natural Aβ”, sequestering Aβ outside lipid rafts and preventing its accumulation at synapses. The presence of monoacylated PrPC reduced the Aβ-induced activation of cytoplasmic phospholipase A2 (cPLA2) and Aβ-induced synapse damage. This protective effect was stimulus specific, as treated neurons remained sensitive to α-synuclein, a protein associated with synapse damage in Parkinson’s disease. In synaptosomes, the aggregation of PrPC by Aβ oligomers triggered the formation of a signaling complex containing the cPLA2.a process, disrupted by monoacylated PrPC. We propose that monoacylated PrPC acts as a molecular sponge, binding Aβ oligomers at the neuronal perikarya without activating cPLA2 or triggering synapse damage.
1. Introduction
Alzheimer’s disease (AD) is a complex neurological disorder that is characterized by a progressive dementia as a consequence of synapse failure [1]. The amyloid hypothesis of AD pathogenesis maintains that the primary event is the cleavage of the amyloid precursor protein by β- and γ-secretases into toxic amyloid-β (Aβ) fragments [2]. The accumulation of Aβ peptides, including C-terminal fragments of 42 residues (Aβ42), is thought to cause the abnormal phosphorylation of tau, synapse dysfunction and ultimately the clinical symptoms of AD. Aβ42 self-aggregates and is found in forms ranging from small soluble oligomers to much larger fibrils and plaques. The soluble Aβ oligomers that can diffuse throughout the brain are regarded as the most potent neurotoxins rather than Aβ fibrils or Aβ plaques [3,4]. For these studies, conditioned media from 7PA2 cells (7PA2-CM) containing natural Aβ oligomers [5] that have similar properties, including potency and stability, as the Aβ oligomers found within the cerebrospinal fluid of Alzheimer’s patients [6] were used.
The degree of dementia in AD correlates closely with the loss of synaptic proteins [7,8]. The process of AD-related synapse damage was examined by incubating cultured neurons with Aβ oligomers. Synaptic density in these neurons was determined by measuring the amounts of synaptophysin, a pre-synaptic membrane protein [9], using an enzyme-linked immunoassay (ELISA) [10]. The addition of Aβ reduced the synaptophysin content of cultured neurons indicative of synapse damage [10]. The loss of synaptophysin from neuronal cultures was accompanied by the loss of other synaptic proteins such as synapsin-1 and vesicle-associated membrane protein (VAMP)-1 [11]. This highly reproducible system was used to examine Aβ-induced synapse damage as a model of the synapse damage that occurs in AD.
Soluble Aβ oligomers are thought to bind to neurons in a receptor-mediated process. The identification of disease-relevant Aβ receptors remains controversial, as Aβ binds to many proteins, including the amyloid precursor protein [12], the receptor for advanced glycation end products (RAGE) [13], the p75 neurotrophin receptor [14], and metabotropic glutamate receptors [15]. Recently, the cellular prion protein (PrPC) was identified as a receptor that mediates Aβ-induced synapse dysfunction [16]. PrPC is expressed at high levels within synapses [17] and aggregation of PrPC by Aβ oligomers results in the activation of cytoplasmic phospholipase A2 (cPLA2) and synapse damage [11]. PrPC is anchored to cell membranes by a glycosylphosphatidylinositol (GPI) anchor [18]. Since PrPC-mediated cell signaling was dependent upon the composition of the GPI anchor [19], the effects of PrPC with a modified GPI anchor on Aβ-induced synapse damage was examined. We show that Aβ oligomers bind to PrPC with a monoacylated GPI anchor (monoacylated PrPC). Pre-treatment of neurons with monoacylated PrPC significantly reduced the Aβ-induced activation of cPLA2 and protected neurons against Aβ-induced synapse damage.
2. Experimental Section
Primary neuronal cultures: Cortical neurons were prepared from the brains of day 15.5 murine embryos derived from Prnp wild type(+/+) or Prnp knockout(0/0) mice. After mechanical dissociation, neurons were plated at 2 × 105 cells/well in 48 well plates (pre-coated with poly-L-lysine) in Ham’s F12 containing 5% fetal calf serum for 2 h. Cultures were shaken (600 r.p.m for 5 min) and non-adherent cells removed by 3 washes in PBS. Neurons were grown in neurobasal medium containing B27 components and nerve growth factor (5 ng/mL) for 10 days. Immunohistochemistry showed that 95% of the cells were neurofilament positive. To determine cell viability thiazolyl blue tetrazolium bromide (MTT) was added to neuronal cultures at a final concentration of 50 µM for 3 h at 37 °C. The supernatant was removed, the formazan product solubilized in 200 μL of dimethyl sulfoxide, transferred to an immunoassay plate and absorbance read at 595 nm. Neuronal survival was calculated with reference to untreated neurons (100% survival).
Cell extracts: Treated neurones were washed 3 times with PBS and homogenized in a buffer containing 150 mM NaCl, 10 mM Tris-HCl, pH 7.4, 10 mM EDTA, 0.2% SDS, mixed protease inhibitors (4-(2-aminoethyl)benzenesulfonyl flouride, Aprotinin, Leupeptin, Bestain, Pepstatin A and E-46) and a phosphatase inhibitor cocktail (PP1, PP2A, microcystin LR, cantharidin and p-bromotetramisole) (Sigma, Poole, UK) at 106 cells/mL. Nuclei and cell debris was removed by centrifugation (300× g for 5 min).
Isolation of synaptosomes: Synaptosomes were prepared on a discontinuous Percoll gradient. Cortical neurons were homogenized at 4 °C in 1 mL of SED solution (0.32 M sucrose, 50 mM Tris-HCl, pH 7.2, 1 mM EDTA, and 1 mM dithiothreitol and centrifuged at 1000× g for 10 min). The supernatant was transferred to a 4-step gradient of 3, 7, 15, and 23% Percoll in SED solution and centrifuged at 16,000× g for 30 min at 4 °C. The synaptosome fractions were collected from the interface of the 15% and 23% Percoll steps, washed twice (16,000× g for 30 min at 4 °C) and suspended in extraction buffer (150 mM NaCl, 10 mM Tris-HCl pH 7.4, 10 mM EDTA, 0.2% SDS and mixed protease/phosphatase inhibitors).
Isolation of DRMs: These membranes were isolated by their insolubility in non-ionic detergents, as previously described [20]. Briefly, samples were homogenized in an ice-cold buffer containing 1% Triton X-100, 10 mM Tris-HCl, pH 7.2, 150 mM NaCl, 10 mM EDTA and mixed protease inhibitors and nuclei and large fragments were removed by centrifugation (300× g for 5 min at 4 °C). The supernatant was incubated on ice (4 °C) for 1 h and centrifuged (16,000× g for 30 min at 4 °C). The supernatant was reserved as the detergent soluble membrane (DSM), while the insoluble pellet was homogenized in an extraction buffer containing 10 mM Tris-HCL, pH 7.4, 150 mM NaCl, 10 mM EDTA, 0.5% Nonidet P-40, 0.5% sodium deoxycholate, 0.2% SDS and mixed protease inhibitors at 106 cells/mL, centrifuged (10 min at 16,000× g) and the soluble material was reserved as the DRM fraction.
Western Blotting: Samples were mixed with Laemmli buffer containing β-mercaptoethanol, heated to 95°C for 5 min and proteins were separated by electrophoresis on 15% polyacrylamide gels (PAGE). Proteins were transferred onto a Hybond-P PVDF membrane by semi-dry blotting. Membranes were blocked using 10% milk powder; synapsin-1 was detected with goat polyclonal (Santa Crux Biotech, London, UK), vesicle-associated membrane protein (VAMP)-1 with mAb 4H302 (Abcam, Cambridge, UK), rabbit polyclonal antibodies to caveolin (Upstate, Damstadt, Germany), cPLA2 with mAb CH-7 (Upstate) and PrPC by mAb 4F2 (Jaques Grassi, Parus, France); these were visualized using a combination of biotinylated anti-mouse/goat/rat/rabbit IgG (Sigma), extravidin-peroxidase and enhanced chemiluminescence.
Isolation of GPI anchored proteins: PrPC and Thy-1 were isolated from GT1 murine neuronal cells, as previously described [21]. Briefly, membranes were homogenized in a buffer containing 10 mM Tris-HCl pH 7.4, 100 mM NaCl, 10 mM EDTA, 0.5% Nonidet P-40, 0.5% sodium deoxycholate and mixed protease inhibitors (as above) and passed over affinity columns loaded with mAbs to PrPC (ICSM18) or anti-Thy-1 (Serotec, Kidlington, UK). PrPC and Thy-1 was eluted using glycine-HCl at pH 2.7, neutralized with 1 M Tris pH 7.4 and desalted (3 kDa filter, Sartorius). Proteins were digested with 100 units/mL bee venom phospholipase A2 (PLA2) (Sigma) to generate monoacylated PrPC and monoacylated Thy-1 (37 °C for 1 h) and isolated via reverse phase chromatography on C18 columns (Waters) using a gradient of propanol in water. PrP containing fractions were pooled, desalted, and concentrated. For high performance thin-layer chromatography (HPTLC) analysis, samples were dissolved in ethanol and separated on silica gel 60 plates using a mixture of chloroform/methanol/water (10/10/3 v/v/v). Plates were soaked in 0.1% polyisobutyl methacrylate in hexane, dried, and blocked with 5% milk powder. PrPC was detected with mAb 4F2. For bioassays, samples were solubilized in culture medium by sonication.
Synaptophysin ELISA: The amounts of synaptophysin in neurons were measured by ELISA [22]. Maxisorb immunoplates (Nunc, Roskilde, Denmark) were coated with a mouse monoclonal antibody (mAb) to synaptophysin MAB368 (Millipore, Damstadt, Germany). Samples were applied and bound, synaptophysin was detected using rabbit polyclonal anti-synaptophysin (Abcam) followed by a biotinylated anti-rabbit IgG, extravidin-alkaline phosphatase and 1 mg/mL 4-nitrophenol phosphate (Sigma). Absorbance was measured on a microplate reader at 405 nm and the synaptophysin content calculated. Samples were expressed as “units synaptophysin”, where 100 units was the amount of synaptophysin in 106 control neurons.
cPLA2 ELISA: The amounts of cPLA2 in extracts was measured by ELISA [21]. Maxisorb immunoplates were coated with 0.5 µg/mL of mouse mAb anti-cPLA2 (clone CH-7—Upstate) and blocked with 5% milk powder. Samples were incubated for 1 h and the amount of bound cPLA2 was detected using a goat polyclonal anti-cPLA2 (Santa-Cruz Biotech, London, UK) followed by biotinylated anti-goat IgG, extravidin-alkaline phosphatase and 1 mg/mL 4-nitrophenol phosphate. Absorbance was measured at 405 nm and the amount of cPLA2 protein expressed in units, 100 units = amount of cPLA2 in control preparations.
Activated cPLA2 ELISA: The activation of cPLA2 is accompanied by the phosphorylation of the 505 serine residue and can be measured by phospho-specific antibodies. Maxisorb immunoplates were coated with 100 nM mAb anti-cPLA2, clone CH-7 (Upstate) and blocked with 10% milk powder. Samples were incubated for 1 h and the amount of activated cPLA2 was detected using a rabbit polyclonal anti-phospho-cPLA2 (Cell Signaling Technology, Cambridge, UK), biotinylated anti-rabbit IgG, extravidin-alkaline phosphatase and 1 mg/mL 4-nitrophenyl phosphate. Absorbance was measured at 405 nm and the amounts of activated cPLA2 present were expressed as “units activated cPLA2”, where 100 units were defined as the amount of activated cPLA2 in control synaptosomes.
PrPC ELISA: The amount of PrPC in samples was determined by [10]. Maxisorb immunoplates were coated with mAb ICSM18 (Dr. Mourad Tayebi). Samples were added and bound PrP was detected with biotinylated mAb ICSM35 (Dr. Mourad Tayebi). Biotinylated mAb was detected using extravidin-alkaline phosphatase and 1 mg/mL 4-nitrophenyl phosphate. Absorbance was measured on a microplate reader at 405 nm and the amount of PrP in samples was calculated by reference to a standard curve of recombinant murine PrP (Prionics, London, UK).
Preparation of Aβ-containing medium: CHO cells stably transfected with a cDNA encoding APP751 (referred to as 7PA2 cells) were cultured in DMEM with 10% fetal calf serum as described [5]. Conditioned medium (CM) from these cells contains Aβ oligomers (7PA2-CM). CM from non-transfected CHO cells (CHO-CM) was used as controls. 7PA2-CM and CHO-CM were centrifuged at 100,000× g for 4 h at 4 °C to remove cell debris and then passed through a 50 kDa filter (Sartorius, Damstadt, Germany). 7PA2-CM contains Aβ monomers and low-n Aβ oligomers [5]. For immunoblot analysis, extracts were concentrated, mixed with an equal volume of 0.5% NP-40, 5 mM CHAPS, 50 mM Tris, pH 7.4 and separated by electrophoresis using Novex, Triz-glycine native running buffer (Life technologies, Paisley, UK). Proteins were transferred onto a PVDF membrane by semi-dry blotting and blocked using 10% milk powder. Aβ was detected by incubation with mAb 6E10 (Covance, Maidenhead, UK), biotinylated anti-mouse IgG, extravidin-peroxidase and enhanced chemiluminescence. The amounts of Aβ42 in preparations were determined by ELISA.
Immunodepletions: 7PA2-CM were incubated with 0.1 μg/mL mAb 4G8 (reactive with amino acids 17–24 of Aβ) or isotype controls (mock-depletion) and incubated at 4 °C on rollers for 24 h. Protein G microbeads were added (10 µL/mL) (Sigma) for 2 h and protein G bound-antibody complexes removed by centrifugation and filtration.
Sample preparation for end-specific ELISAs: To detach Aβ42 from cellular components that could occlude specific epitopes samples (50 µL) were mixed with 250 µL of 70% formic acid and sonicated. A 50 µL aliquot was added to 50 µL of 10M Tris-HCl with protease inhibitors (as above) and sonicated before addition to ELISA.
Aβ42 ELISA: Maxisorb immunoplates were coated with mAb 4G8 (epitope 17–24) (Covance). Plates were blocked with 5% milk powder and samples were applied. The detection antibody was an Aβ42 selective rabbit mAb BA3-9 (Covance) followed by biotinylated anti-rabbit IgG and extravidin alkaline phosphatase. Total Aβ was visualized by addition of 1mg/mL 4-nitrophenol phosphate solution and optical density was read in a spectrophotometer at 405 nm.
PrPC-Aβ ELISA: Maxisorb immunoplates were coated with 10 nM PrPC, monoacylated PrPC or monoacylated Thy-1 and blocked with 5% milk powder. Samples were added for 1 h and bound Aβ was detected with biotinylated mAb 4G8 (epitope 17–24 of Aβ) (Covance), followed by extravidin-alkaline phosphatase and 1 mg/mL 4-nitrophenol phosphate solution. Optical density was read in a spectrophotometer at 405 nm.
Peptides: Recombinant human αSN was obtained from Sigma. Peptides were thawed on the day of use and mixed in neurobasal medium containing B27. Mixtures were subjected to sonication and vigorous shaking (disruptor genie, full power for 10 min) before they were added to neurons.
Statistical Analysis: Comparison of treatment effects was carried out using Student’s paired t-tests.
3. Results and Discussion
Monoacylated PrPC is stable within neuronal membranes: Monoacylated PrPC eluted from C18 columns at lower concentrations of propanol than PrPC (Figure 1A). Western blots demonstrated that there was no obvious difference in the molecular weight of PrPC and monoacylated PrPC (Figure 1B), which is consistent with the loss of an acyl chain with a molecular mass of ~0.2 kDa. The loss of a hydrophobic acyl chain resulted in monoacylated PrPC migrating differently from PrPC in HPTLC (Figure 1C). Many GPI-anchored proteins bind to recipient cells [23] and both PrPC and monoacylated PrPC bound to Prnp(0/0) neurons in neurons in a dose-dependent manner (Figure 1D). Whereas PrPC was found within DRMs (lipid rafts), the monoacylated PrPC was found within DSMs (normal cell membrane) [21] (Figure 1E). In neurons from Prnp(0/0), mice PrPC had a half-life of less than 24 h in accordance with previous reports [24], whereas monoacylated PrPC remained in neurons far longer and had a half-life of greater than four days (Figure 1F).
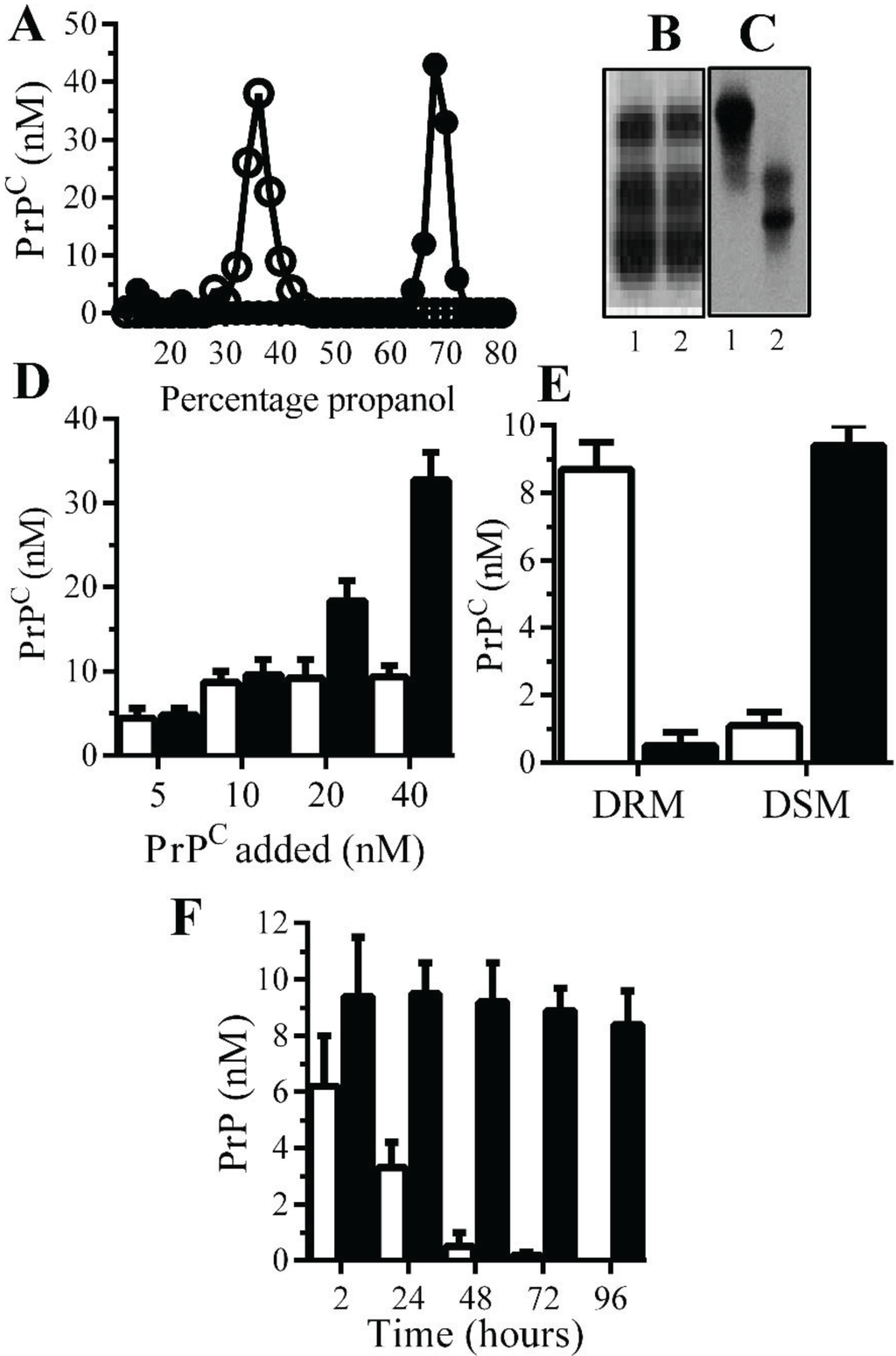
Figure 1.
Monoacylated PrPC is expressed in neurons: (A) The concentrations of PrPC (●) or monoacylated PrPC (○) in fractions eluted from C18 columns. Values are means of duplicates; PrPC (1) and monoacylated PrPC (2) separated by PAGE (B) or HPTLC (C). (D) The concentrations of PrPC in Prnp(0/0) neurons treated with PrPC (□) and monoacylated PrPC (■), as shown for 2 h. Values are means ± SD from triplicate experiments performed four times (n = 12). (E) The concentrations of PrPC (□) and monoacylated PrPC (■) in DRM (rafts) or DSMs in Prnp(0/0) neurons pulsed with 10 nM PrPC preparations for 2 h. Values are means ± SD from triplicate experiments performed four times (n = 12). (F) The concentrations of PrPC (□) and monoacylated PrPC (■) in Prnp(0/0) neurons at different time periods after being pulsed with 10 nM PrPC preparations. Values are means ± SD from triplicate experiments performed four times (n = 12).
Figure 1.
Monoacylated PrPC is expressed in neurons: (A) The concentrations of PrPC (●) or monoacylated PrPC (○) in fractions eluted from C18 columns. Values are means of duplicates; PrPC (1) and monoacylated PrPC (2) separated by PAGE (B) or HPTLC (C). (D) The concentrations of PrPC in Prnp(0/0) neurons treated with PrPC (□) and monoacylated PrPC (■), as shown for 2 h. Values are means ± SD from triplicate experiments performed four times (n = 12). (E) The concentrations of PrPC (□) and monoacylated PrPC (■) in DRM (rafts) or DSMs in Prnp(0/0) neurons pulsed with 10 nM PrPC preparations for 2 h. Values are means ± SD from triplicate experiments performed four times (n = 12). (F) The concentrations of PrPC (□) and monoacylated PrPC (■) in Prnp(0/0) neurons at different time periods after being pulsed with 10 nM PrPC preparations. Values are means ± SD from triplicate experiments performed four times (n = 12).
Natural Aβ binds to monoacylated PrPC: PrPC acts as a receptor for Aβ and mediated Aβ-induced synapse damage [11,16]. Since the presence of a GPI anchor affected the structure of some proteins [25], the binding of Aβ to monoacylated PrPC was examined. An immunoblot showed that 7PA2-CM contained several forms of Aβ that were not found in CHO-CM (Figure 2A). When immunoplates coated with 10 nM PrPC, 10 nM monoacylated PrPC or monoacylated Thy-1 PrPC were incubated with 7PA2-CM, Aβ bound to both PrPC and monoacylated PrPC without binding to monoacylated Thy-1 (Figure 2B).
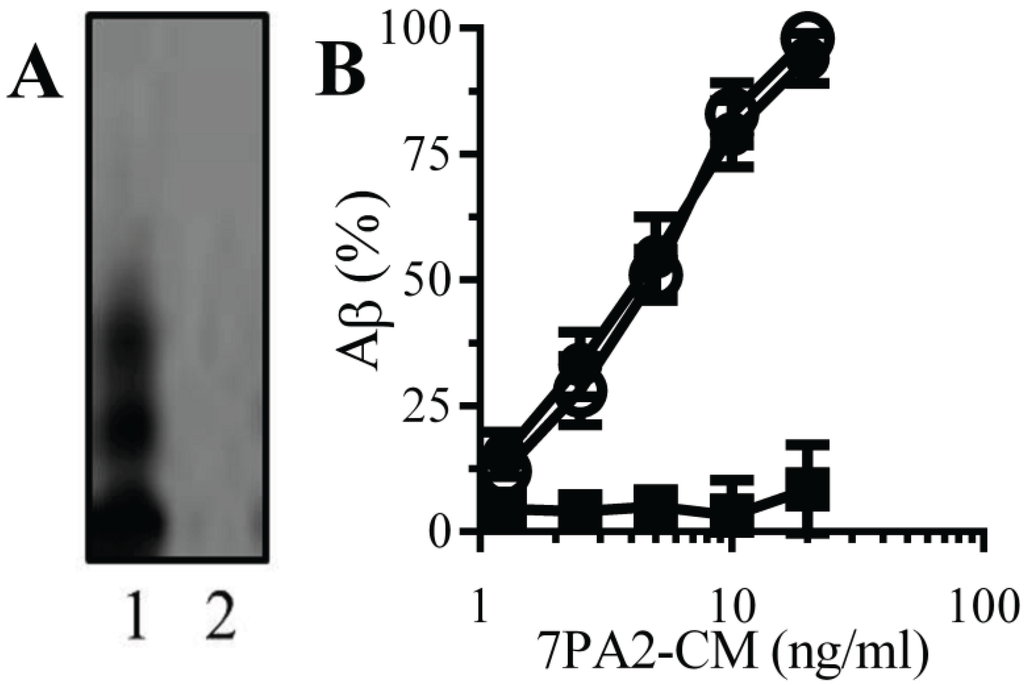
Figure 2.
Aβ binds to monoacylated PrPC: (A) Immunoblots showing forms of Aβ in 7PA2-CM (1) and CHO-CM (2). (B) The amounts of Aβ detected in immunoplates coated with 10 nM PrPC (●), 10 nM monoacylated PrPC (○) or 10 nM monoacylated Thy-1 (■) and incubated with 7PA2-CM as shown. Values are means ± SD from triplicate experiments performed 4 times (n = 12).
Figure 2.
Aβ binds to monoacylated PrPC: (A) Immunoblots showing forms of Aβ in 7PA2-CM (1) and CHO-CM (2). (B) The amounts of Aβ detected in immunoplates coated with 10 nM PrPC (●), 10 nM monoacylated PrPC (○) or 10 nM monoacylated Thy-1 (■) and incubated with 7PA2-CM as shown. Values are means ± SD from triplicate experiments performed 4 times (n = 12).
Aβ oligomers trigger synapse damage: Since the loss of synaptic proteins is a feature of AD that strongly correlates with cognitive decline [7,8] the amounts of synaptophysin in neurons incubated with Aβ was studied. 7PA2-CM, but not CHO-CM, reduced the synaptophysin content of neurons indicative of synapse damage (Figure 3A) [11]. Immunoblots showed that 7PA2-CM also caused the loss of synapsin-1 and VAMP-1 from cultured neurons but without affecting the amounts of caveolin (Figure 3B). The addition of CHO-CM to neurons did not significantly affect levels of synaptic proteins. These concentrations of 7PA2-CM did not significantly reduce cell viability as measured by the MTT method (98% ± 4% cell survival, compared to 100% ± 5%, p = 0.45, n = 9). The addition of 7PA2-CM that had been depleted of Aβ did not trigger the loss of synaptophysin from neurons (Figure 3C) indicating that Aβ was responsible for synapse damage.
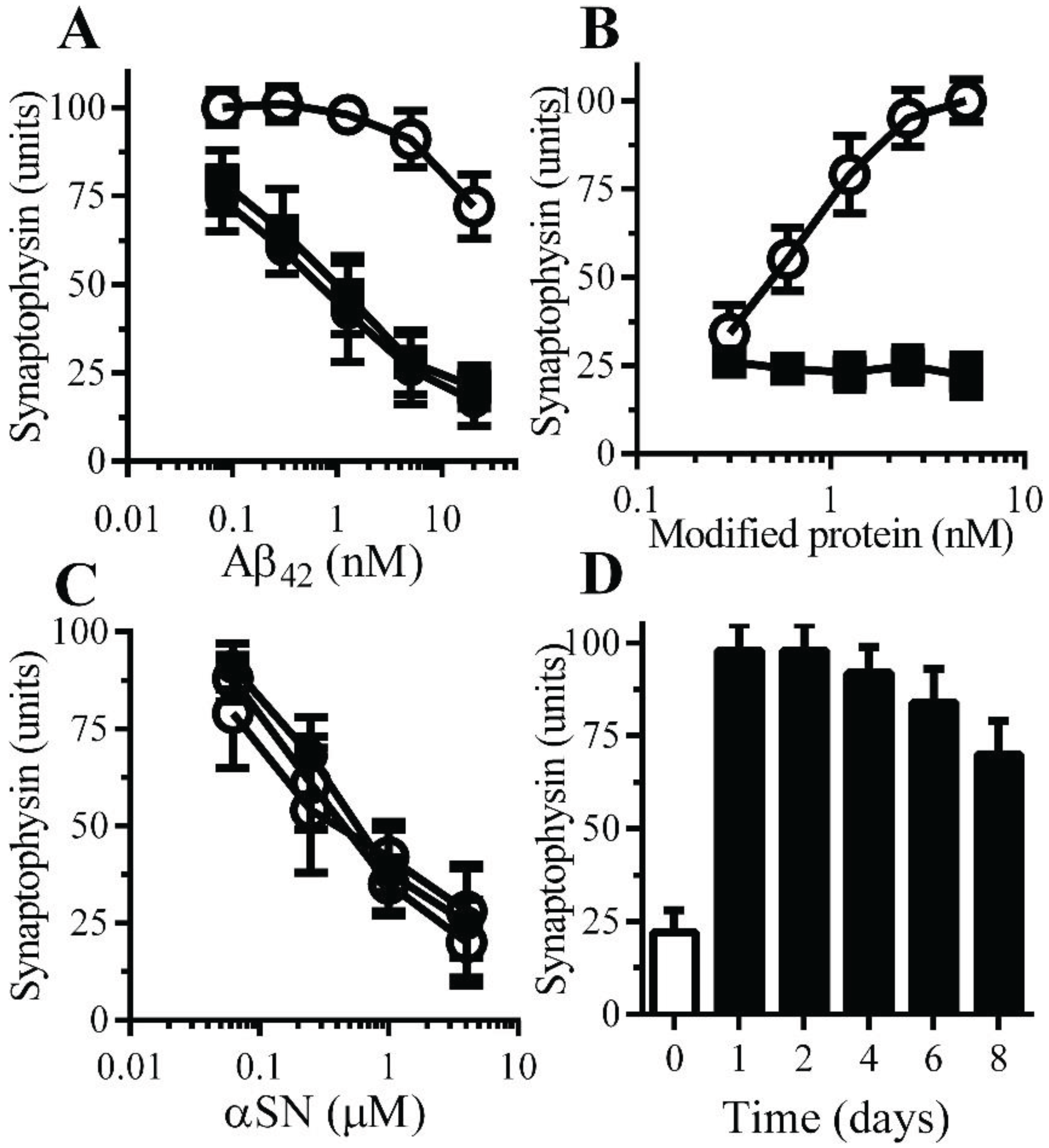
Monoacylated PrPC reduced Aβ-induced synapse damage: The polymorphic nature of Aβ aggregates indicates that there are disease-relevant conformational forms of Aβ, while other conformations are less toxic [26]. The possibility that it was mainly the non-toxic conformations of Aβ that bound to monoacylated PrPC was tested by examining the effects of monoacylated PrPC upon Aβ-induced synapse damage. The addition of either monoacylated PrPC or monoacylated Thy-1 did not cause synapse damage as determined by the loss of synaptophysin from neurons. However, pre-treatment of neurons with 10 nM monoacylated PrPC, but not with 10 nM monoacylated Thy-1, reduced the Aβ-induced synapse damage (Figure 4A). The presence of monoacylated PrPC protected neurons against Aβ-induced synapse damage in a dose-dependent manner (Figure 4B). The synapse damage in Parkinson’s disease (PD) and dementia with Lewy bodies is associated with the accumulation of α-synuclein (αSN) at synapses [27] and the addition of recombinant human αSN triggered synapse damage in neurons [28]. Pre-treatment of neurons with 10 nM monoacylated PrPC did not affect αSN-induced synapse damage (Figure 4C). To determine whether the protective effect of monoacylated PrPC was long lived, neurons were pulsed with 10 nM monoacylated PrPC for 1 h and 10 nM Aβ42 was added at time points thereafter. Neurons treated with monoacylated PrPC remained resistant to Aβ-induced synapse damage for eight days (Figure 4D).
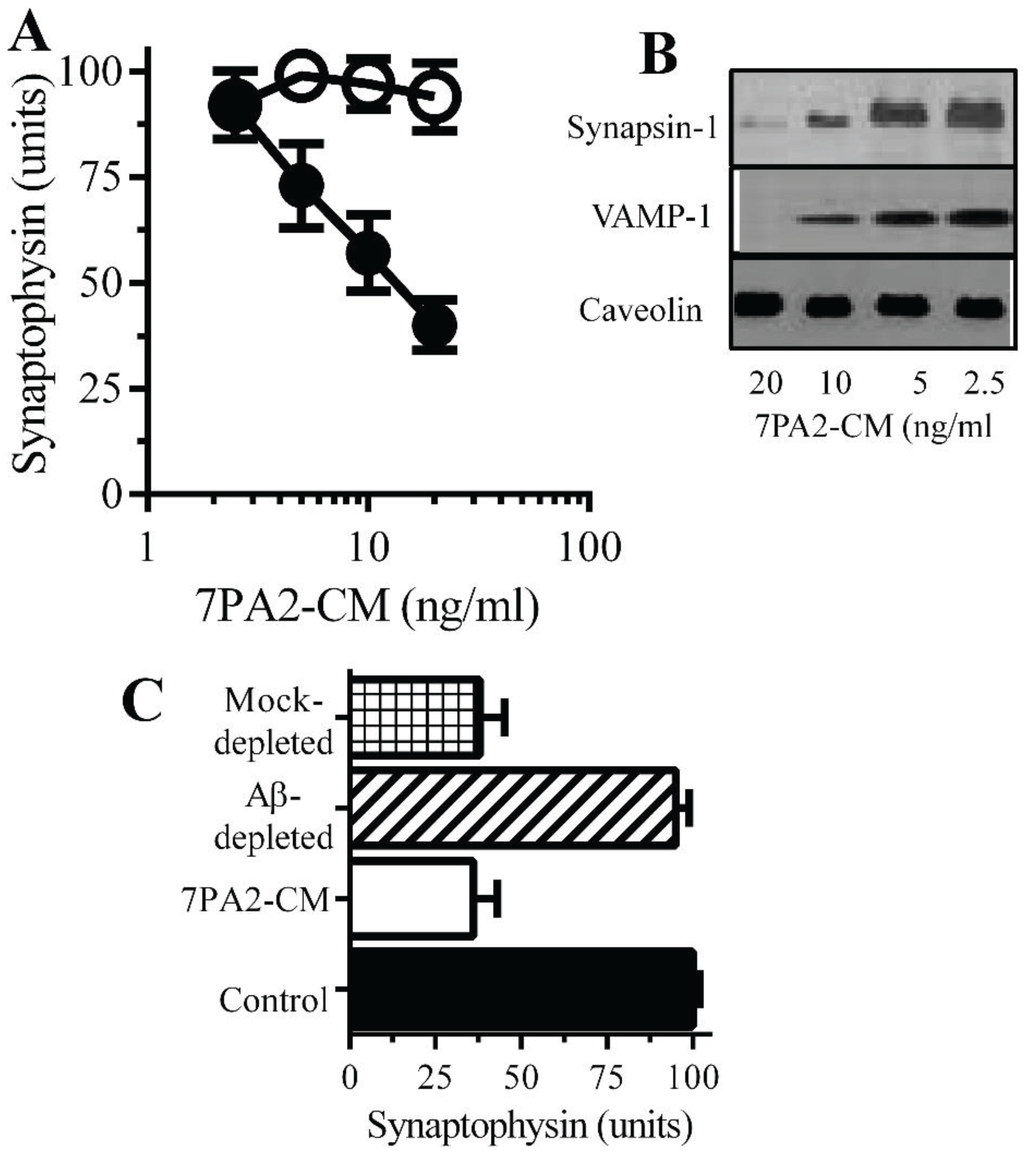
Figure 3.
Aβ oligomers cause synapse damage in neurons: (A) The amounts of synaptophysin in cultured neurons incubated with 7PA2-CM (●) or CHO-CM (○) for 24 h. Values are means ± SD from triplicate experiments performed 4 times (n = 12). (B) Immunoblots showing the amount of synapsin-1, VAMP-1 and caveolin in neurons incubated with 7PA2-CM for 24 h. (C) The amounts of synaptophysin in neurons incubated with control medium (■), 7PA2-CM (□), Aβ-depleted 7PA2-CM (striped bar) or mock-depleted 7PA2-CM (hatched bar) for 24 h. Values are means ± SD from triplicate experiments performed four times (n = 12).
Figure 3.
Aβ oligomers cause synapse damage in neurons: (A) The amounts of synaptophysin in cultured neurons incubated with 7PA2-CM (●) or CHO-CM (○) for 24 h. Values are means ± SD from triplicate experiments performed 4 times (n = 12). (B) Immunoblots showing the amount of synapsin-1, VAMP-1 and caveolin in neurons incubated with 7PA2-CM for 24 h. (C) The amounts of synaptophysin in neurons incubated with control medium (■), 7PA2-CM (□), Aβ-depleted 7PA2-CM (striped bar) or mock-depleted 7PA2-CM (hatched bar) for 24 h. Values are means ± SD from triplicate experiments performed four times (n = 12).
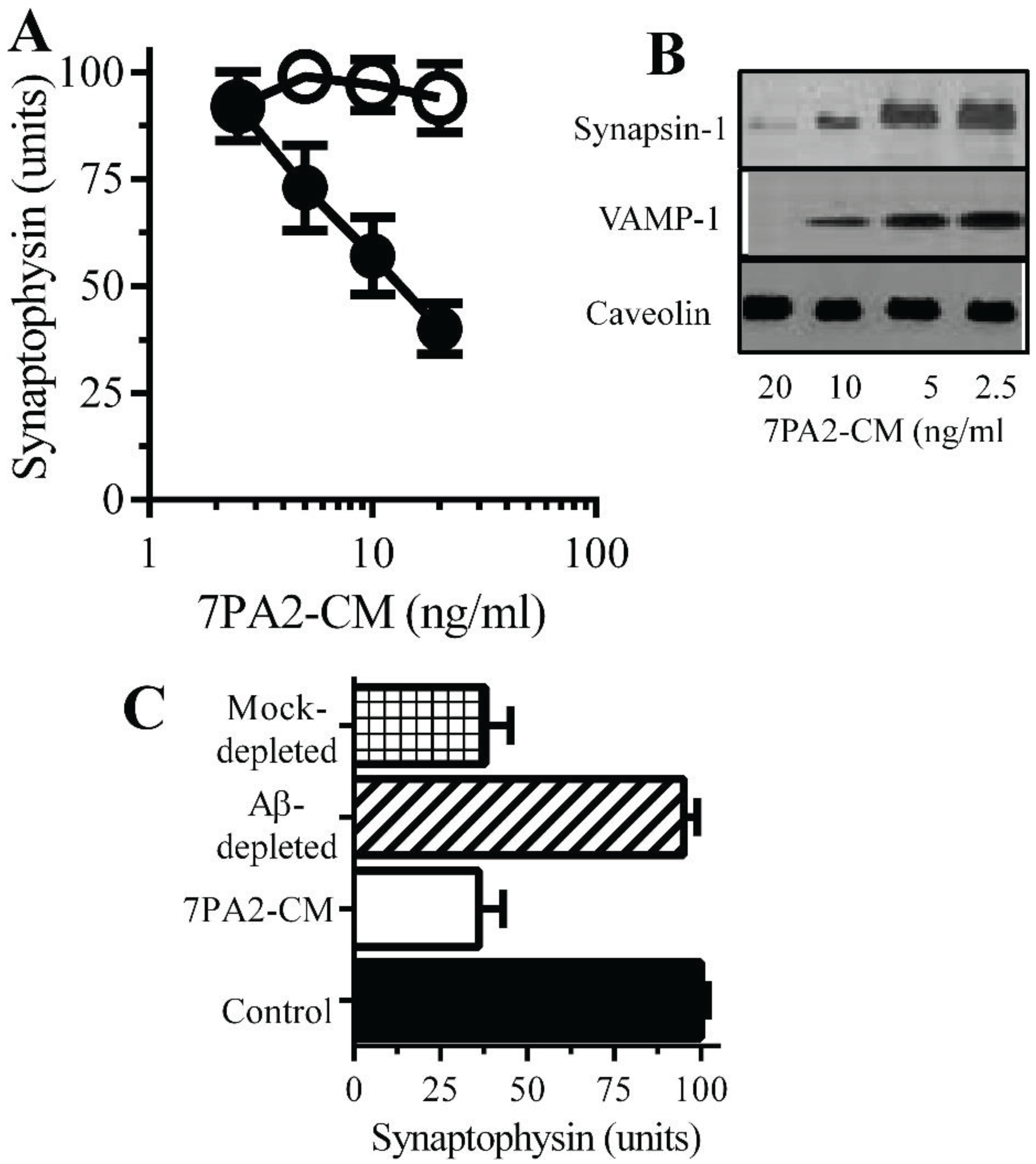
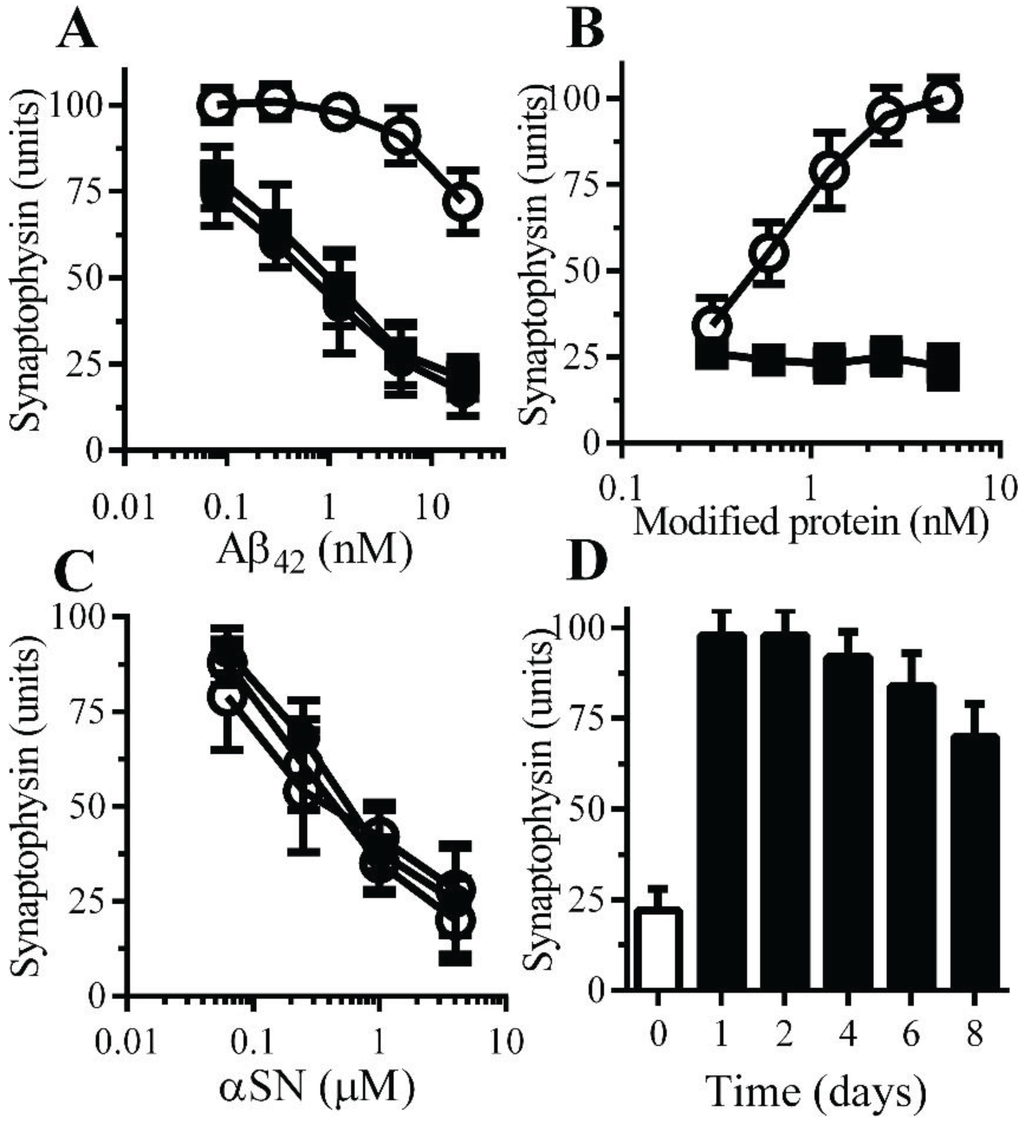
Figure 4.
Monoacylated PrPC protected neurons against Aβ-induced synapse damage: (A) The amounts of synaptophysin in neurons pre-treated with control medium (●), 10 nM monoacylated PrPC (○) or 10 nM monoacylated Thy-1 (■) and incubated with Aβ42. Values are means ± SD, from triplicate experiments performed four times, n = 12. (B) The amounts of synaptophysin in neurons pre-treated with monoacylated PrPC (○) or monoacylated Thy-1 (■) as shown and incubated with 10 nM Aβ42. Values are means ± SD, from triplicate experiments performed four times, n = 12; (C) The amounts of synaptophysin in neurons pre-treated with control medium (●), 10 nM monoacylated PrPC (○) or 10 nM monoacylated Thy-1 (■) and incubated with αSN. Values are means ± SD, from triplicate experiments performed four times, n = 12. (D) The amounts of synaptophysin in neurons pre-treated with control medium (□) or 10 nM monoacylated PrPC for different time periods as shown (■) and incubated with 10 nM Aβ42. Values are means ± SD, from triplicate experiments performed three times, n = 9.
Figure 4.
Monoacylated PrPC protected neurons against Aβ-induced synapse damage: (A) The amounts of synaptophysin in neurons pre-treated with control medium (●), 10 nM monoacylated PrPC (○) or 10 nM monoacylated Thy-1 (■) and incubated with Aβ42. Values are means ± SD, from triplicate experiments performed four times, n = 12. (B) The amounts of synaptophysin in neurons pre-treated with monoacylated PrPC (○) or monoacylated Thy-1 (■) as shown and incubated with 10 nM Aβ42. Values are means ± SD, from triplicate experiments performed four times, n = 12; (C) The amounts of synaptophysin in neurons pre-treated with control medium (●), 10 nM monoacylated PrPC (○) or 10 nM monoacylated Thy-1 (■) and incubated with αSN. Values are means ± SD, from triplicate experiments performed four times, n = 12. (D) The amounts of synaptophysin in neurons pre-treated with control medium (□) or 10 nM monoacylated PrPC for different time periods as shown (■) and incubated with 10 nM Aβ42. Values are means ± SD, from triplicate experiments performed three times, n = 9.
Monoacylated PrPC reduced the accumulation of Aβ42 within synapses: The presence of 10 nM monoacylated PrPC on neurons did not affect the binding of Aβ to neurons; 2 h after the addition of 10 nM Aβ42 there were no significant differences between control and treated neurons (9.2 nM Aβ42 ± 0.8 compared with 8.9 nM ± 1, n = 9, p = 0.4). Whereas the majority of Aβ42 added to control neurons was found within DRMs (rafts), consistent with reports [29], in neurons pre-treated with 10 nM monoacylated-PrPC significantly less Aβ42 was found within DRMs and more with the DSMs (Figure 5A). The targeting of Aβ42 to rafts may affect the subsequent trafficking of Aβ42, which accumulates within synapses in control neurons [11]. To determine whether Aβ42 had the same fate in treated neurons they were pulsed with 10 nM monoacylated PrPC or monoacylated Thy-1 and incubated with 10 nM of Aβ42 for 2 h and synaptosomes isolated. Pre-treatment with monoacylated PrPC significantly reduced the concentrations of Aβ42 found in synaptosomes compared to control neurons or neurons pre-treated with 10 nM monoacylated Thy-1 (Figure 5B).
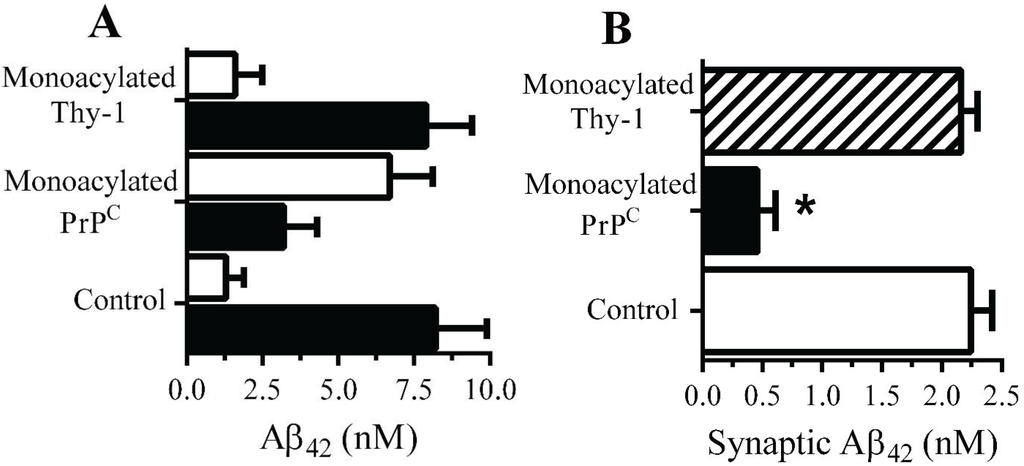
Figure 5.
Monoacylated PrPC reduced the accumulation of Aβ42 at synapses: (A) The concentrations of Aβ42 in DRMs (■) or DSMs (□) of neurons pre-treated with control medium, 10 nM monoacylated PrPC or 10 nM monoacylated Thy-1 as shown and incubated with 10 nM Aβ42. Values are means ± SD from triplicate experiments performed three times (n = 9). (B) The concentrations of Aβ42 in synaptosomes derived from neurons pre-treated with control medium (□), 10 nM monoacylated PrPC (■) or 10 nM monoacylated Thy-1 (striped bar) and incubated with 10 nM Aβ42 for 2 h. Values are means ± SD from triplicate experiment performed four times, n = 4.
Figure 5.
Monoacylated PrPC reduced the accumulation of Aβ42 at synapses: (A) The concentrations of Aβ42 in DRMs (■) or DSMs (□) of neurons pre-treated with control medium, 10 nM monoacylated PrPC or 10 nM monoacylated Thy-1 as shown and incubated with 10 nM Aβ42. Values are means ± SD from triplicate experiments performed three times (n = 9). (B) The concentrations of Aβ42 in synaptosomes derived from neurons pre-treated with control medium (□), 10 nM monoacylated PrPC (■) or 10 nM monoacylated Thy-1 (striped bar) and incubated with 10 nM Aβ42 for 2 h. Values are means ± SD from triplicate experiment performed four times, n = 4.
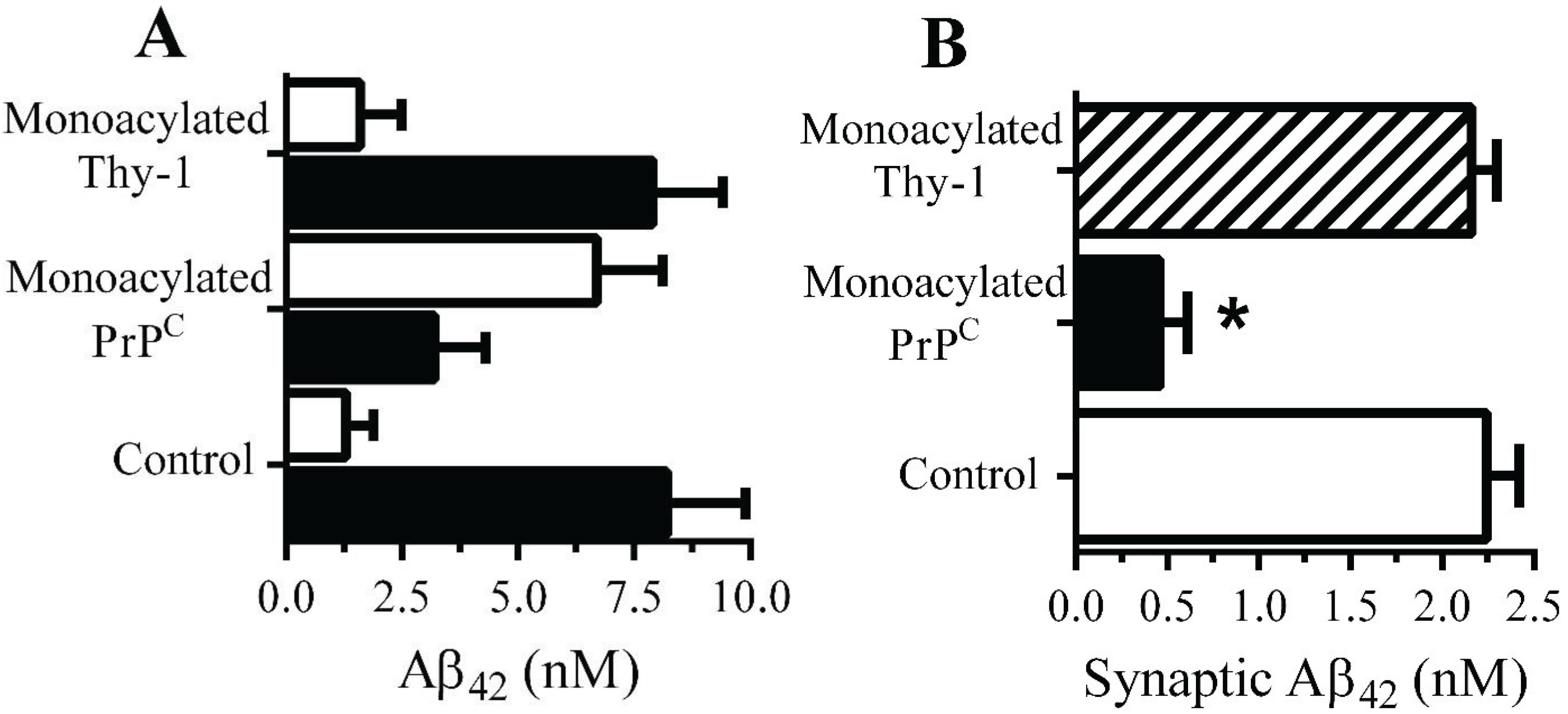
Monoacylated PrPC reduced Aβ-induced activation of cPLA2 in synapses: There is evidence that Aβ-induced aberrant activation of cell signaling pathways is involved in synapse damage. Since Aβ activates cPLA2[10,30], and pharmacological inhibition of PLA2 protected against Aβ-induced synapse damage [10], the effect of monoacylated PrPC on the activation of cPLA2 was examined. The amount of activated cPLA2 in synaptosomes was not affected by addition of 10 nM monoacylated PrPC (108 units activated cPLA2 ± 12 compared with 100 units ± 14, n = 9, p = 0.4) or monoacylated Thy-1 (104 units activated cPLA2 ± 12 compared with 100 units ± 14, n = 9, p = 0.6). However, pre-treatment of neurons with 10 nM monoacylated PrPC, but not 10 nM monoacylated Thy-1, significantly reduced the Aβ-induced activation of cPLA2 in synaptosomes (Figure 6A). In contrast, pre-treatment with 10 nM monoacylated PrPC did not affect αSN-induced activation of cPLA2 (Figure 6B). Activation of cPLA2 is associated with its translocation to specific membrane micro-domains by an N-terminal lipid-binding motif [31]. Sucrose density gradients showed that in synaptosomes, the addition of Aβ results in the migration of cPLA2 to DRMS [11]. In control synaptosomes, incubated with 1 nM Aβ42 approximately 50% of cPLA2 was found within DRMs (Figure 6C). In synaptosomes pre-treated with 10 nM monoacylated PrPC and incubated with 1 nM Aβ42, significantly less cPLA2 was found within DRMs. The addition of 10 nM monoacylated Thy-1 did not affect the Aβ-induced translocation of cPLA2 to DRMs. The aggregation of PrPC by Aβ oligomers results in the formation of a signaling complex containing cPLA2 [11]. To determine whether monoacylated PrPC interfered with the formation of these complexes synaptosomes were pre-treated with 10 nM monoacylated PrPC or 10 nM monoacylated Thy-1 and incubated with 10 nM mAb 4F2 for 1 h. The presence of monoacylated PrPC resulted in complexes that did not contain cPLA2 (Figure 6D).
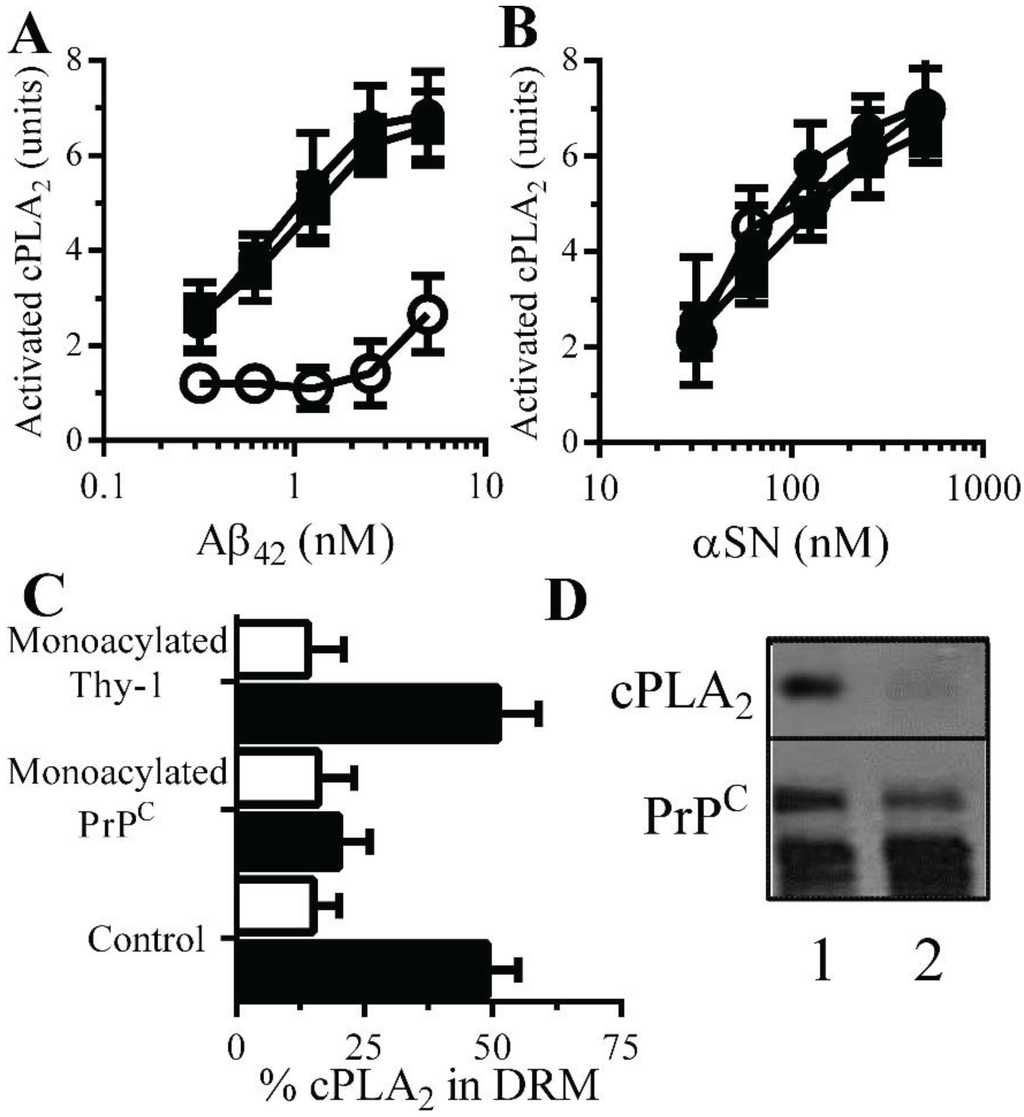
Figure 6.
Monoacylated PrPC reduced Aβ-induced activation of cPLA2 in synaptosomes: The amounts of activated cPLA2 in synaptosomes pre-treated with control medium (●), 10 nM monoacylated PrPC (○) or 10 nM monoacylated Thy-1 (■) and incubated with Aβ42 (A) or αSN (B) for 1 h. Values are means ± SD from triplicate experiments performed three times (n = 9). (C) The amounts of cPLA2 in DRMs (rafts) from synaptosomes pre-treated with control medium, 10 nM monoacylated PrPC or 10 nM monoacylated Thy-1 and incubated with control medium (□) or 10 nM Aβ42 (■) for 1 h. (D) Blot showing the amounts of cPLA2 and PrPC in immunoprecipitates from synaptosomes treated with control medium (1) or 10 nM monoacylated PrPC (2) and incubated with the PrPC-reactive mAb (4F2) for 1 h.
Figure 6.
Monoacylated PrPC reduced Aβ-induced activation of cPLA2 in synaptosomes: The amounts of activated cPLA2 in synaptosomes pre-treated with control medium (●), 10 nM monoacylated PrPC (○) or 10 nM monoacylated Thy-1 (■) and incubated with Aβ42 (A) or αSN (B) for 1 h. Values are means ± SD from triplicate experiments performed three times (n = 9). (C) The amounts of cPLA2 in DRMs (rafts) from synaptosomes pre-treated with control medium, 10 nM monoacylated PrPC or 10 nM monoacylated Thy-1 and incubated with control medium (□) or 10 nM Aβ42 (■) for 1 h. (D) Blot showing the amounts of cPLA2 and PrPC in immunoprecipitates from synaptosomes treated with control medium (1) or 10 nM monoacylated PrPC (2) and incubated with the PrPC-reactive mAb (4F2) for 1 h.
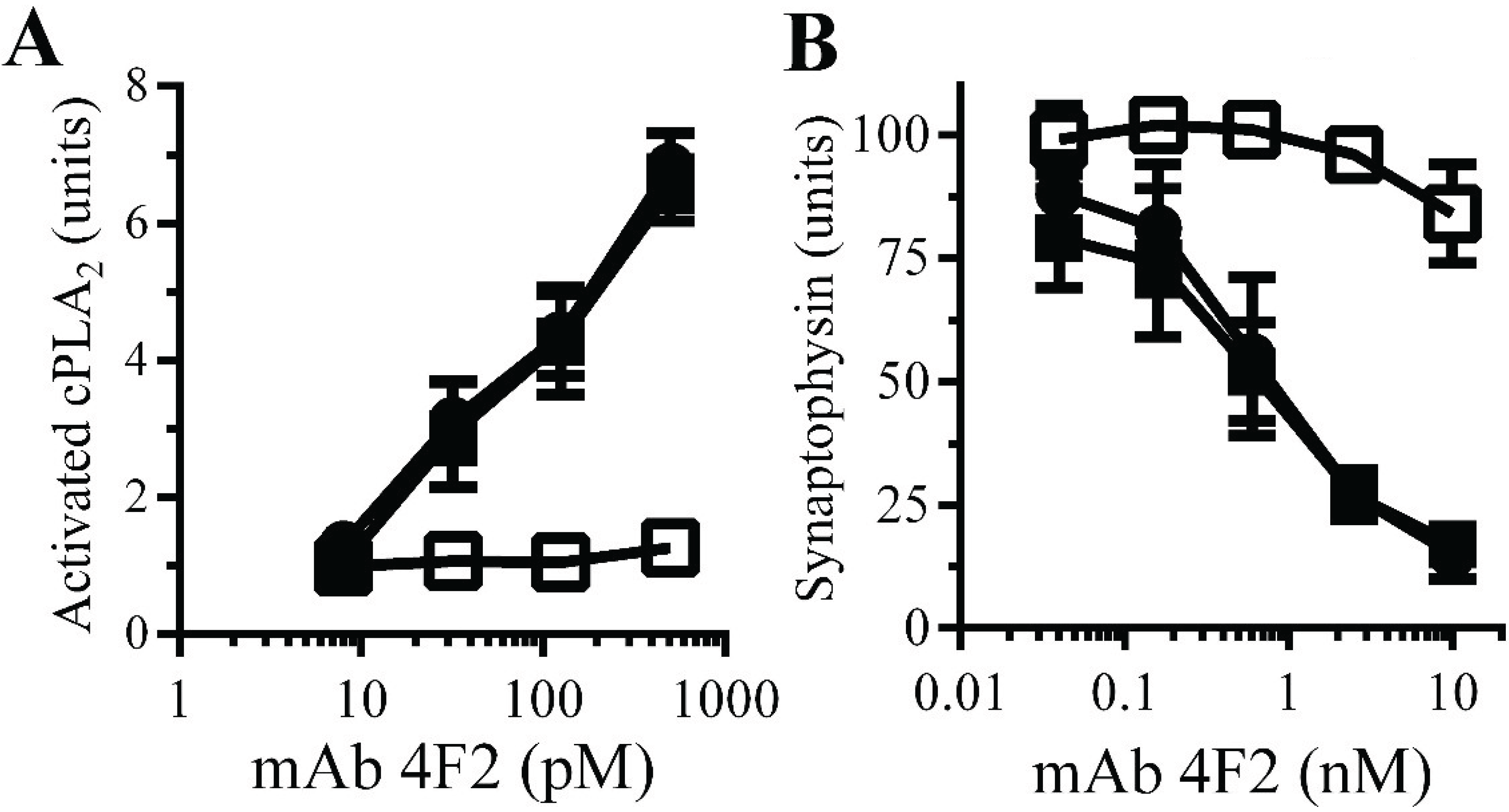
Monoacylated PrPC reduced synapse damage induced by the PrPC-reactive mAb 4F2: The observations that Aβ oligomers trigger neurodegeneration and that Aβ oligomers cross-link PrPC at synapses [11] suggested that aggregation of PrPC by Aβ oligomers. PrPC-reactive mAbs cause neurodegeneration in vivo [32] and trigger synapse damage in vitro [11]. The PrPC-reactive mAb 4F2 mimicked some of the effects of Aβ upon synaptosomes, including increasing the activation of cPLA2. Here we show that pre-treatment of synaptosomes with 10 nM monoacylated PrPC, but not monoacylated Thy-1, significantly reduced the mAb 4F2-induced activation of cPLA2 (Figure 7A). In addition, pre-treatment of neurons with 10 nM monoacylated PrPC, but not 10 nM monoacylated Thy-1, significantly reduced mAb 4F2-induced synapse damage (Figure 7B).
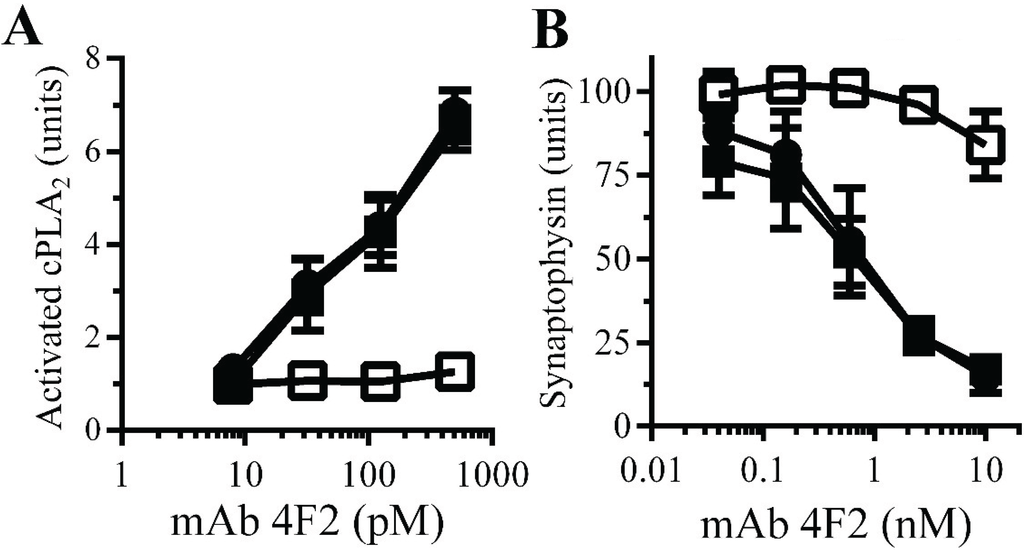
Figure 7.
Monoacylated PrPC reduced mAb 4F2-induced synapse damage: (A) The amounts of activated cPLA2 in synaptosomes pre-treated with control medium (●), 10 nM monoacylated PrPC (□) or 10 nM monoacylated Thy-1 (■) and incubated with mAb 4F2. Values are means ± SD from triplicate experiments performed three times (n = 9). (B) The amounts of synaptophysin in neurons pre-treated with control medium (●), 10 nM monoacylated PrPC (□) or 10 nM monoacylated Thy-1 (■), incubated with mAb 4F2. Values are means ± SD from triplicate experiments performed three times, n = 9.
Figure 7.
Monoacylated PrPC reduced mAb 4F2-induced synapse damage: (A) The amounts of activated cPLA2 in synaptosomes pre-treated with control medium (●), 10 nM monoacylated PrPC (□) or 10 nM monoacylated Thy-1 (■) and incubated with mAb 4F2. Values are means ± SD from triplicate experiments performed three times (n = 9). (B) The amounts of synaptophysin in neurons pre-treated with control medium (●), 10 nM monoacylated PrPC (□) or 10 nM monoacylated Thy-1 (■), incubated with mAb 4F2. Values are means ± SD from triplicate experiments performed three times, n = 9.
4. Discussion
PrPC acts as a receptor for the Aβ oligomers that cause cognitive impairment in a model of AD [16] and mediated Aβ-induced synapse damage in cultured neurons [11]. The key finding of this study was that the presence of monoacylated PrPC significantly reduced Aβ-induced synapse damage. The protective effects of monoacylated PrPC were related to two interrelated activities, the disruption of Aβ-induced cell signaling and the sequestration of Aβ outside lipid rafts.
Although PrPC is associated with several signaling pathways, it lacks a transmembrane component. The GPI attached to PrPC targets the protein to lipid rafts [33] in which signaling complexes, often called signalosomes, assemble [34]. PrPC is thought to act as a “scaffold protein” which organizes the composition and function of signalosomes. Observations that Aβ is found within lipid rafts [29] and that Aβ-induced synapse damage is sensitive to raft disruption [35] suggest that the events leading to synapse degeneration are initiated from within lipid rafts. Consistent with this theory, aggregation of PrPC by Aβ oligomers induced activation of cPLA2 and led to synapse degeneration [11].
We demonstrate that the properties of monoacylated PrPC were different from those of PrPC. Perhaps the key observation was that monoacylated PrPC was not targeted to membrane rafts [21]. PrPC is a recycling protein that like many raft-associated proteins traffics to and from the plasma membrane [36], whereas monoacylated PrPC was found within the normal cell membrane. While the loss of an acyl chain from PrPC affected membrane targeting it did not affect the binding of soluble Aβ suggesting that the protein structure was not altered. Therefore it was not surprising to find that in neurons decorated with monoacylated PrPC a significant percentage of Aβ42 was found outside rafts consistent with the hypothesis that monoacylated PrPC sequestered Aβ42 into non-signaling membrane domains. The targeting of proteins to rafts also affects the trafficking of proteins [37] and monoacylated PrPC reduced the accumulation of Aβ at synapses. These results are consistent with the hypothesis that monoacylated PrPC acts as a molecular sponge which adsorbs Aβ in specific cell compartments, in the process preventing Aβ binding to native PrPC and triggering the raft-dependent signaling that leads to synapse damage.
The key finding, that neurons decorated with monoacylated PrPC were less susceptible to Aβ-induced synapse damage, was stimulus specific; these neurons were not protected against αSN-induced synapse damage. The protective effect of monoacylated PrPC was long lasting and was related to the long half-life of monoacylated PrPC in neurons. Although neurons decorated with monoacylated PrPC bound similar amounts of Aβ as control neurons the Aβ did not cause synapse damage indicating that the presence of Aβ alone does not cause synapse damage and that synapse damage is mediated via specific mechanisms.
PrPC has been associated with the activation of cPLA2, which occurs within rafts [11,38]. Some clues about how the binding of Aβ to PrPC activates cPLA2 can be gathered from the prion literature, where aggregation of PrPC caused synapse damage in neurons similar to that seen with aggregates of PrPSc [19].The observations that Aβ oligomers that can cross-link PrPC are toxic, but Aβ monomers are not, indicate that the clustering of PrPC is key to cell signaling and link prion and Alzheimer’s diseases to a common pathway leading to neurodegeneration. In this regard, it is of interest that in a transgenic mouse model of AD containing APPPS1+ Prnp°/° and crossed with mice producing anchorless PrPC [39] the APPPS1-related suppression of LTP was inhibited; an effect that was independent of any effects upon the production of Aβ42 [40]. The oligomerization of GPI-anchored proteins stimulates raft formation [41] and the clustering of specific GPI anchors leads to activation of cPLA2 and synapse damage [19]. In this study, the presence of monoacylated PrPC reduced Aβ-induced activation of cPLA2 in synapses as complexes formed by the aggregation of monoacylated PrPC did not contain cPLA2.
5. Conclusions
We report that monoacylated PrPC bound natural Aβ and that neurons decorated with monoacylated PrPC were protected against Aβ-induced synapse damage. These studies support the hypothesis that the GPI anchor attached to PrPC plays a role in mediating the effects of Aβ on neurons. The protective effect of monoacylated PrPC was two fold: firstly, monoacylated PrPC sequestered Aβ into cellular compartments not associated with cell signaling; and secondly, we demonstrate that monoacylated PrPC reduced the Aβ-induced translocation and subsequent activation of cPLA2 that leads to synapse damage. Cell signaling by GPI-anchored proteins is a poorly understood process; proteins with modified GPI anchors may help explain this process.
Acknowledgments
This work was supported by the European Commission FP6 “Neuroprion”—Network of Excellence and the Royal Veterinary College, Bioveterinary Science undergraduate research project funds. We also thank Amijee and Treherne (Senexis) for supplying 7PA2-CM and CHO-CM.
Author Contributions
Ewan West, Craig Osborne, William Nolan and Clive Bate all contributed to the design and performance of experiments and analysis of results. Clive Bate was responsible for writing the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [PubMed]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Ab1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [PubMed]
- Klein, W.L.; Krafft, G.A.; Finch, C.E. Targeting small Ab oligomers: The solution to an Alzheimer’s disease conundrum? Trends Neurosci. 2001, 24, 219–224. [Google Scholar] [CrossRef]
- Podlisny, M.B.; Ostaszewski, B.L.; Squazzo, S.L.; Koo, E.H.; Rydell, R.E.; Teplow, D.B.; Selkoe, D.J. Aggregation of secreted amyloid b-protein into sodium dodecyl sulfate-stable oligomers in cell culture. J. Biol. Chem. 1995, 270, 9564–9570. [Google Scholar] [PubMed]
- Walsh, D.M.; Selkoe, D.J. Ab oligomers—A decade of discovery. J. Neurochem. 2007, 101, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Terry, R.D.; Alford, M.; deTeresa, R.; Hansen, L.A. Cortical and subcortical patterns of synaptophysinlike immunoreactivity in Alzheimer’s disease. Am. J. Path. 1991, 138, 235–246. [Google Scholar] [PubMed]
- Reddy, P.H.; Mani, G.; Park, B.S.; Jacques, J.; Murdoch, G.; Whetsell, W., Jr.; Kaye, J.; Manczak, M. Differential loss of synaptic proteins in Alzheimer’s disease: Implications for synaptic dysfunction. J. Alzheimers Dis. 2005, 7, 103–117. [Google Scholar] [PubMed]
- Daly, C.; Sugimori, M.; Moreira, J.E.; Ziff, E.B.; Llinas, R. Synaptophysin regulates clathrin—Independent endocytosis of synaptic vesicles. Proc. Natl. Acad. Sci. USA 2000, 97, 6120–6125. [Google Scholar] [CrossRef] [PubMed]
- Bate, C.; Tayebi, M.; Williams, A. Phospholipase A2 inhibitors protect against prion and Ab mediated synapse degeneration. Mol. Neurodegener. 2010. [Google Scholar] [CrossRef]
- Bate, C.; Williams, A. Amyloid-b-induced synapse damage is mediated via cross-linkage of the cellular prion protein. J. Biol. Chem. 2011, 286, 37955–37963. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, A.; Yuan, M.; Zhang, Z.; Paganetti, P.A.; Sturchler-Pierrat, C.; Staufenbiel, M.; Mautino, J.; Vigo, F.S.; Sommer, B.; Yankner, B.A. Amyloid beta interacts with the amyloid precursor protein: A potential toxic mechanism in Alzheimer’s disease. Nat. Neurosci. 2000, 3, 460–464. [Google Scholar] [PubMed]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Knowles, J.K.; Rajadas, J.; Nguyen, T.V.; Yang, T.; LeMieux, M.C.; vander Griend, L.; Ishikawa, C.; Massa, S.M.; Wyss-Coray, T.; Longo, F.M. The p75 neurotrophin receptor promotes amyloid-β1–42-induced neuritic dystrophy in vitro and in vivo. J. Neurosci. 2009, 29, 10627–10637. [Google Scholar] [CrossRef] [PubMed]
- Renner, M.; Lacor, P.N.; Velasco, P.T.; Xu, J.; Contractor, A.; Klein, W.L.; Triller, A. Deleterious effects of amyloid β oligomers acting as an extracellular scaffold for mGluR5. Neuron 2010, 66, 739–754. [Google Scholar] [CrossRef] [PubMed]
- Lauren, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Herms, J.; Tings, T.; Gall, S.; Madlung, A.; Giese, A.; Siebert, H.; Schurmann, P.; Windl, O.; Brose, N.; Kretzschmar, H. Evidence of presynaptic location and function of the prion protein. J. Neurosci. 1999, 19, 8866–8875. [Google Scholar] [PubMed]
- Stahl, N.; Baldwin, M.A.; Hecker, R.; Pan, K.M.; Burlingame, A.L.; Prusiner, S.B. Glycosylinositol phospholipid anchors of the scrapie and cellular prion proteins contain sialic acid. Biochemistry 1992, 31, 5043–5053. [Google Scholar] [CrossRef] [PubMed]
- Bate, C.; Williams, A. Neurodegeneration induced by the clustering of sialylated glycosylphosphatidylinositols of prion proteins. J. Biol. Chem. 2012, 287, 7935–7944. [Google Scholar] [CrossRef] [PubMed]
- London, E.; Brown, D.A. Insolubility of lipids in Triton X-100: Physical origin and relationship to sphingolipid/cholesterol membrane domains (rafts). Biochim. Biophys. Acta 2000, 1508, 182–195. [Google Scholar] [CrossRef]
- Bate, C.; Williams, A. Monoacylated cellular prion protein modifies cell membranes, inhibits cell signaling and reduces prion formation. J. Biol. Chem. 2011, 286, 8752–8758. [Google Scholar] [CrossRef] [PubMed]
- Lipton, A.M.; Cullum, C.M.; Satumtira, S.; Sontag, E.; Hynan, L.S.; White, C.L., Ш; Bigio, E.H. Contribution of asymmetric synapse loss to lateralizing clinical deficits in frontotemporal dementias. Arch. Neurol. 2001, 58, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Li, R.; Pan, T.; Liu, D.; Petersen, R.B.; Wong, B.S.; Gambetti, P.; Sy, M.S. Intercellular transfer of the cellular prion protein. J. Biol. Chem. 2002, 277, 47671–47678. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Race, R.E.; Ernst, D.; Buchmeier, M.J.; Chesebro, B. Prion protein biosynthesis in scrapie-infected and uninfected neuroblastoma cells. J. Virol. 1989, 63, 175–181. [Google Scholar] [PubMed]
- Butikofer, P.; Malherbe, T.; Boschung, M.; Roditi, I. GPI-anchored proteins: Now you see ’em, now you don’t. FASEB J. 2001, 15, 545–548. [Google Scholar] [CrossRef] [PubMed]
- Glabe, C.G. Structural Classification of Toxic Amyloid Oligomers. J. Biol. Chem. 2008, 283, 29639–29643. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.L.; Schulz-Schaeffer, W.J. Presynaptic a-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J. Neurosci. 2007, 27, 1405–1410. [Google Scholar] [CrossRef] [PubMed]
- Bate, C.; Gentleman, S.; Williams, A. α-Synuclein induced synapse damage is enhanced by amyloid-β1–42. Mol. Neurodegener. 2010. [Google Scholar] [CrossRef] [PubMed]
- Oshima, N.; Morishima-Kawashima, M.; Yamaguchi, H.; Yoshimura, M.; Sugihara, S.; Khan, K.; Games, D.; Schenk, D.; Ihara, Y. Accumulation of amyloid b-protein in the low-density membrane domain accurately reflects the extent of β-amyloid deposition in the brain. Am. J. Pathol. 2001, 158, 2209–2218. [Google Scholar] [CrossRef]
- Shelat, P.B.; Chalimoniuk, M.; Wang, J.H.; Strosznajder, J.B.; Lee, J.C.; Sun, A.Y.; Simonyi, A.; Sun, G.Y. Amyloid beta peptide and NMDA induce ROS from NADPH oxidase and AA release from cytosolic phospholipase A2 in cortical neurons. J. Neurochem. 2008, 106, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Nalefski, E.A.; Sultzman, L.A.; Martin, D.M.; Kriz, R.W.; Towler, P.S.; Knopf, J.L.; Clark, J.D. Delineation of two functionally distinct domains of cytosolic phospholipase A2, a regulatory Ca2+-dependent lipid-binding domain and a Ca2+-independent catalytic domain. J. Biol. Chem. 1994, 269, 18239–18249. [Google Scholar] [PubMed]
- Solforosi, L.; Criado, J.R.; McGavern, D.B.; Wirz, S.; Sanchez-Alavez, M.; Sugama, S.; DeGiorgio, L.A.; Volpe, B.T.; Wiseman, E.; Abalos, G.; et al. Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science 2004, 303, 1514–1516. [Google Scholar] [CrossRef] [PubMed]
- Taraboulos, A.; Scott, M.; Semenov, A.; Avrahami, D.; Laszlo, L.; Prusiner, S.B.; Avraham, D. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J. Cell Biol. 1995, 129, 121–132. [Google Scholar] [CrossRef]
- Pike, L.J. Lipid rafts: Heterogeneity on the high seas. Biochem. J. 2004, 378, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Bate, C.; Williams, A. Squalestatin protects neurons and reduces the activation of cytoplasmic phospholipase A2 by Aβ1–42. Neuropharmacology 2007, 53, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Shyng, S.L.; Huber, M.T.; Harris, D.A. A prion protein cycles between the cell surface and an endocytic compartment in cultured neuroblastoma cells. J. Biol. Chem. 1993, 268, 15922–15928. [Google Scholar] [PubMed]
- Nichols, B.J.; Kenworthy, A.K.; Polishchuk, R.S.; Lodge, R.; Roberts, T.H.; Hirschberg, K.; Phair, R.D.; Lippincott-Schwartz, J. Rapid cycling of lipid raft markers between the cell surface and Golgi complex. J. Cell Biol. 2001, 153, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Gaudreault, S.B.; Chabot, C.; Gratton, J.P.; Poirier, J. The caveolin scaffolding domain modifies 2-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor binding properties by inhibiting phospholipase A2 activity. J. Biol. Chem. 2004, 279, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Chesebro, B.; Trifilo, M.; Race, R.; Meade-White, K.; Teng, C.; LaCasse, R.; Raymond, L.; Favara, C.; Baron, G.; Priola, S.; et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 2005, 308, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- Calella, A.M.; Farinelli, M.; Nuvolone, M.; Mirante, O.; Moos, R.; Falsig, J.; Mansuy, I.M.; Aguzzi, A. Prion protein and Aβ-related synaptic toxicity impairment. EMBO Mol. Med. 2010, 2, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).